

In order to elucidate biological processes, precise control over these processes is required. Light represents an ideal external control element as it can be easily regulated in a spatial and a temporal fashion, conveying spatiotemporal control of biological activity to the system under study.[1] The photochemical regulation of oligonucleotide function via the installation of light-removable protecting groups (caging groups) on either the phosphate or the nucleotide base has recently received considerable attention.[2–4] Important applications of this technology involve the transient disruption of DNA hybridization to photochemically control DNAzyme activity, the polymerase chain reaction, antisense activity, as well as inhibition of transcription.[4, 5] In this context, we demonstrated that a single caging group installed on one base of a typical oligonucleotide 20mer still enables DNA/DNA and DNA/RNA hybridization, but could disrupt processing of the oligomer by polymerases and inactivate the catalytic ability of DNAzymes.[2] As a result, we became interested in exploring other biologically relevant processes with photocaged DNA that did not involve perturbation of hybridization. Due to the prevalence of DNA/protein interactions both in vivo and in vitro,[6] we hypothesized that it may be feasible to photochemically control such an interaction for restriction endonucleases via incorporation of our NPOM caged thymidine nucleotide (Scheme 1) into DNA. Very few studies have been conducted involving the effects of non-natural nucleotides on the fidelity and functionality of restriction enzymes. Those that have, primarily involve the effects of endogenous base mutations (e.g. methylation events) that do not drastically affect hydrogen bonding and base pair recognition. In many of these cases the catalytic capabilities of the restriction endonucleases are dramatically decreased, if not abrogated.[7, 8]
Scheme 1.

NPOM caged, 5′ dimethyltrityl (DMT) protected thymidine phosphoramidite and its incorporation into synthetic DNA. The caged DNA can be effectively decaged through a brief irradiation with UV light of 365 nm.
Restriction endonucleases are enzymes which are capable of the site-specific recognition and cleavage of double stranded DNA. Based on their unique activity, they have been employed extensively in molecular biology and have facilitated the development of recombinant DNA technology and cloning. To date over 3500 restriction enzymes are known, and the number that are commercially available are growing (>600).[9] There are three major classes of restriction enzymes, which differ in their utilization of cofactors, their target sequence, and the location of the cleavage site relative to their target sequence. The most commonly employed restriction endonucleases are from the Type II family, which typically only require Mg2+ as a cofactor, recognize a 4–8 base dsDNA sequence, and cleave directly within that sequence. Based on their extensive use in the manipulation of DNA and the site-specific mechanism of action,[8] we investigated the photochemical regulation of these enzymes using our developed nucleobase caging technology.
Thus, we designed a DNA construct, which possesses multiple restriction sites and thymidine residues at various positions relative to the site of cleavage (Table 1). Ideally, this should afford a means to probe the effects of the caging group on restriction endonuclease recognition and function. The EcoRI, BglII, and BamHI, sites were selected for this study as they represent commonly employed restriction endonucleases, and have thymidine residues at various positions relative to their cleavage site. The non-caged DNA and its complement were synthesized and an initial study was performed to demonstrate the efficient cleavage of the substrate by the three enzymes (see Supporting Information). Additionally, to ensure that hybridization to the complementary sequence was occurring despite the presence of the caging group, the melting temperature (Tm) of each caged oligonucleotide Tn in presence of the complementary strand was determined on a BioRad MyiQ RT-PCR thermocycler by conducting a sequence of 3 heating and cooling cycles (Table 1). It appears that the presence of a single caging group reduces the melting temperature of this DNA sequence by approximately 5 °C, while two-three caged thymidines reduce the melting temperature by 9–18 °C; however, hybridization was detected for all constructs, and in no case was the disruption sufficient enough to prevent hybridization at 37 °C, the temperature at which the restriction enzyme assays were conducted.
Table 1.
Synthesized caged and non-caged restriction enzyme templates.[a]
 | ||
|---|---|---|
| DNA | Sequence | Tm/°C |
| T0 | 5’ GGGTGAATTCAGATCTGGATCCAAAAG 3’ | 68.0 ± 0.7 |
| T1 | 5’ GGGTGAATTCAGATCTGGATCCAAAAG 3’ | 62.8 ± 1.1 |
| T2 | 5’ GGGTGAATTCAGATCTGGATCCAAAAG 3’ | 62.5 ± 0.8 |
| T3 | 5’ GGGTGAATTCAGATCTGGATCCAAAAG 3’ | 62.5 ± 0.7 |
| T4 | 5’ GGGTGAATTCAGATCTGGATCCAAAAG 3’ | 63.5 ± 0.4 |
| T5 | 5’ GGGTGAATTCAGATCTGGATCCAAAAG 3’ | 63.3 ± 0.3 |
| T2,3,6 | 5’ GGGTGAATTCAGATCTGGATCCAAAAG 3’ | 50.8 ± 1.2 |
| T4,5 | 5’ GGGTGAATTCAGATCTGGATCCAAAAG 3’ | 59.5 ± 0.7 |
Melting temperature of PS DNA/DNA hybrids. T denotes the caged thymidine.
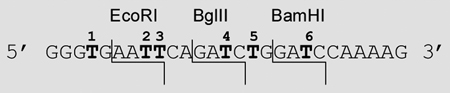
Initially we explored the propensity of the caging group to inhibit EcoRI function based on the position of the caged thymidine residue relative to the site of cleavage using oligomers T0-T3, T5, and T2,3,6. The complementary sequence was end labelled with 32P, and hybridized with the corresponding oligomer. The resulting double-stranded DNA, both non-irradiated and irradiated (5 min, 25 W, 365 nm), was digested with the restriction enzyme (New England Biolabs) for 1 hour at 37 °C following the manufacturers protocol. The cleavage was then analyzed via polyacrylamide gel electrophoresis (Figure 1).
Figure 1.
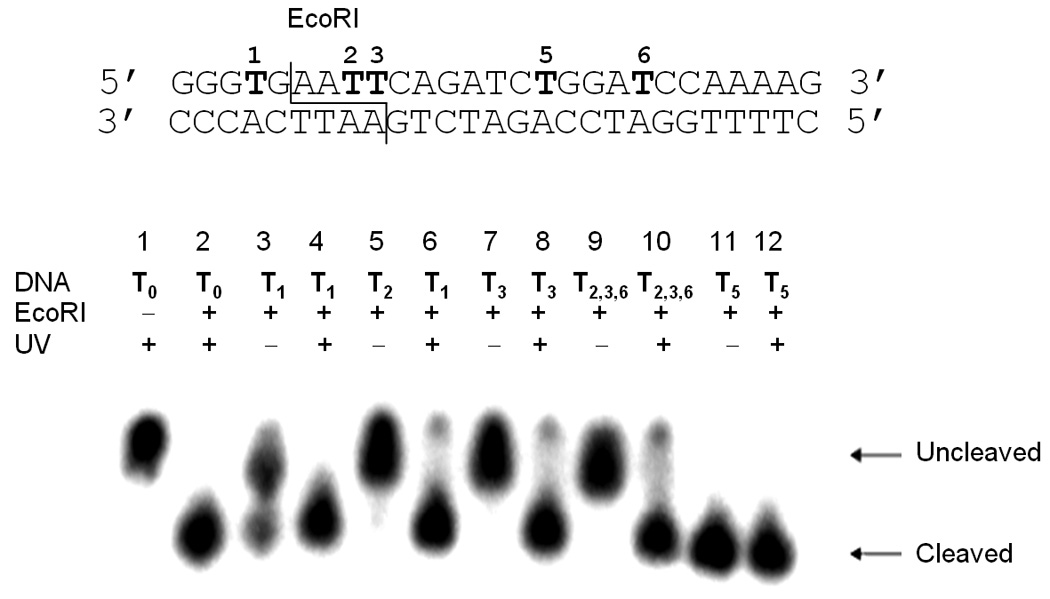
EcoRI digest of caged constructs. Six substrates with caging groups in different positions were digested with EcoRI (1 h, 37 oC) to assess the ability of the caging group to inhibit enzymatic activity. Reactions were irradiated at 365 nm (25 W) for 5 minutes. Timecourses for the digestions are shown in the Supporting Information.
A control reaction in the absence of the restriction enzyme lead to no cleavage of the double-stranded DNA (Lane 1, Figure 1), while the enzyme in the absence of any caged oligonucleotides resulted in complete cleavage of the substrate T0 within 1 hour (Lane 2, Figure 1). The T1 construct contains a single caged thymidine two bases upstream of the EcoRI cleavage site. Interestingly, the digest of the non-irradiated substrate lead to a mixture of cleaved and uncleaved substrate (Lane 3, Figure 1), which suggests that the reaction is inhibited by the presence of the caging group. Upon irradiation of T1, complete enzymatic cleavage is observed (Lane 4, Figure 1). Substrates T2, T3, and T2,3,6 possess either one or two caging groups within the recognition site of the enzyme, which are either 3 or 4 residues downstream from the cleavage site. The non-irradiated double-stranded oligonucleotides are completely resistant to EcoRI, however, complete cleavage is observed after a brief UV irradiation (Lanes 5–10, Figure 1; Figure 2) removing the caging groups. Substrate T5 possesses a caging group outside the enzymatic recognition and cleavage site, which had no effect on DNA cleavage, and does not slow the rate of cleavage (Lanes 11–12, Figure 1). Conversely, substrate T4 affords complete cleavage, albeit at a slower rate (see Supporting Information). This suggests that the caging group does not significantly affect the general binding of the enzyme to the substrate, but instead may inhibit specific recognition of the restriction site. Identical results were observed when labelling the caged DNA strand followed by digestion under identical conditions (data not shown).
Figure 2.
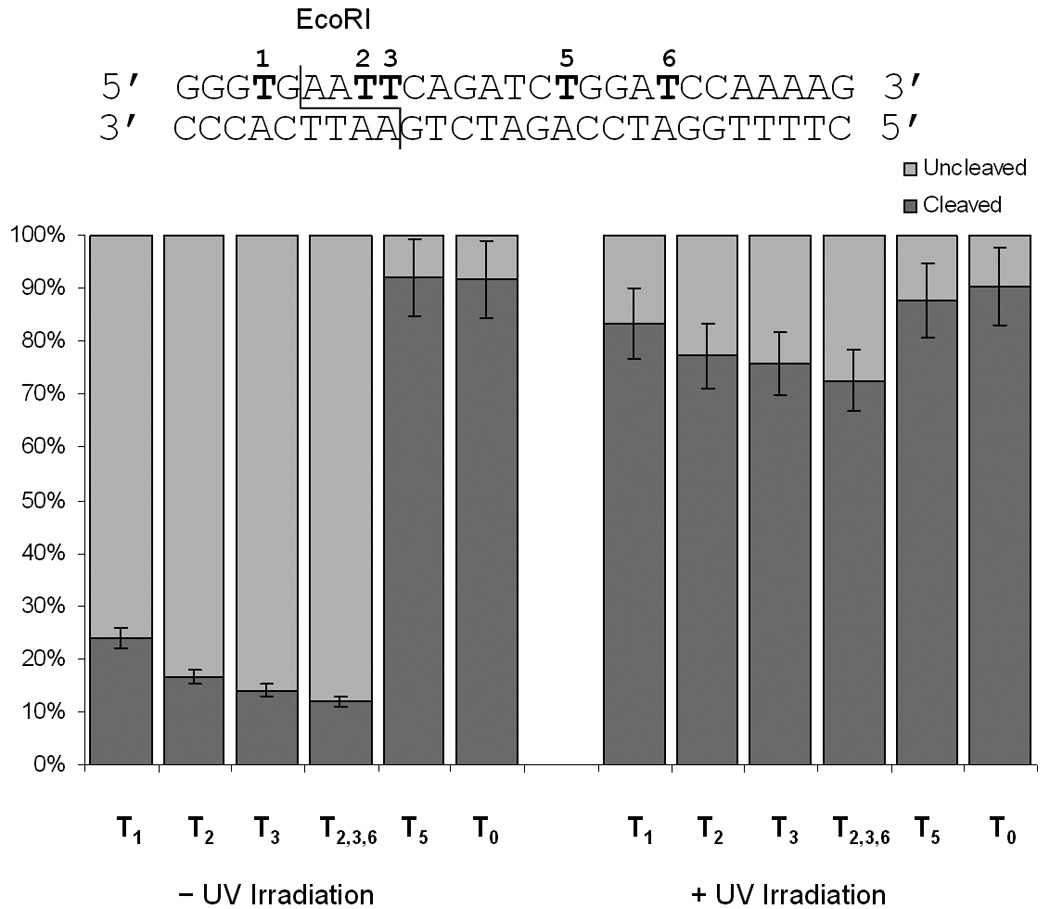
Quantitiative measurement of the EcoRI DNA cleavage with different caged substrates. Reactions were irradiated at 365 nm (25 W) for 5 minutes. All digests were performed in triplicate and the error bars represent the standard deviation.
In order to thoroughly assess the binding of the enzyme to the substrate we conducted a gel shift assay to ascertain the different binding constants for the caged and non-caged double-stranded DNA. Oligomers T0 and T2 were hybridized with the radioactively labelled complement, followed by incubation with EcoRI (1 h, 37 °C) at different concentrations in the absence of Mg2+ to prevent cleavage activity. This takes advantage of the strong dependence of cleavage activity but not binding on Mg2+.[10, 11] The incubations were then analyzed by polyacrylamide gel electrophoresis for the presence of a gel shift and quantitated using ImageQuant software. The data was plotted (see Supporting Information) and analyzed to determine binding constants of 0.54 µM for the non-caged substrate T0 and 0.71 µM for the caged substrate T2. These values are in agreement with literature binding constants which were performed under similar conditions (i.e. no Mg2+) .[8, 11] This suggests that it is feasible for the enzyme to bind the DNA substrate irrespective of the presence of a caging group, albeit at a what appears to be only a slightly lower affinity. The mechanism of many Type II restriction endonucleases involves the initial non-specific binding to DNA, followed by the electrostatic influenced scanning for the restriction site, and ultimately the site-specific cleavage.[8] Thus, our results suggest that the caging group does not significantly inhibit the non-specific binding (based on the gel shift assay), but does appear to affect the specific recognition of the binding site (based on the cleavage assays). Additionally, since in some cases the caged thymidine is located outside of the recognition site of the enzyme, it does not directly inhibit the recognition of the substrate, but leads to a slower cleavage of the substrate. We speculate that this may possibly be a result of steric interference of enzyme recognition as it scans the DNA for the target sequence. This hypothesis is consistent with literature reports of the crystal structures of restriction enzymes as the active site of restriction endonucleases is often responsible for base pair recognition, and the presence of the caged thymidine would represent a significant perturbation of this event system.[12]
We next investigated the cleavage reaction of BglII. Here, we employed oligomers T3-T5 and T4,5 to probe the effect of the caging group. Oligomer T4 possesses a caging group within the enzyme recognition site that is 3 nucleotides downstream from the cleavage site. The T5 oligomer is also caged within the enzyme recognition site, 5 bases downstream from the cleavage site; however is located directly in the cleavage site of the opposite strand. Finally, both residues are caged in the T4,5 oligomer. As a control oligomer T3 was also used, which contains a caging group outside the recognition site of this enzyme. As with the EcoRI digest, each double-stranded DNA was digested for 1 hour at 37 °C, then analyzed by polyacrylamide gel electrophoresis (Figure 3, and see Supporting Information). As observed with EcoRI, the digestion of the substrate with BglII can be regulated photochemically via the installation of caged bases. A single caged thymidine completely inhibits enzymatic cleavage if it is located within the enzyme recognition site, as observed with the T4 and T5 double-stranded DNA. This principle logically extends to the installation of two caging groups, in the case of T4,5, in which enzymatic activity is abrogated completely prior to irradiation. In all cases, enzymatic activity is restored upon brief irradiation with UV light at 365 nm (Figure 3). However, complete deactivation of the substrate towards cleavage is not observed in the case of T3. The degree of DNA cleavage was again analyzed by quantification using ImageQuant software.
Figure 3.
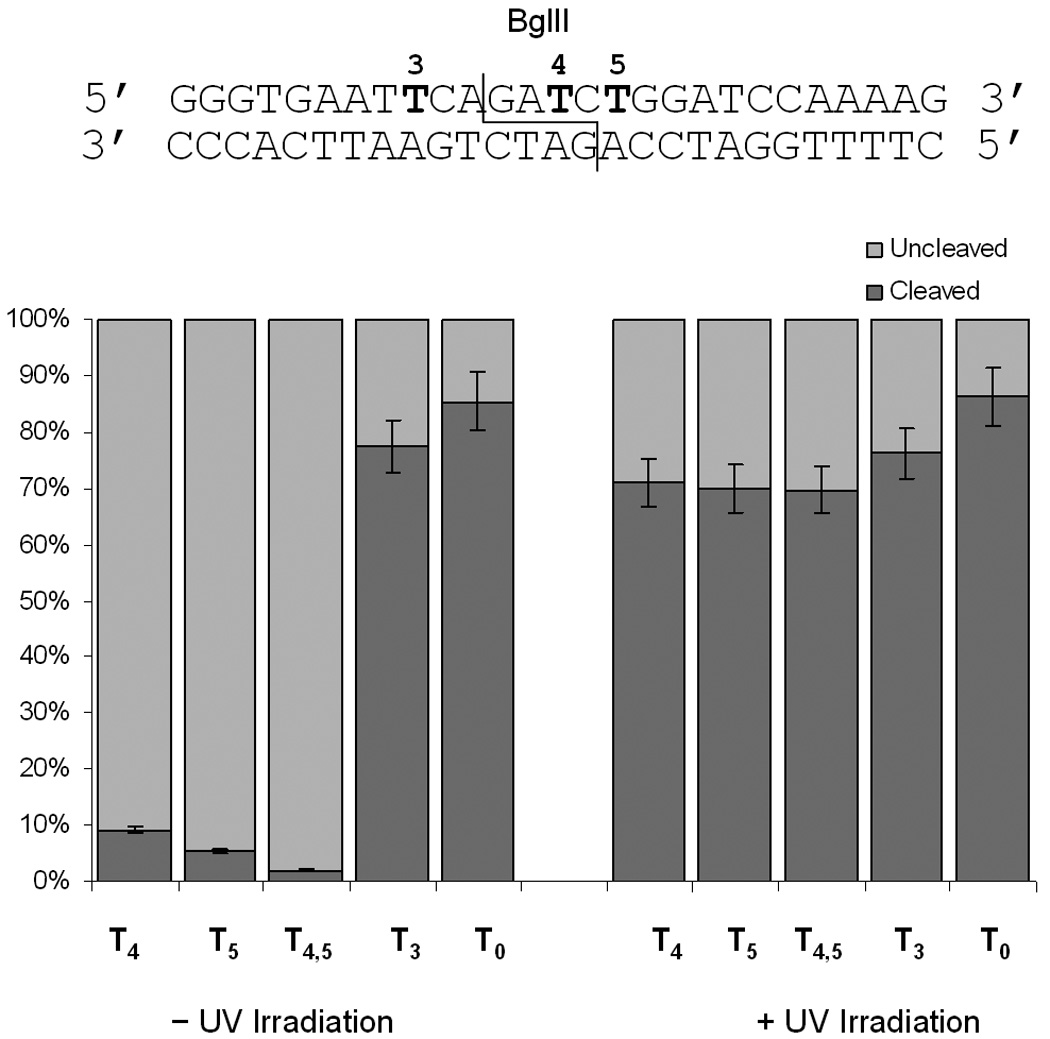
Quantitative assessment of the BglII DNA cleavage with different caged substrates. Reactions were irradiated at 365 nm (25 W) for 5 minutes. All digests were performed in triplicate and the error bars represent the standard deviation.
Next we examined the application of the nucleobase caging methodology towards the regulation of BamHI on the same substrate. In this investigation we employed the T4-T5 and T2,3,6 oligomers. Here, only the T2,3,6 oligomer possessed a caging group within the recognition site of the enzyme, 3 bases from the cleavage site. Oligomer T5 contained a caging group outside of the recognition site, but only 2 bases away from the cleavage site. The T4 oligomer was used as a control caged sequence, in which the caging group was located substantially further away from the enzyme recognition and cleavage site. Again, the enzymatic digestions were incubated for 1 hour at 37 °C, prior to analysis by gel electrophoresis and quantification using ImageQuant (Figure 4). As observed for the other enzymes, deactivation of DNA cleavage strongly depends on the proximity of the caging group to the cleavage site with complete inhibition in the case of a caged thymidine located directly in the recognition site of BamHI. In all cases, DNA cleavage could be activated via a brief irradiation with UV light of 365 nm. Based on these observations the photochemical regulation of restriction endonuclease activity appears to be generally applicable, as digestion could be regulated via light irradiation for all three enzymes.
Figure 4.
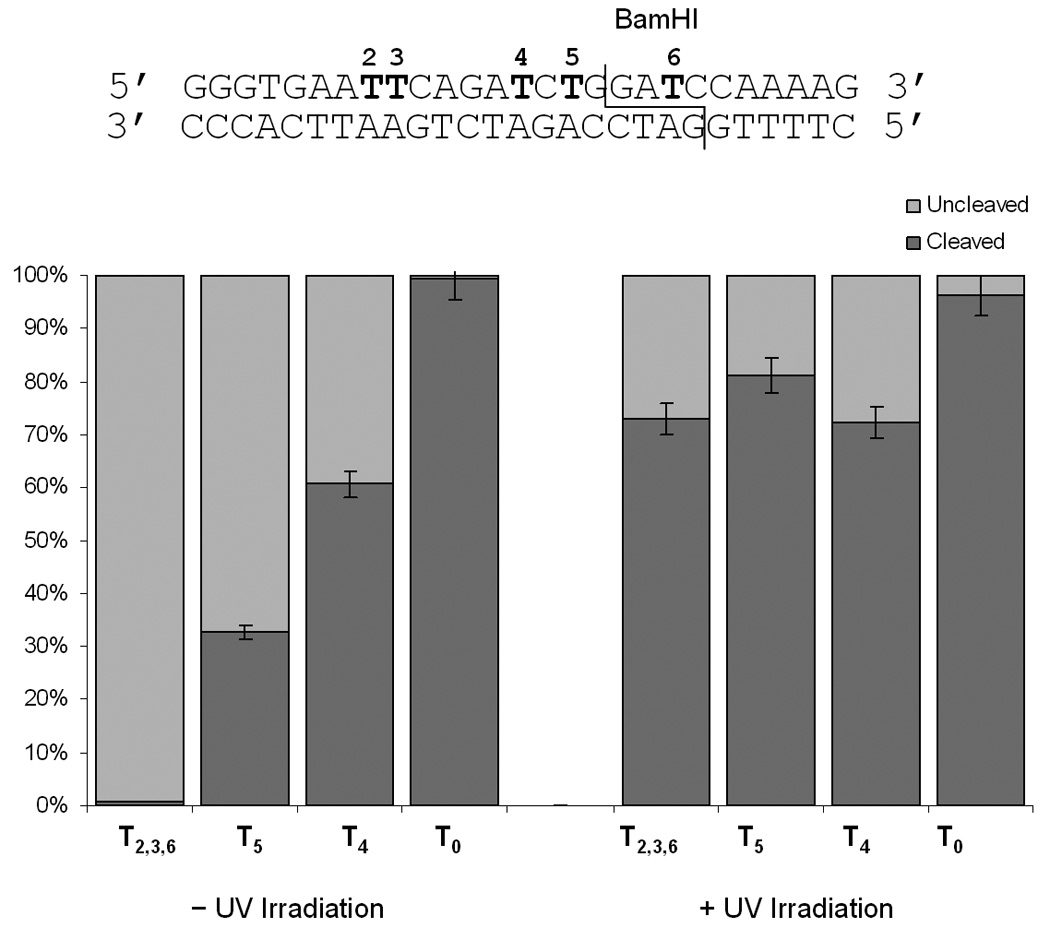
Quantitative assessment of the BamHI DNA cleavage with different caged substrates. Reactions were irradiated at 365 nm (25 W) for 5 minutes. All digests were performed in triplicate and the error bars represent the standard deviation.
Finally, we prepared a substrate for digestion that possessed a restriction site for TaqαI, a hyperthermophilic restriction endonuclease with an optimal activity at 65 °C. We were interested in exploring the scope of the technology by using this enzyme, which is active at high temperatures where DNA hybridization is weaker.[13, 14] While we examined enzymatic activity at the optimal temperature, we also probed enzymatic activity at 37 °C. The caged substrate was again hybridized with its radioactively labeled complement, then subjected to a 2 hour digestion (Figure 5).
Figure 5.
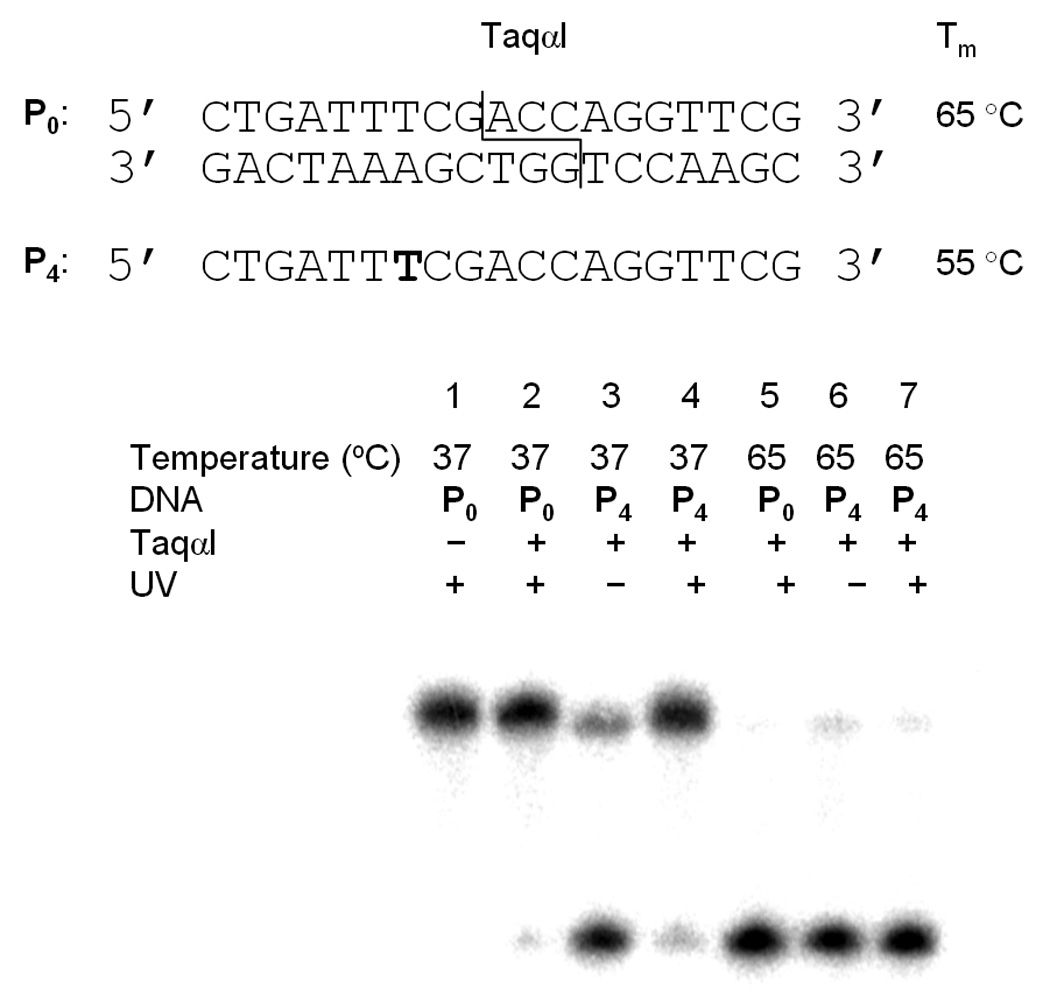
Investigation of the effect of a caging group on the hyperthermophilic restriction enzyme TaqαI. Reactions were irradiated at 365 nm (25 W) for 5 minutes.
Interestingly, in the case of TaqαI, the presence of one caging group in P4 was found to actually activate enzymatic cleavage of the substrate at 37 °C. At this temperature the non-caged substrate P0 remains uncleaved as the enzyme is not active (Lane 1, Figure 5); however, the caged substrate prior irradiation affords a substantial amount of substrate cleavage (Lane 3, Figure 5). This activation is abrogated upon irradiation, as very little substrate cleavage is observed in the absence of a caging group (Lanes 2 and 4, Figure 5). This unexpected result may occur due to the fact that this enzyme possesses a different mechanism of action than the previously employed endonucleases, and base modifications have little effect on enzyme binding to the DNA target sequence.[13] While only the complementary strand is labelled in Figure 5, both strands are cleaved in the presence of the caging group (see Supporting Information). At the optimal temperature, the caging group has no effect on the enzymatic cleavage as virtually all substrate is cleaved irrespective of the presence of a caging group (Lanes 4–6, Figure 5). This is most likely due to the ability of the TaqαI enzyme to ignore base modifications.[13] To ascertain if this effect is specific to the enzyme, or if this substrate is simply prone to non-enzyme specific degradation as a result of the caging group, we conducted the enzymatic digest with the caged substrate and a variety of restriction endonucleases (see Supporting Information). However, this effect was only observed in the presence of the TaqαI enzyme, as no other enzyme was capable of cleaving the substrate, even after 24 hours of incubation.
Based on the enzymatic digests and measured binding constants, we have developed a working hypothesis for the role of the caged thymidine residue on restriction endonuclease activity as shown in Figure 6. It appears that the enzymes are capable of nonspecifically binding the caged DNA substrate and subsequently scanning for the restriction site.[8] This is confirmed by gel shift assays (see Supporting Information), as EcoRI was indeed able to bind DNA, despite the caging of the restriction site. However, the presence of a caging group inhibits proper identification of this site, preventing dsDNA cleavage. This is confirmed via the enzymatic digest experiments that demonstrate that the degree of substrate cleavage is dependent upon the position of the caging group relative to the restriction site. Upon irradiation with UV light, the caging group is removed, affording site specific binding and DNA cleavage by the restriction endonuclease (Figure 6).
Figure 6.
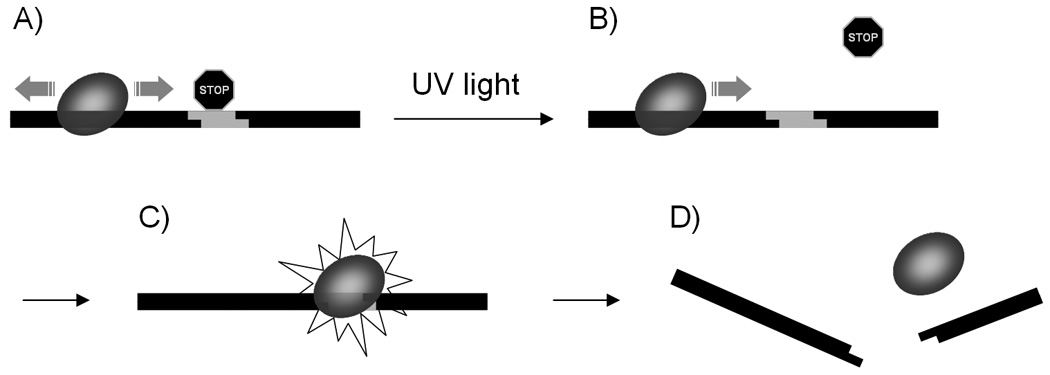
Schematic of the light-triggered DNA cleavage by restriction endonucleases: a) the restriction enzyme is bound to the DNA and scans it for the restriction site which is blocked by a caging group; b) UV irradiation removes the caging group and allows access by the restriction enzyme; c) the enzymes finds the restriction site and cleaves it, leading to d) degraded DNA.
In summary, we have effectively demonstrated both the activation and deactivation of restriction endonucleases via the installation of a photolabile protecting group on the DNA substrate of these enzymes. The results suggest that interplay of enzyme recognition and cleavage inhibition gives rise to this phenomenon. Based on a developed model, we hypothesize that enzyme recognition plays a larger role in restriction enzyme regulation by caging groups, than inhibition of catalysis, as cleavage occurs even when caging groups are distanced from the site of cleavage, however, the rate of catalysis is prolonged. Gratifyingly, in all cases, normal endonuclease activity is completely restored upon the photochemical removal of the caging group. These results indicate the possibility of a differential digestion of two cleavage sites with the same restriction enzyme prior and after light irradiation. Moreover, protection of restriction sites by photocaging groups may have implications on the stability of caged DNA in a cellular environment.
Experimental Section
Light-activated Restriction Enzyme Digests
Oligonucleotides were end labeled using γ32P-ATP (MP Biomedicals) and T4 Kinase (New England Biolabs) at 37 °C for 1 hour, and then purified using TE Midi Select-D, G25 microcentrifuge spin columns (Shelton Scientific). The γ32P-end labeled substrate (10 µL, 1 nmol) was incubated with its complementary strand (10 µL, 1 nmol) at 90 °C for 1 min, and then gradually cooled to 4 °C over 2 hours. The dsDNA construct (2 µL, 0.1 nmol) was then subjected to an enzymatic digest (50 µL total volume) according to manufacturer’s protocols with the appropriate buffer (New England Biolabs). Upon completion, the enzyme was deactivated (70 °C, 20 min), and digests were analyzed on a 20% polyacrylamide gel (400V, 40 min). Acrylamide gels were visualized using a Storm phosphorimaging system, and radioactive band intensities were quantified using Image Quant 5.2.
Supplementary Material
Acknowledgements
We thank Hrvoje Lusic for the preparation of the NPOM-caged thymidine phosphoramidite. DDY acknowledges a graduate research fellowship from the ACS Medicinal Chemistry Division. AD is a Beckman Young Investigator and a Cottrell Scholar. DNA synthesis was conducted in the Biomolecular Resource Facility of the Comprehensive Cancer Center of Wake Forest University, supported in part by NIH grant P30 CA-12197-30.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.a) Young DD, Deiters A. Org. Biomol. Chem. 2007;5:999–1005. doi: 10.1039/b616410m. [DOI] [PubMed] [Google Scholar]; b) Tang XJ, Dmochowski IJ. Molecular Biosystems. 2007;3:100–110. doi: 10.1039/b614349k. [DOI] [PubMed] [Google Scholar]; c) Mayer G, Heckel A. Angew. Chem. Int. Ed. 2006;45:4900–4921. doi: 10.1002/anie.200600387. [DOI] [PubMed] [Google Scholar]
- 2.a) Lusic H, Young DD, Lively MO, Deiters A. Org. Lett. 2007;9:1903–1906. doi: 10.1021/ol070455u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Young DD, Edwards WF, Lusic H, Lively MO, Deiters A. Chem. Commun. 2008:462–464. doi: 10.1039/b715152g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Lusic H, Lively MO, Deiters A. Mol. Biosys. 2008;4:508–511. doi: 10.1039/b800166a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tanaka K, Katada H, Shigi N, Kuzuya A, Komiyama M. Chembiochem. 2008;9:2120–2126. doi: 10.1002/cbic.200800285. [DOI] [PubMed] [Google Scholar]; c) Dmochowski IJ, Tang XJ. Biotechniques. 2007;43:161. doi: 10.2144/000112519. [DOI] [PubMed] [Google Scholar]; d) Tang X, Swaminathan J, Gewirtz AM, Dmochowski IJ. Nucleic Acids Res. 2008;36:559–569. doi: 10.1093/nar/gkm1029. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Tang XJ, Dmochowski IJ. Nature Prot. 2006;1:3041–3048. doi: 10.1038/nprot.2006.462. [DOI] [PubMed] [Google Scholar]; f) Tang XJ, Maegawa S, Weinberg ES, Dmochowski IJ. J. Am. Chem. Soc. 2007;129:11000. doi: 10.1021/ja073723s. [DOI] [PubMed] [Google Scholar]; g) Liu Y, Sen D. J. Mol. Biol. 2004;341:887–892. doi: 10.1016/j.jmb.2004.06.060. [DOI] [PubMed] [Google Scholar]
- 4.Shestopalov IA, Sinha S, Chen JK. Nat. Chem. Biol. 2007;3:650–651. doi: 10.1038/nchembio.2007.30. [DOI] [PubMed] [Google Scholar]
- 5.a) Krock L, Heckel A. Angew. Chem. Int. Ed. 2005;44:471–473. doi: 10.1002/anie.200461779. [DOI] [PubMed] [Google Scholar]; b) Mayer G, Krock L, Mikat V, Engeser M, Heckel A. Chembiochem. 2005;6:1966–1970. doi: 10.1002/cbic.200500198. [DOI] [PubMed] [Google Scholar]; c) Ting R, Lermer L, Perrin DM. J. Am. Chem. Soc. 2004;126:12720–12721. doi: 10.1021/ja046964y. [DOI] [PubMed] [Google Scholar]
- 6.a) Kim TH, Ren B. Annu. Rev. Genomics Hum. Genet. 2006;7:81–102. doi: 10.1146/annurev.genom.7.080505.115634. [DOI] [PubMed] [Google Scholar]; b) Vigneault F, Guerin SL. Expert Rev. Proteomics. 2005;2:705–718. doi: 10.1586/14789450.2.5.705. [DOI] [PubMed] [Google Scholar]; c) von Hippel PH. Annu. Rev. Biophys. Biomol. Struct. 2007;36:79–105. doi: 10.1146/annurev.biophys.34.040204.144521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Zebala J, Choi J, Barany F. Faseb Journal. 1992;6:A358–a358. [Google Scholar]; b) Huang LH, Farnet CM, Ehrlich KC, Ehrlich M. Nucleic Acids Res. 1982;10:1579–1591. doi: 10.1093/nar/10.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pingoud A, Jeltsch A. Eur. J. Biochem. 1997;246:1–22. doi: 10.1111/j.1432-1033.1997.t01-6-00001.x. [DOI] [PubMed] [Google Scholar]
- 9.a) Roberts RJ. Crit. Rev. Biochem. 1976;4:123–164. doi: 10.3109/10409237609105456. [DOI] [PubMed] [Google Scholar]; b) Martin P, Boulukos KE, Pognonec P. BMC Bioinformatics. 2006;7:98. doi: 10.1186/1471-2105-7-98. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Williams RJ. Mol. Biotechnol. 2003;23:225–243. doi: 10.1385/mb:23:3:225. [DOI] [PubMed] [Google Scholar]
- 10.Erskine SG, Halford SE. J. Mol. Biol. 1998;275:759–772. doi: 10.1006/jmbi.1997.1517. [DOI] [PubMed] [Google Scholar]
- 11.Sidorova NY, Rau DC. Proc. Natl. Acad. Sci. U. S. A. 1996;93:12272–12277. doi: 10.1073/pnas.93.22.12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Kim YC, Grable JC, Love R, Greene PJ, Rosenberg JM. Science. 1990;249:1307–1309. doi: 10.1126/science.2399465. [DOI] [PubMed] [Google Scholar]; b) Lukacs CM, Kucera R, Schildkraut I, Aggarwal AK. Nat. Struct. Biol. 2001;8:126–130. doi: 10.1038/84111. [DOI] [PubMed] [Google Scholar]; c) Newman M, Strzelecka T, Dorner LF, Schildkraut I, Aggarwal AK. Nature. 1994;368:660–664. doi: 10.1038/368660a0. [DOI] [PubMed] [Google Scholar]
- 13.Zebala JA, Choi J, Barany F. J. Biol. Chem. 1992;267:8097–8105. [PubMed] [Google Scholar]
- 14.Zebala JA, Choi J, Trainor GL, Barany F. J. Biol. Chem. 1992;267:8106–8116. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.