

Abstract
Heterozygous mutations in FOXP2, which encodes a forkhead transcription factor, have been shown to cause developmental verbal dyspraxia and language impairment. FOXP2 and its closest homolog, FOXP1, are coexpressed in brain regions that are important for language and cooperatively regulate developmental processes, raising the possibility that FOXP1 may also be involved in developmental conditions that are associated with language impairment. In order to explore this possibility, we searched for mutations in FOXP1 in patients with intellectual disability (ID; mental retardation) and/or autism spectrum disorders (ASD). We first performed array-based genomic hybridization on sporadic nonsyndromic ID (NSID) (n = 30) or ASD (n = 80) cases. We identified a de novo intragenic deletion encompassing exons 4–14 of FOXP1 in a patient with NSID and autistic features. In addition, sequencing of all coding exons of FOXP1 in sporadic NSID (n = 110) or ASD (n = 135) cases, as well as in 570 controls, revealed the presence of a de novo nonsense mutation (c.1573C>T [p.R525X]) in the conserved forkhead DNA-binding domain in a patient with NSID and autism. Luciferase reporter assays showed that the p.R525X alteration disrupts the activity of the protein. Formal assessments revealed that both patients with de novo mutations in FOXP1 also show severe language impairment, mood lability with physical aggressiveness, and specific obsessions and compulsions. In conclusion, both FOXP1 and FOXP2 are associated with language impairment, but decrease of the former has a more global impact on brain development than that of the latter.
Main Text
Developmental language disorders represent a heterogeneous group of conditions that are frequent but poorly understood. Although these disorders show high heritability, very little is known about the causative genes. FOXP2 (MIM 605317) represents an important entry point into the molecular basis of developmental language disorders.1,2 Patients heterozygous for mutations in FOXP2 show verbal dyspraxia (MIM 602081), which is characterized by impaired coordinated mouth movements that are required for speech, as well as variable impairment of expressive and receptive language, without consistent nonverbal cognitive deficits. Recent studies suggest that FOXP2 is also required for vocal learning in birds and that it might have played a role during the evolution of human language.3–5
FOXP2 belongs to a subgroup of the FOX family of winged-helix/forkhead transcription factors, which also includes FOXP1 (MIM 605515), FOXP3 (MIM 300292), and FOXP4 (MIM 608924).6 Several observations suggest that FOXP2 and its closest relative, FOXP1, may regulate common processes. For instance, FOXP1 and FOXP2 are expressed in distinct but also in overlapping regions of the developing bird, mouse, and human brain, including areas associated with the production and processing of vocalization and language.7–10 FOXP proteins act as transcriptional repressors by forming homo- or heterodimers.9,11,12 Interestingly, FOXP1 and FOXP2 can physically interact in vitro, can repress the transcription of common targets in vivo by occupying the same binding sites, and cooperatively regulate lung and esophageal development in mice.11,13
Because FOXP1 and FOXP2 cooperate to control developmental processes, it was hypothesized that mutations in FOXP1 could likewise be associated with language impairment.10,14 However, sequencing of FOXP1 in a cohort of individuals with developmental verbal dyspraxia did not reveal any pathogenic mutations.14 Another possibility is that FOXP1 disruption causes other developmental conditions that are associated with language impairment, such as intellectual disability (ID; mental retardation) and autism spectrum disorders (ASD [MIM 209850]).15 In order to explore this possibility, we performed mutation analyses of FOXP1 in patients with ID and/or ASD. Because FOXP2 heterozygous mutations are sufficient to disrupt language development, we decided, likewise, to focus our attention on heterozygous mutations, which in the case of ID are more likely to arise de novo than to be transmitted. By analogy to the FOXP2 paradigm, we also restricted our analysis to patients without specific morphological abnormalities (referred to herein as having the nonsyndromic form of ID and ASD).
We studied sporadic cases with nonsyndromic ID (NSID), ASD, or both NSID and ASD, most of which were of French Canadian origin. We chose to study sporadic cases to increase the likelihood of identifying de novo mutations. The NSID cases were selected with the use of previously described criteria.16 The sporadic ASD patients were diagnosed according to DSM-IV criteria and were selected on the basis of a positive Autism Diagnostic Interview-Revised (ADI-R) and/or Autism Diagnostic Observation Schedule-Generic (ADOS-G). We used 570 healthy individuals as controls, including 285 French Canadians and 285 European individuals who were evaluated and found not to have cognitive dysfunction, neuropsychiatric symptoms, or family history of neuropsychiatric problems, as detailed elsewhere.17 Blood samples were collected from all members of these cohorts and from their parents, with informed consent and after approval of the study by institutional ethics committees. Genomic DNA was extracted from blood samples with the Puregene kit (Gentra). Paternity and maternity of each NSID and ASD patient proband trio were confirmed with the use of six informative microsatellite markers.
We initially determined whether FOXP1 is affected by copy number changes in 80 individuals from the sporadic ASD cohort (including 27 with documented ID), using Genome-Wide Human Affymetrix 5.0 SNP arrays, and in 30 individuals from the sporadic NSID cohort, using Affymetrix 6.0 SNP arrays. Both parents of each case were also studied with these arrays. We identified a de novo intragenic FOXP1 deletion in an NSID female patient (R0031608) (patient A). This ∼390 kb deletion (genomic region on chromosome 3:71114875–71504640; hg18) encompasses exons 4–14 of the longest isoform of FOXP1 (FOXP1a; Refseq no. NM_032682.4), including sequence corresponding to its translation initiation site as well as to leucine zipper and zinc finger domains that are important for FOXP1 dimerization and regulation of transcriptional activity (Figure 1A).11,13 Multiplex ligation-dependent probe amplification (MLPA) analysis of FOXP1 in patient A confirmed the loss of these exons (Figure 1B). No deletions encompassing FOXP1 exons were identified in the other tested patients or their parents, nor have any been reported in the Database of Genomic Variants, a large public repository of structural variations in population controls.18
Figure 1.
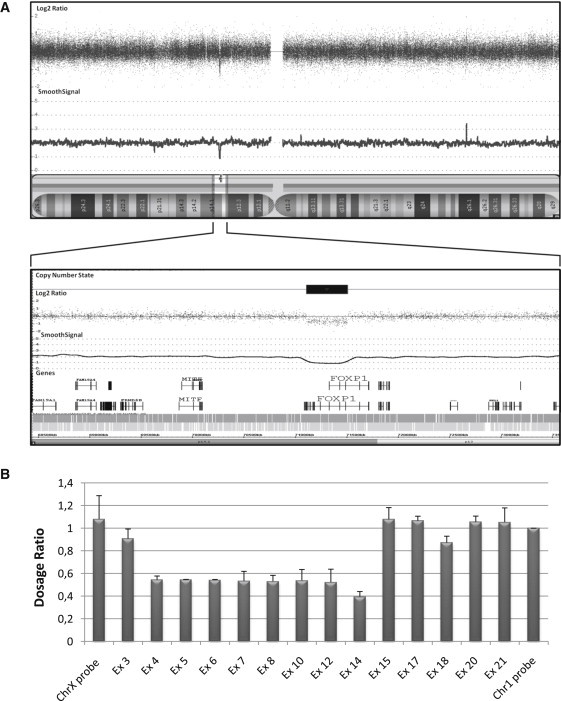
De Novo FOXP1 Deletion at 3p14.1 in Patient A
(A) The Affymetrix Genome-Wide Human SNP 6.0 Array result for patient A is shown above the ideogram of chromosome 3 (Netaffx version 28). Each dot represents a SNP or a copy number marker, with normal copy number having a log2 ratio of ∼0 and deleted regions less than −1. The CNV analysis was performed according to the manufacturer's procedure with the use of the Affymetrix reference library, GenomeWideSNP_6.hapmap270.na30r1.a5, with a regional GC correction. The de novo deletion (chr3: 71109689–71508061; hg18) is boxed, and the only gene affected with this deletion, FOXP1, is shown.
(B) Mapping of FOXP1 deleted exons in patient A. Using MLPA analysis,35 we mapped the deletion breakpoints between exon 4 and exon 14 of FOXP1. MLPA probes targeting 14 exons (Ex) of FOXP1 were custom designed with the ProSeek software.36 These 14 probes were compared to two control probes on chromosome X and chromosome 1. MLPA was performed on 50 ng of genomic DNA. Probe amplification products were run on an ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA). The data were analyzed with the GeneMapper software version 4.0 (Applied Biosystems). The dosage ratio (DR) was calculated as follows: DR = (peak height FOXP1 / peak height chromosome 1 probe) in the patient carrying the deletion / (peak height FOXP1 / peak height chromosome 1 probe) in her unaffected mother, who has no copy number variation in FOXP1. The DR has a theoretical value of ≤ 0.7 for a deletion, between 0.7 and 1.3 for a normal situation, and ≥ 1.3 for a duplication. Values represent mean of samples ± standard deviation of samples run in triplicate. Data show that exons 4–14 are deleted in patient A.
We next sequenced all the coding exons and intron-exon boundaries of the longest FOXP1 isoform (FOXP1a; 16 coding exons) in 110 cases with NSID, 84 cases with ASD, and 51 cases with both NSID and ASD, as well as in 570 controls. We identified a de novo nonsense mutation, c.1573C>T (p.R525X), in a male patient with NSID and autism (R0024121) (patient B). This alteration abolishes the last 152 amino acids of FOXP1, including part of the forkhead DNA-binding domain (FHD) and a conserved nuclear localization signal (NLS) (Figure 2A).19,20 The FHDs of other FOXP proteins share more than 90% sequence identity with that of FOXP1 (Figure 2B).21 Alteration of the corresponding arginine residue in FOXP3 (c.1189C>T [p.R397W]), as well as several other residues in the FOXP3 FHD, has been reported to cause a lethal X-linked neonatal autoimmune disease known as IPEX (MIM 304790).22,23 Furthermore, high-resolution analysis of the crystal structure of the human FOXP2 FHD revealed that the residue homologous to p.R525, as well as residues downstream of it, plays an important role in DNA binding.21 Indeed, pathogenic alterations in FOXP2 that abolish the FHD (p.R328X) or that are located in proximity to the residue homologous to p.R525 (p.R553H) have been shown to cause verbal dyspraxia (Figure 2B).24,25
Figure 2.
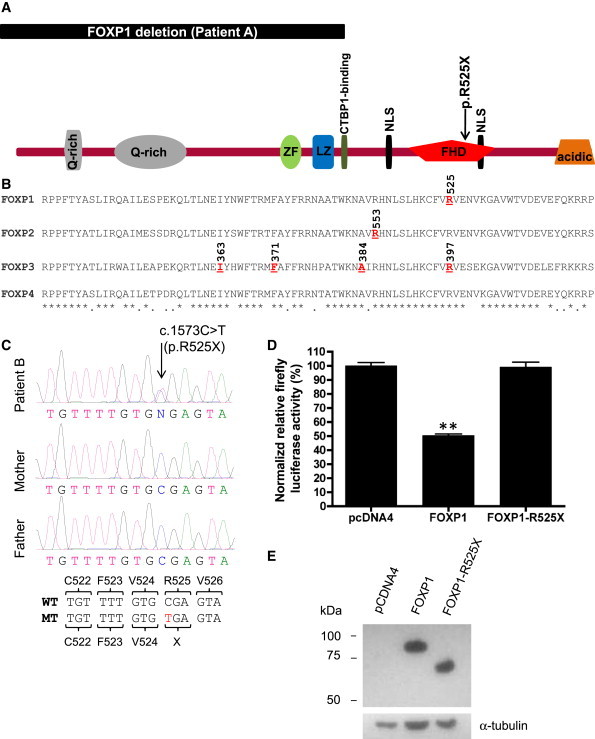
Localization and Characterization of FOXP1 De Novo Mutations
(A) Localization of the de novo mutations identified herein with respect to FOXP1 known protein domains (Uniprot no. Q9H334; 677 amino acids [aa]). The deletion in patient A is predicted to affect the first half of FOXP1, including important functional domains such as a zinc finger (ZF; aa 306–331), a leucine zipper (LZ; aa 348–369), and CTBP1-binding (amino acids 382–386) domains. The nonsense p.R525X alteration affects the conserved forkhead DNA-binding domain (FHD, aa 465–555). Also shown are two glutamine rich (Q-rich) regions (aa 55–77, aa 110–194) at the N terminus of the protein and an acidic-rich region (aa 637–677) at its C terminus. The positions of the two nuclear localization signals (NLS; aa 434–440, aa 543–546) are indicated.
(B). Alignment of the conserved FHD region of the four members of the human FOXP family. Highlighted and underlined in red are residues that when mutated lead to human disease: p.R525X (current study) in NSID, autism, and language impairment; p.R553H in FOXP2 in severe congenital speech disorder;24 and p.I363V, p.F371C/L, p.A384T, and p.R397T in FOXP3 in IPEX syndrome.21 Amino acid identity and similarity are represented by asterisks (∗) and dots (.), respectively.
(C) Chromatograms corresponding to the FOXP1 de novo mutation (c.1573C>T [p.R525X]) identified in patient B. Wild-type (WT) and mutant (MT) FOXP1 DNA sequences are shown along with the corresponding amino acids.
(D) Luciferase reporter assay assessing the impact of p.R525X on FOXP1 transactivation activity in transfected HEK293 cells. FOXP1 significantly inhibited pGL3-promoter (SV40) transcriptional activity (p < 0.001) as compared to empty vector (pcDNA4)-transfected cells only. In contrast, FOXP1-R525X failed to repress the pGL3-promoter activity. Results (mean ± SEM of three independent experiments, each performed in triplicate) are shown as relative firefly luciferase activity, which is driven by SV40 promoter, normalized with Renilla luciferase, and driven by HSV-TK promoter activity. The control signal value (set to 100%) was obtained with cells transfected with the empty expression vector (pcDNA4HisMax) and both pGL3-promoter (firefly luciferase) and pRL-TK (Renilla luciferase) constructs.
(E) Immunoblot performed on total protein extracts from HEK293 cells transfected with pcDNA4-HisMax-based constructs expressing FOXP1 or FOXP1-R525X in frame with an N-terminal Xpress tag, as described in the text. Proteins were resolved on SDS-PAGE, and FOXP1 and FOXP1-R525X were detected with the use of a monoclonal antibody against the N-terminal Xpress tag (Invitrogen). The blot was stripped and probed with an anti-alpha-tubulin antibody (Abcam) as an internal loading control.
Sequencing also revealed a heterozygous missense mutation, c.643C>A (p.P215A), in both an NSID patient (n = 1/110) and a healthy individual (n = 1/570). In the case of the NSID patient, the variant was transmitted from an unaffected parent, further supporting the notion that it is not pathogenic. This variant was also previously reported in an individual with developmental verbal dyspraxia and in a control sample.14 Three additional heterozygous missense variants (c.1333G>A [p.V445M], n = 1/570; c.1838C>A [p.T613N], n = 1/570; c.1709A>G [p.N570S], n = 2/570) were detected in FOXP1, but these did not affect any known functional domains and were present only in our cohort of controls, indicating that they are not pathogenic. No other amino-acid-altering variants were identified in any of the screened individuals, including the 570 healthy controls.
In order to test the effect of the p.R525X mutation on FOXP1 activity, we took advantage of the well-documented ability of FOXP1 to repress transcription from the SV40 promoter, which can be easily monitored with the use of a standard luciferase assay.9,20 The p.R525X mutation was introduced into the full-length coding sequence of the FOXP1 longest isoform (FOXP1a; obtained from Kazusa DNA Research Institute, Japan) and subcloned into the mammalian expression vector pcDNA4HisMax (Invitrogen). Human embryonic kidney (HEK) 293 cells were then cotransfected with the use of Fugene 6 (Roche) in 24-well plates, with 400 ng of pcDNA4HisMax without an insert or containing either the wild-type (WT) FOXP1 or the FOXP1-R525X cDNA, along with 50 ng of pGL3-promoter construct (Promega) (in which the SV40 promoter drives a firefly luciferase reporter). In order to account for variations in transfection efficiency and variation in cell number, cells were also cotransfected with 50 ng of a Renilla luciferase construct (pRL-TK; Promega) driven by the HSV-thymidine kinase promoter, which is not affected by FOXP1. Cells were lysed 48 hr after transfection and quantified for firefly and Renilla luciferase activities with the use of the Dual Luciferase Reporter Assay System (Promega). As shown in Figure 2D, WT FOXP1 was found to significantly inhibit the firefly luciferase signal (∼50% reduction; p < 0.001), as compared with an empty vector, whereas FOXP1-R525X failed to induce any reduction in luciferase levels, indicating that the p.R525X alteration impaired the FOXP1 ability to repress the SV40 promoter. Collectively, these results, along with the position of the p.R525X alteration in the FHD, strongly suggest that the de novo p.R525X alteration disrupts FOXP1 function. The mechanism(s) underlying this loss of FOXP1 activity could involve altered nuclear localization, as suggested by the disruption of one of the NLS in the FOXP1 mutant protein, impaired DNA binding, as suggested from the crystal structure of the FOXP2 FHD, or a combination of both. Although mutant and WT proteins were detected at comparable levels in transfected HEK293 cells, we cannot also exclude the possibility that the p.R525X mutation induces nonsense-mediated decay of FOXP1 mRNA in the patient's cells (Figure 2E). Additional work is needed to address these mechanistic questions.
The phenotypes of both patients with de novo mutations in FOXP1 are summarized in Table 1. Patients A and B were born from nonconsanguineous French Canadian parents after uneventful pregnancies and deliveries. Patient B was operated on for atresia of the midjejunum and midileum gut during the neonatal period. Their initial development was characterized by global delay, with severe language impairment. They started to walk between the ages of 18 and 20 mo. Patient A was not clearly pronouncing any word until 3 yrs of age and started to associate words at 4 yrs of age, whereas patient B said his first words at the age of 6 yrs. The patients exhibited no deficits of oromotor coordination that could explain this delay in learning to talk.
Table 1.
Clinical Phenotype of the Patients with De Novo FOXP1 Mutations
| Patient A (R0031608) | Patient B (R0024121) | |
|---|---|---|
| FOXP1 de novo mutation | del (exons 4–14) | c.1573C>T (p.R525X) |
| Age (yrs: mo) / sex | 15:11 / F | 9:11 / M |
| ADI-R / ADOS-G | −/− | +/+ |
| Leiter International Performance Scale-R | ||
| BRIEF IQ: standard score (percentile) | 58 (0.3) | 48 (0.1) |
| Pre-school Language Scale: Age Equivalent (Yrs: Mo) | ||
| Auditory comprehension | ND | 3:7 |
| Expressive communication | ND | 1:11 |
| Total language score | ND | 2:7 |
| Clinical Evaluation of Language Fundamentals: Age Equivalent (Yrs:Mo) | ||
| Expressive subtests | ||
| Morphology | < 4:0 | ND |
| Recalling sentences | < 4:0 | ND |
| Expressive vocabulary | < 4:5 | ND |
| Receptive subtests | ||
| Concepts and following directions | 4:3 | ND |
| Basic concepts | > 4:0 | ND |
| Sentence structure | < 4:0 | ND |
| Vineland Adaptive Behavior Scales: Percentile Rank | ||
| Communication | 1 | < 1 |
| Daily living | 2 | < 1 |
| Socialization | < 1 | 1 |
| Adaptive behavior composite | 1 | < 1 |
| Internalizing behavioral abnormalities | clinically significanta | ND |
| Externalizing behavioral abnormalities | clinically significanta | ND |
| Maladaptive behavior index | clinically significanta | ND |
| Aberrant Behavior Checklist: Raw Score (SD above Normal Mean)b | ||
| Irritability subscale | 26 (2.3) | 31 (2.9) |
| Lethargy subscale | 19 (1.7) | 8 (0.2) |
| Stereotypy subscale | 7 (1.6) | 14 (2.9) |
| Hyperactivity subscale | 24 (1.6) | 42 (2.6) |
| Inappropriate speech subscale | 6 (1.6) | 6 (1.5) |
| Repetitive Behavior Scale-Revised: Raw Scoresc | ||
| Stereotyped behavior | 3 | 7 |
| Self-injurious behavior | 1 | 16 |
| Compulsive behavior | 3 | 6 |
| Ritualistic behavior | 6 | 6 |
| Sameness behavior | 6 | 11 |
| Restricted behavior | 4 | 10 |
| Other Tests | ||
| Fragile X testing | negative | negative |
| AGH | del FOXP1 (exons 4–14) | normal |
| CT scan | normal | ND |
ND, not determined; AGH, array genomic hybridization. Affymetrix SNP arrays (5.0 or 6.0) were used for AGH.
Clinically significant behavior problems on these scales correspond to the extreme top 2% of children of the same age.
Clinically elevated: 1.5–1.9 SDs, significantly elevated: ≥ 2.0 SDs, where reported SDs are standard deviations from the normative ABC mean (community versus score).33
All subscale scores correspond to significant case identification, except for the “self-injurious behavior” for patient A, as previously outlined.34
Patients A and B were assessed with the ADI-R and the ADOS-G at 6 yrs 8 mo and at 2 yrs 7 mo, respectively. Despite some autistic features in reciprocal social interaction (social avoidance with peers and decreased inhibition with adults) and restricted interests and repetitive behaviors (delayed echolalia and stereotyped language, self-injury, perceptual fixations), patient A would not have been considered as being autistic because of subthreshold scores in the communication area. In contrast, patient B showed scores above the diagnostic threshold for autism in all areas.
Patients A and B were evaluated with a series of cognitive and behavioral assessment tools at 15 and 9 yrs of age, respectively. Assessment with the Leiter International Performance Scale-R (Brief IQ), an estimate of nonverbal intellectual functioning, revealed ID in the mild to moderate range. Results from the Vineland Adaptive Behavior Scale were consistent with the presence of ID in both patients, showing severe deficits in adaptive behaviors across the range of subscales (communication, daily living skills, socialization). Formal testing of patient A via the Clinical Evaluation for Language Fundamentals confirmed the presence of severe language impairment. Specifically, expressive language was severely limited, with oral communication characterized by single words or very short, simple sentences. Expressive vocabulary, equivalent to about 4.5 yrs, was slightly better than other expressive skills, such as morphology and repetition of increasingly complex sentences (both below 4 yrs) but was nonetheless mostly limited to relatively simple, frequent words. In several tests, she gave responses to expressive language items by miming the required words, indicating good comprehension of the question but an inability to produce the answer verbally. In support of this, receptive language abilities were somewhat more developed, as indicated by her understanding of a number of more complex relational and numerical concepts. Assessment of patient B with the Preschool Language Scale also confirmed severe language impairment. In terms of expressive communication, he scored at an age equivalence of 1 yr 11 mo. He had a range of about 100 words in his vocabulary and communicated with single words or short sentences (two to three words). In contrast, he was able to understand more complex relational concepts and performed at an age equivalence of 3 yrs 7 mo in auditory comprehension tasks.
Both patients also showed a particular behavioral profile. Clinically significant problems were noted on the Vineland Maladaptive Behavior scale for patient A and were characterized by both internalizing (e.g., social withdrawal, anxiety) and externalizing (e.g., impulsivity, mood lability, temper tantrums, sulking, disobedience, physical aggression) behaviors. Parent responses on the Aberrant Behavior Checklist also indicated the presence of behavioral problems in both patients. In patient A, irritability was rated as the most significant problem, with all other problem scales (lethargy, stereotypy, hyperactivity, inappropriate speech) reaching clinically significant levels. Similarly, patient B had significantly elevated problems on the irritability, stereotypy, and hyperactivity subscales and clinically significant speech problems. A number of specific obsessions and compulsions (e.g., looking in mirrors, touching people's hair, nail biting, removing silicone in the house, hoarding objects) were described by the parents and confirmed via the Repetitive Behavior Scale. On this scale, ritualistic, restricted, and “sameness” behavior were most often reported for patient A (e.g., mealtime and bedtime rituals, rigid routines, listens to the same music continuously, strong attachment to specific objects). Stereotyped and self-injurious behavior (e.g., turning in circles, spinning objects, hitting and biting self) were also reported for patient B.
In summary, we report two patients with de novo deleterious intragenic mutations in FOXP1. These patients share strikingly similar phenotypes, including mild to moderate ID with severe language impairment, autism and /or autistic features, mood lability with physical aggressiveness, and specific obsessions and compulsions. The patients with FOXP1 disruption described herein do not show verbal dyspraxia, but their phenotype nevertheless overlaps with that of patients with FOXP2 disruption to the extent that they also show language impairment. Two other patients with de novo deletions encompassing FOXP1 were recently described.26,27 These patients, who were less than 4 yrs of age when studied, showed developmental and speech delay, but formal cognitive or behavioral evaluation could not be performed. In one case, the deletion spans 750 kb and affects three other genes whereas in the other case, the deletion spans 1 Mb and extends over 500 kb on the 3′ end of FOXP1. Even though this latter deletion does not affect any other known genes, the possibility that it encompasses elements that are important for the expression of genes located in its vicinity cannot be ruled out. Nevertheless, these cases reinforce our conclusion that FOXP1 haploinsufficiency affects cognitive development. Although an association between vitiligo (MIM 193200), an autoimmune form of skin depigmentation, and an intronic variant in FOXP1 was recently reported,28 none of our patients displayed signs of such a condition.
Although FOXP1 and FOXP2 can cooperate to regulate transcription and are both associated with language impairment, FOXP1 disruption appears to have a more global impact on brain development than FOXP2 disruption. The difference between the phenotypic consequence of FOXP1 and FOXP2 haploinsufficiency might be explained at least in part by some difference in their expression patterns.7,9 Alternatively, FOXP heterodimers and homodimers may have different biochemical properties. For instance, FOXP1-FOXP2 heterodimers may regulate targets that are involved in language impairment, whereas the respective FOXP homodimers may regulate additional targets that are associated with other developmental processes. Vernes et al. have found that FOXP2 directly regulates the expression of CNTNAP2, a gene that encodes a member of the neurexin superfamily of transmembrane proteins implicated in neuronal recognition and cell adhesion.29 CNTNAP2 has been associated with language impairment and autism.29–32 It is tempting to speculate that FOXP1 homodimers or FOXP1-FOXP2 heterodimers also regulate CNTNAP2 expression and that FOXP1 haploinsufficiency affects language development and possibly causes ID and autism by disrupting this regulatory interaction. FOXP1 and FOXP2 thus represent interesting entry points for dissecting the molecular mechanisms underlying neurodevelopmental disorders such as ID, autism, and language impairment.
Acknowledgments
This work was supported by grants from the Canadian Institute of Health Research (CIHR) (J.L.M., G.A.R., J.-C.L.), Réseau de Génétique Médicale Appliquée (RMGA) / Fonds de la Recherche en Santé du Québec (FRSQ) (J.L.M. and M.S.P.), Genome Canada and Genome Quebec, and cofunding by Université de Montréal for the Synapse to Diseases (S2D) project (G.A.R. and P.D.). J.L.M. is the recipient of a Clinical Investigatorship Award of the CIHR (Institute of Genetics) and a Senior Scientist Award from FRSQ. E.F. holds the CIHR-funded Canada Research Chair in Child Psychiatry. H.D. is a recipient of a CIHR postdoctoral fellowship award. We thank the patients and their parents for participating in this study. We are grateful for the dedicated work of members of the S2D team (CHUM Research Center, Montreal), including Management (Claude Marineau), Bioinformatics (Edouard Henrion, Ousmane Diallo, and Dan Spiegelman), and the Genetic Screening divisions (Annie Levert, Annie Raymond, Pascal Thibodeau, Sandra Laurent, and Karine Lachapelle). We are thankful for the efforts of the members of McGill University and Génome Québec Innovation Centre Sequencing (Pierre Lepage, Sébastien Brunet, and Hao Fan Yam) and Bioinformatics (Louis Létourneau and Louis Dumond Joseph) groups.
Contributor Information
Guy A. Rouleau, Email: guy.rouleau@umontreal.ca.
Jacques L. Michaud, Email: jacques.michaud@recherche-ste-justine.qc.ca.
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants, http://projects.tcag.ca/variation/
Online Mendelian Inheritance of Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
UCSC Genome Browser, http://www.genome.ucsc.edu/
Uniprot, http://www.uniprot.org/
References
- 1.Fisher S.E., Lai C.S., Monaco A.P. Deciphering the genetic basis of speech and language disorders. Annu. Rev. Neurosci. 2003;26:57–80. doi: 10.1146/annurev.neuro.26.041002.131144. [DOI] [PubMed] [Google Scholar]
- 2.Fisher S.E., Scharff C. FOXP2 as a molecular window into speech and language. Trends Genet. 2009;25:166–177. doi: 10.1016/j.tig.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Enard W., Gehre S., Hammerschmidt K., Hölter S.M., Blass T., Somel M., Brückner M.K., Schreiweis C., Winter C., Sohr R. A humanized version of Foxp2 affects cortico-basal ganglia circuits in mice. Cell. 2009;137:961–971. doi: 10.1016/j.cell.2009.03.041. [DOI] [PubMed] [Google Scholar]
- 4.Haesler S., Rochefort C., Georgi B., Licznerski P., Osten P., Scharff C. Incomplete and inaccurate vocal imitation after knockdown of FoxP2 in songbird basal ganglia nucleus Area X. PLoS Biol. 2007;5:e321. doi: 10.1371/journal.pbio.0050321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konopka G., Bomar J.M., Winden K., Coppola G., Jonsson Z.O., Gao F., Peng S., Preuss T.M., Wohlschlegel J.A., Geschwind D.H. Human-specific transcriptional regulation of CNS development genes by FOXP2. Nature. 2009;462:213–217. doi: 10.1038/nature08549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hannenhalli S., Kaestner K.H. The evolution of Fox genes and their role in development and disease. Nat. Rev. Genet. 2009;10:233–240. doi: 10.1038/nrg2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferland R.J., Cherry T.J., Preware P.O., Morrisey E.E., Walsh C.A. Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J. Comp. Neurol. 2003;460:266–279. doi: 10.1002/cne.10654. [DOI] [PubMed] [Google Scholar]
- 8.Lai C.S., Gerrelli D., Monaco A.P., Fisher S.E., Copp A.J. FOXP2 expression during brain development coincides with adult sites of pathology in a severe speech and language disorder. Brain. 2003;126:2455–2462. doi: 10.1093/brain/awg247. [DOI] [PubMed] [Google Scholar]
- 9.Shu W., Yang H., Zhang L., Lu M.M., Morrisey E.E. Characterization of a new subfamily of winged-helix/forkhead (Fox) genes that are expressed in the lung and act as transcriptional repressors. J. Biol. Chem. 2001;276:27488–27497. doi: 10.1074/jbc.M100636200. [DOI] [PubMed] [Google Scholar]
- 10.Teramitsu I., Kudo L.C., London S.E., Geschwind D.H., White S.A. Parallel FoxP1 and FoxP2 expression in songbird and human brain predicts functional interaction. J. Neurosci. 2004;24:3152–3163. doi: 10.1523/JNEUROSCI.5589-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li S., Weidenfeld J., Morrisey E.E. Transcriptional and DNA binding activity of the Foxp1/2/4 family is modulated by heterotypic and homotypic protein interactions. Mol. Cell. Biol. 2004;24:809–822. doi: 10.1128/MCB.24.2.809-822.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang B., Lin D., Li C., Tucker P. Multiple domains define the expression and regulatory properties of Foxp1 forkhead transcriptional repressors. J. Biol. Chem. 2003;278:24259–24268. doi: 10.1074/jbc.M207174200. [DOI] [PubMed] [Google Scholar]
- 13.Shu W., Lu M.M., Zhang Y., Tucker P.W., Zhou D., Morrisey E.E. Foxp2 and Foxp1 cooperatively regulate lung and esophagus development. Development. 2007;134:1991–2000. doi: 10.1242/dev.02846. [DOI] [PubMed] [Google Scholar]
- 14.Vernes S.C., MacDermot K.D., Monaco A.P., Fisher S.E. Assessing the impact of FOXP1 mutations on developmental verbal dyspraxia. Eur. J. Hum. Genet. 2009;17:1354–1358. doi: 10.1038/ejhg.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaufman L., Ayub M., Vincent J.B. The genetic basis of non-syndromic intellectual disability: a review. J. Neurodev. Disord. 2010 doi: 10.1007/s11689-010-9055-2. Published online July 28 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamdan F.F., Gauthier J., Spiegelman D., Noreau A., Yang Y., Pellerin S., Dobrzeniecka S., Côté M., Perreau-Linck E., Perreault-Linck E., Synapse to Disease Group Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N. Engl. J. Med. 2009;360:599–605. doi: 10.1056/NEJMoa0805392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gauthier J., Champagne N., Lafrenière R.G., Xiong L., Spiegelman D., Brustein E., Lapointe M., Peng H., Côté M., Noreau A., S2D Team De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. USA. 2010;107:7863–7868. doi: 10.1073/pnas.0906232107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J., Feuk L., Duggan G.E., Khaja R., Scherer S.W. Development of bioinformatics resources for display and analysis of copy number and other structural variants in the human genome. Cytogenet. Genome Res. 2006;115:205–214. doi: 10.1159/000095916. [DOI] [PubMed] [Google Scholar]
- 19.Banham A.H., Beasley N., Campo E., Fernandez P.L., Fidler C., Gatter K., Jones M., Mason D.Y., Prime J.E., Trougouboff P. The FOXP1 winged helix transcription factor is a novel candidate tumor suppressor gene on chromosome 3p. Cancer Res. 2001;61:8820–8829. [PubMed] [Google Scholar]
- 20.Vernes S.C., Nicod J., Elahi F.M., Coventry J.A., Kenny N., Coupe A.M., Bird L.E., Davies K.E., Fisher S.E. Functional genetic analysis of mutations implicated in a human speech and language disorder. Hum. Mol. Genet. 2006;15:3154–3167. doi: 10.1093/hmg/ddl392. [DOI] [PubMed] [Google Scholar]
- 21.Stroud J.C., Wu Y., Bates D.L., Han A., Nowick K., Paabo S., Tong H., Chen L. Structure of the forkhead domain of FOXP2 bound to DNA. Structure. 2006;14:159–166. doi: 10.1016/j.str.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 22.Bennett C.L., Christie J., Ramsdell F., Brunkow M.E., Ferguson P.J., Whitesell L., Kelly T.E., Saulsbury F.T., Chance P.F., Ochs H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 23.Wildin R.S., Ramsdell F., Peake J., Faravelli F., Casanova J.L., Buist N., Levy-Lahad E., Mazzella M., Goulet O., Perroni L. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 24.Lai C.S., Fisher S.E., Hurst J.A., Vargha-Khadem F., Monaco A.P. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001;413:519–523. doi: 10.1038/35097076. [DOI] [PubMed] [Google Scholar]
- 25.MacDermot K.D., Bonora E., Sykes N., Coupe A.M., Lai C.S., Vernes S.C., Vargha-Khadem F., McKenzie F., Smith R.L., Monaco A.P., Fisher S.E. Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. Am. J. Hum. Genet. 2005;76:1074–1080. doi: 10.1086/430841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carr C.W., Moreno-De-Luca D., Parker C., Zimmerman H.H., Ledbetter N., Martin C.L., Dobyns W.B., Abdul-Rahman O.A. Chiari I malformation, delayed gross motor skills, severe speech delay, and epileptiform discharges in a child with FOXP1 haploinsufficiency. Eur. J. Hum. Genet. 2010 doi: 10.1038/ejhg.2010.96. Published online June 23, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pariani M.J., Spencer A., Graham J.M., Jr., Rimoin D.L. A 785kb deletion of 3p14.1p13, including the FOXP1 gene, associated with speech delay, contractures, hypertonia and blepharophimosis. Eur. J. Med. Genet. 2009;52:123–127. doi: 10.1016/j.ejmg.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin Y., Birlea S.A., Fain P.R., Mailloux C.M., Riccardi S.L., Gowan K., Holland P.J., Bennett D.C., Wallace M.R., McCormack W.T. Common variants in FOXP1 are associated with generalized vitiligo. Nat. Genet. 2010;42:576–578. doi: 10.1038/ng.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vernes S.C., Newbury D.F., Abrahams B.S., Winchester L., Nicod J., Groszer M., Alarcón M., Oliver P.L., Davies K.E., Geschwind D.H. A functional genetic link between distinct developmental language disorders. N. Engl. J. Med. 2008;359:2337–2345. doi: 10.1056/NEJMoa0802828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alarcón M., Abrahams B.S., Stone J.L., Duvall J.A., Perederiy J.V., Bomar J.M., Sebat J., Wigler M., Martin C.L., Ledbetter D.H. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 2008;82:150–159. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arking D.E., Cutler D.J., Brune C.W., Teslovich T.M., West K., Ikeda M., Rea A., Guy M., Lin S., Cook E.H., Chakravarti A. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 2008;82:160–164. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bakkaloglu B., O'Roak B.J., Louvi A., Gupta A.R., Abelson J.F., Morgan T.M., Chawarska K., Klin A., Ercan-Sencicek A.G., Stillman A.A. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 2008;82:165–173. doi: 10.1016/j.ajhg.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown E.C., Aman M.G., Havercamp S.M. Factor analysis and norms for parent ratings on the Aberrant Behavior Checklist-Community for young people in special education. Res. Dev. Disabil. 2002;23:45–60. doi: 10.1016/s0891-4222(01)00091-9. [DOI] [PubMed] [Google Scholar]
- 34.Bodfish J.W., Symons F.J., Parker D.E., Lewis M.H. Varieties of repetitive behavior in autism: comparisons to mental retardation. J. Autism Dev. Disord. 2000;30:237–243. doi: 10.1023/a:1005596502855. [DOI] [PubMed] [Google Scholar]
- 35.Schouten J.P., McElgunn C.J., Waaijer R., Zwijnenburg D., Diepvens F., Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pantano L., Armengol L., Villatoro S., Estivill X. ProSeeK: a web server for MLPA probe design. BMC Genomics. 2008;9:573. doi: 10.1186/1471-2164-9-573. [DOI] [PMC free article] [PubMed] [Google Scholar]