

Abstract
High throughput screening of 66,000 compounds using competitive binding of peptides comprising the BH3 domain to anti-apoptotic Bfl-1 led to the identification of fourteen validated “hits” as inhibitors of Bfl-1. N-Aryl maleimide 1 was among the validated “hits”. A chemical library encompassing over 280 analogs of 1 was prepared following a two-step synthesis. Structure-activity studies for inhibition of Bfl-1 by analogs of N-aryl maleimide 1 revealed a preference for electron-withdrawing substituents in the N-aryl ring and hydrophilic amines appended to the maleimide core. Inhibitors of Bfl-1 are potential development candidates for anti-cancer therapeutics.
Keywords: Bfl-1 inhibitors, N-aryl maleimides, high-throughput screening, anti-cancer agents
The B-cell lymphoma/leukemia-2 (Bcl-2) family of proteins plays a prominent role in regulating programmed cell death (apoptosis). In humans, proteins within the Bcl-2 family having anti-apoptotic activity include Bcl-2, Bcl-XL, Mcl-1, Bcl-W, Bcl-B, and Bfl-1.1 Members of this group of proteins are commonly over-expressed in human cancers thereby promoting tumorigenesis.2, 3 Additional groups of proteins within the Bcl-2 family have pro-apoptotic activity and include Bak, Bax, Bad, Bim, and Bid. Anti-apoptotic and pro-apoptotic proteins can dimerize, either activating or antagonizing each other.4
Heterodimerization occurs at a hydrophobic pocket on the surface of anti-apoptotic proteins and a region of pro-apoptotic proteins called the BH3 region to neutralize the cytoprotective function of anti-apoptotic Bcl-2 family proteins. The BH3 region is comprised of an amphipathic α-helix of roughly 16-25 amino acids that binds to the hydrophobic region of the anti-apoptotic Bcl-2 family proteins.1 Agents that mimic the BH3 region of pro-apoptotic proteins may be used to antagonize anti-apoptotic Bcl-2 proteins by binding to the hydrophobic pocket thus promoting cell death. Synthetic peptides mimicking the BH3 region have been shown to bind to the anti-apoptotic Bcl-2 proteins and initiate apoptosis.5, 6
Small molecule inhibitors of anti-apoptotic Bcl-2 members have been reported.7 Some natural product inhibitors include gossypol8 and epigallocatechine (EGCG).9 Some synthetic and semisynthetic inhibitors include ABT-737,10 GX15-070,11 apogossypol12 and the commercially available thioxothiazolidine, BH3I-1.13, 14 These agents act by binding to the hydrophobic pocket of anti-apoptotic Bcl-2 proteins, mimicking BH3 peptides to block their cytoprotective effects. Some small molecules inhibit a broad range of the anti-apoptotic Bcl-2 proteins while others are more selective.
Bfl-1 is a NF-κB-inducible member of the Bcl-2 family that is highly expressed in immune cells15. Defective apoptosis has been implicated in several autoimmune diseases, thus suggesting a therapeutic opportunity for agents that neutralize Bfl-1. In addition, Bfl-1 is highly expressed in some cancers accounting for resistance to selective Bcl-2/Bcl-XL inhibitors that fail to neutralize this anti-apoptosis member of the Bcl-2 family. Based on these results, we decided to focus on small molecule inhibitors of Bfl-1 that interact at the BH3 binding site.
Herein, we describe the process used to identify and synthesize promising lead compounds for inhibition of the anti-apoptotic protein, Bfl-1. A high-throughput fluorescence polarization (FP) assay was developed to detect inhibition of Bfl-1 by small molecules.14 In this assay, inhibitors were incubated with glutathione-S-transferase (GST)-Bcl-2 anti-apoptotic fusion proteins.16 FP was measured after addition of an N-terminal fluoroscein isothiocyanate (FITC) BH3 peptide (i.e., FITC-Ahx-EDIIRNIARHLAQVGDSMDR-CO2H).5, 17 Inhibitory potency was confirmed using a LanthaScreen time-resolved fluorescence resonance energy transfer (TR FRET) assay that examined the same molecular interaction between the FITC-peptide and GST-tagged protein.18 An initial “hit” was selected, and exploratory libraries based on the “hit” were generated and tested to identify structural features necessary for inhibition of Bfl-1.
The NIH Roadmap Small Molecule Library (at the time, representing approximately 66,000 compounds from BioFocus, San Francisco, CA) was screened for inhibition of GST-Bfl-1. Twenty compounds were identified in the initial FP assay to inhibit GST-Bfl-1 with potency of 20 μM or less. Of those twenty “hits”, fourteen showed similar GST-Bfl-1 inhibition in the TR-FRET assay. Overall, there was reasonably good agreement between the FP and TR-FRET assay for potency of Bfl-1 inhibition on the basis of IC50 values (shown in Tables 1 and 2). The most potent “hit” was compound 1 (Figure 1) with submicromolar potency in both assays.
Table 1.
Effect of N-aryl maleimide ring substitution on GST-Bfl-1 inhibition
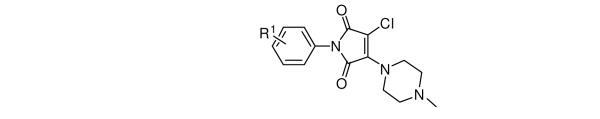 | |||
|---|---|---|---|
| Compound # |
R1 | Bfl-1 FPa IC50 (μM) |
Bfl-1 TR-FRETa IC50 (μM) |
| 1 | 3,4-dichloro-phenyl | 0.6±0.1 | 0.8±0.1 |
| 8 | 3,5-dichloro-phenyl | 2.4±0.4 | 1.9±0.2 |
| 9 | 2,4-dichloro-phenyl | 2.6±0.2 | 1.4±0.1 |
| 10 | 2,3-dichloro-phenyl | 8.7±1.7 | 2.6±0.1 |
| 11 | 2,6-dichloro-3-methyl-phenyl | 23.9±3.9 | 11.1±2.3 |
| 12 | 2-chloro-phenyl | 3.3±0.3 | 1.0±0.1 |
| 13 | 3-chloro-phenyl | 2.1±0.2 | 1.2±0.1 |
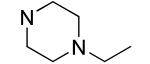
| 14 | 3-methyl-phenyl | 12.6±2.3 | 10.8±0.9 |
| 15 | 3-nitro-phenyl | 6.8±1.4 | 3.9±0.4 |
| 16 | 3-Chloro-pyridin-4yl | 29±3.9 | 35.6±3.9 |
Data are the mean ± SEM.
Table 2.
SAR for analogs of “hit” 1 with varied Amines.
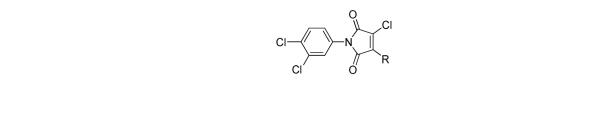 | |||
|---|---|---|---|
| Compound # |
R1 | Bfl-1 FPa IC50 (μM) |
Bfl-1 TR-FRETa IC50 (μM) |
| 17 |
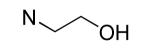
|
>50 | >50 |
| 18 |
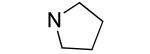
|
>50 | >50 |
| 19 |
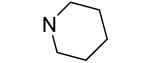
|
>50 | >50 |
| 20 |
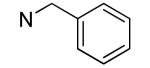
|
>50 | >50 |
| 21 |
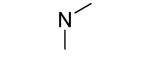
|
0.3±0.2 | 0.4±0.1 |
| 22 |
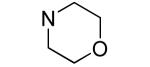
|
0.8±0.1 | 0.45±0.03 |
| 23 |
|
1.7±0.2 | 1.0±0.1 |
| 24 |
|
0.6±0.3 | 0.8±0.1 |
| 25 |
|
2.2±0.4 | 1.1±0.1 |
| 26 |
|
5.6±0.4 | 2.0±0.4 |
| 27 |
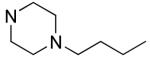
|
10.5±1.7 | 3.3±0.3 |
| 28 |
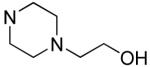
|
0.24±0.03 | 0.62±0.03 |
| 29 |
|
0.3±0.2 | 0.40±0.04 |
| 30 |
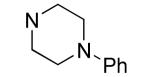
|
4.9±0.8 | 4.1±0.5 |
| 31 |
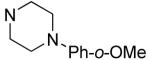
|
9.6±2.0 | 6.2±0.3 |
| 32 |
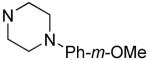
|
1.2±0.4 | 5.0±0.8 |
| 33 |
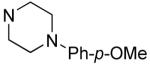
|
>10 | 8.3±2.1 |
| 34 |
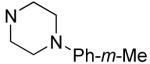
|
>10 | >10 |
| 35 |
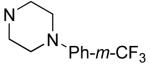
|
>10 | >10 |
| 36 |
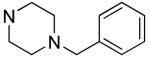
|
2.4±0.2 | 1.3±0.1 |
| 37 |
|
4.1±0.4 | 2.1±0.2 |
| 38 |
|
0.25±0.05 | 0.69±0.03 |
Data are the mean ± SEM.
Figure 1.

Chemical structure of “hit” 1 from a high-throughput screen for inhibition of GST-Bfl-1 and regions of “hit” amenable to SAR work.
“Hit” 1 was amenable to structure-activity relationship (SAR) studies. Three regions of the molecule were examined in an iterative process of chemical synthesis and in both FP and TR-FRET vitro binding assays to define Bfl-1 inhibition SAR potency. The regions for SAR studies included: 1) the chloro substituent(s) and unsaturation of the maleimide ring, 2) substituents on the N-aryl ring, and 3) the amine functionality of the maleimide ring (Figure 1). More than 280 analogs of 1 were prepared to optimize selectivity and potency for inhibition of Bfl-1.
We first investigated the necessity of the chloride substitution and the requirement of unsaturation of the maleimide ring system for potency of Bfl-1 inhibition. Two commercially available analogs of 1 were purchased (ChemBridge, San Diego, CA) and tested as inhibitors of GST-Bfl-1 (Figure 2). Compound 2 did not have the maleimide chloride substituent but retained the maleimide ring double bond while compound 3 possessed neither the chloride nor the ring double bond. Both compounds were not potent (i.e., IC50 > 10 μmol) in both the FP and TR-FRET assays showing the necessity of each moiety for potent inhibition of GST-Bfl-1.
Figure 2.

Analogs of 1 without the maleimide chloro atom and double bond moieties necessary for potent GST-Bfl-1 inhibition.
N-Aryl maleimide analogs were prepared using a convenient two-step process starting from commercially available starting materials (Scheme 1).19 In the first step, substituted aniline 4 was heated with one equivalent of dichloromaleic anhydride 5 in acetic acid for one hour. After extraction and purification by silica gel chromatography, N-aryl dichloromaleimide 6 was isolated. Monosubstitution of the dichloromaleimide with amines was done in a medium throughput format. Dichloromaleimides 6 dissolved in dioxane were combined with amines dissolved in tetrahydrofuran at room temperature to afford 3-chloro-1-aryl-4-amino-1H-pyrrole-2,5-dione analogs of 7. Reaction progress was monitored by TLC and reactions were heated if necessary. Products 7 were purified by preparative TLC and verified by electrospray mass spectrometry. Reactions were conducted on a 5 mg scale. Amounts of product isolated ranged from 3-5 mg.
Scheme 1.

Synthesis of maleimides following a two-step procedure.
Initially, SAR studies of Bfl-1 inhibition were conducted with compounds of structure 7 where the N-methylpiperazine of the side chain amine was kept constant and substituents on the N-phenyl ring were varied. A total of 10 anilines (4) were used (Table 1). Because the original “hit” contained 3,4-dichloro substituents in the N-aryl ring, maleimides with different permutations of mono- and dichloro-substituted N-aryl rings were synthesized. Additional compounds with N-aryl methyl, and nitro substituents were prepared to examine possible electronic substituent effects of the N-aryl ring on GST-Bfl-1 inhibition. From among compounds 1, and 8-16 (Table 1) the original “hit” 1 was the most potent GST-Bfl-1 inhibitor in both the FP and TR-FRET assays (IC50 = 0.6±0.1 μM and 0.8±0.1 μM, respectively). Of the dichlorophenyl compounds examined, both the 3,5- (8) and 2,4-dichlorophenyl (9) substituents afforded IC50 values of 2.4±0.4 μM and 2.6±0.2 μM, respectively, and had similar activities but were three-fold less potent than the original “hit” 1. Other disubstituted dichloro compounds (i.e., 10) had much lower inhibitory potency (i.e., IC50 values of 8.7 μM). Compared to compounds 8 and 9, the 2-chloro or 3-chloro monosubstituted compounds (i.e., 12 and 13, with IC50 values of 3.3±0.3 μM and 2.1±0.2 μM, respectively) were not significantly different in the inhibition potency of GST-Bfl-1. Monosubstituted N-aryl maleimides with either methyl (14) or nitro (15) groups afforded GST-Bfl-1 inhibitors with much greater IC50 values (i.e., 12.6±2.3 μM and 6.8±1.4 μM, respectively) indicating that alkyl groups or polar groups on the N-aryl portion of the maleimide did not increase GST-Bfl-1 inhibition potency. Addition of a 3-methyl group to the 2,6-dichloro aryl group (11) further decreased potency 2-3 fold. Pyridine rings in place of the aryl ring also had decreased potency (16). Based on these results, the 3,4-dichlorophenyl group was chosen as the optimal substituent of the N-phenyl ring for further SAR studies on the piperazine portion of the molecule.
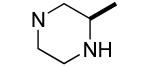
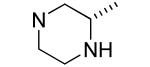
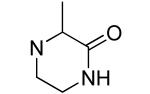
An additional sublibrary of compounds was prepared that retained the 3,4-dichlorophenyl substituent in the N-aryl portion of the molecule while the amine group on the maleimide ring was varied (17-26). Thus, 3,4-dichloro-1-(3,4-dichlorophenyl)-1H-pyrrole-2,5-dione 6 was treated with amines to examine the effect of substitution on the maleimide ring itself. Amines used included morpholines, piperidines, pyrrolidines, cyclohexylamine, ethanolamine, benzylamine, substituted piperazines, piperazinone and dimethylamine. Dimethylamino 21, morpholino 22 and 3-methyl-piperazino 24 afforded compounds equipotent to original hit 1. The remainder of the amine substituents examined (17-20, 23, 25-26) afforded poorly potent compounds. N-terminal piperazines with a center of chirality was tested to examine the stereoselectivity of Bfl-1 inhibition (Table 2).
Compounds 24 and 25 have an unsubstituted nitrogen atom but include a methyl group in the α position resulting in a center of chirality. Compound 24 with R stereochemistry at C-3 of piperazine showed considerable Bfl-1 inhibitory potency (i.e., IC50 values for FP and TR-FRET assays of 0.6±0.3 and 0.8±0.1 μM, respectively). In contrast, compound 25 with the S stereochemistry had considerably less GST-Bfl-1 inhibitory potency (i.e., four-fold greater IC50 value for the FP assay and approximately the same IC50 value for the TR-FRET assay).
Accordingly, another sublibrary of compounds based on compound 1 was prepared that incorporated the 3,4-dichlorophenyl moiety and modification of substituted piperazines to the maleimide structure. Modifications of the 4-N-substituent of the piperazine would allow for properties adjustment such as solubility, metabolic stability in further studies. A complete list of 4-N-substituent tested is available in the supplemental information section. Compounds 27-38 (Table 2) are the most representatives of the SAR trends for this region of the molecule. N-substitution included alkyls, hydroxyl, amine and ether groups, non substituted and substituted phenyl, benzyl, cinnamyl groups.
Hydrophilic hydroxy ethyl groups at the terminal piperazine nitrogen atom were associated with the most potent GST-Bfl-1 inhibitory functional activity in this series (i.e., compounds 28 and 29 with IC50 values 0.24±0.03 and 0.3±0.2 μM, respectively from the FP assay). Compared to “hit” 1, carbon chains longer that methyl on the piperazine terminal nitrogen decreased potency (i.e., 20 and 21 with 1.7±0.2 and 10.5±1.7 μM, respectively). Compared to 1, N-phenylpiperazine (i.e., 24 with IC50 4.9±0.8 μM) had decreased potency and substitution around the terminal phenyl ring in the ortho-, meta- and para-positions (i.e., 31, 33-35 with IC50 >9 μM) showed lower potency in the FP assay. M-methoxy phenyl was the exception 32 showing a moderate potency (i.e., IC50 (FP) value 1.2±0.4 μM) but much improved compared to the m-methyl (33) and m-CF3 (34) equivalents. Both compounds (i.e., 27 and 28) had IC50 values >10 μM for GST-Bfl-1 inhibition in the FP assay. The conclusion was that a hydrophilic pocket was present in GST-Bfl-1 or a hydrogen bonding interaction was occurring with GST-Bfl-1 and 32 between the meta-position to increase inhibitory potency. Compared with 1, maleimides with N-terminal piperazine benzyl or 3,4-methylenedioxybenzyl substituents (i.e., 35 and 36, IC50 values of 2.4±0.2 and 4.1±0.4 μM, respectively) were less potent GST-Bfl-1 inhibitors by two- and four-fold, respectively. Maleimide 38 with a cinnamyl group on the N-terminal piperazine was a potent GST-Bfl-1 inhibitor and comparable to compounds 28 and 29 (i.e., IC50 values of 0.25±0.05 μM versus 0.3±0.2 and 0.3±0.2 μM). Data from the TR-FRET assay were similar for all three compounds (0.4-0.69 μM). It is postulated that the BH3 region of Bfl-1 where the amine functionality of the inhibitor resides is large enough to accommodate larger groups on the maleimide ring. A cinnamyl group may induce additional pi-pi aromatic interactions with Bfl-1 to increased inhibitory potency.
Other combinations of anilines and amines were prepared and tested in order to look at structural synergistic effects (see Table 3 in supplemental information): 3-methoxy, 4-nitro, 3-nitro, 4-methyl, 3-trifluoro, 4-trifluoro anilines in combination of the amines used for compounds from table 2. None of them showed submicromolar potencies.
A potent agent (i.e., 1) and two less potent agents (i.e.,19 and 21) were examined in cell-based viability studies or assays using cancer cell lines or other mammalian cell lines 21. For compound 1, inhibition of human H69AR small cell lung tumor cell growth was observed at a concentration of 10 μM. Compound 21 was not inhibitory to cell viability in a mammalian cell line but compound 19 decreased cell viability at 15 μg/mL. Thus, as a class it does not appear that the compounds possess universal toxicity but depending on the structure, certain N-aryl maleimide Bfl-1 inhibitors can decrease cancer cell viability or cause toxicity to other mammalian cell lines.
In summary, more than 280 substituted maleimides were prepared in a medium throughput format from readily available starting materials. SAR analysis revealed the effects of substitution on the N-phenyl ring and variation of amines on the maleimide ring system, and the necessity of a chloro substituent and a double bond in the maleimide ring for inhibition of GST-Bfl-1. The N-3,4-dichloroaryl moiety of the original “hit” 1 provided the optimal substitution pattern on the N-aryl ring. Optimal amines for substitution of one maleimide chloride atom included hydrophilic amines or amines that could participate in hydrogen bonding or pi-pi interactions. Submicromolar IC50 values for inhibition of Bfl-1 were observed for maleimides substituted with dimethylamine, N-methylpiperazine and piperazines (i.e., 21, 22, 24, 28, 29 and 38) containing water-soluble groups or a cinnamyl group on the terminal nitrogen atom. For one subset of piperazines possessing a center of chirality, considerable stereoselectivity of Bfl-1 inhibition was observed (i.e., 24 > 25). The SAR studies reported herein provide valuable information for the structural requirements for inhibition of Bfl-1 by maleimides and may provide insight into development candidates for anti-cancer therapeutics.
Supplementary Material
Acknowledgments
This work was funded by grant 1 X01 MH077632-01 and UO1-CA-113318 to JCR. We thank Drs. Russell Dahl and Ying Su for their help with the work. We thank Alyssa M. Morgosh for her help in providing analytical data for the manuscript. We thank Dr. Marion Lanier for her careful editing of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Reed JC. Nat. Rev. Drug Discovery. 2002;1:111. doi: 10.1038/nrd726. [DOI] [PubMed] [Google Scholar]
- 2.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M. Nature. 2010;463:899. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnstone RW, Ruefli AA, Lowe SW. Cell. 2002;108:153. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 4.Chen L, Willis SN, Wei A, J. SB, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DCS. Mol. Cell. 2005;7:393. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 5.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. Mol. Cell. 2005;7:525. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 6.Vogler M, Dinsdale D, Dyer MJS, Cohen GM. Cell Death and Differentiation. 2009;16:360. doi: 10.1038/cdd.2008.137. [DOI] [PubMed] [Google Scholar]
- 7.Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. J. Med. Chem. 2003;46 doi: 10.1021/jm030190z. [DOI] [PubMed] [Google Scholar]
- 8.Leone M, Zhai D, Sareth S, Kitada S, Reed JC, Pellecchia M. Cancer Res. 2003;63:8118. [PubMed] [Google Scholar]
- 9.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li Chi, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendy MD, Zhang H, Fesik SW, Rosenberg SH. Nature. 2005;435:677. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 10.Pellecchia M, Reed JC. Curr. Pharm Design. 2004;10:1387. doi: 10.2174/1381612043384880. [DOI] [PubMed] [Google Scholar]
- 11.Wei J, Kitada S, Rega MF, Stebbins JL, Zhai D, Cellitti J, Yuan H, Emdadi A, Dahl R, Zhang Z, Yang L, Reed JC, Pellecchia M. J. Med. Chem. 2009;52:4511. doi: 10.1021/jm900472s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan J. Nat. Cell Biol. 2001;3:173. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- 13.Zhai D, Jin C, Satterthwait AC, Reed JC. Cell Death and Differentiation. 2006;13:1419. doi: 10.1038/sj.cdd.4401937. [DOI] [PubMed] [Google Scholar]
- 14.Holmgreen SP, Huang DCS, Adams JM, Cory S. Cell Death and Differerentiation. 1999;6:525. doi: 10.1038/sj.cdd.4400519. [DOI] [PubMed] [Google Scholar]
- 15.Lee HH, Dadgostar H, Cheng Q, Shu J, Cheng G. Proc. Natl. Acad. Sci. U.S.A. 1999;96:9136. doi: 10.1073/pnas.96.16.9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Werner AB, de Bries E, Tait SWG, Bontjer I, Borst J. J. Biol. Chem. 2002;277:22781. doi: 10.1074/jbc.M201469200. [DOI] [PubMed] [Google Scholar]
- 17.Day CL, Chen L, Richardson SJ, Harrison PJ, Huang DCS, Hings MG. J. Biol. Chem. 2005;280 doi: 10.1074/jbc.M411434200. High throughput FP assay: Chemical inhibitors were dissolved in 10% DMSO at 100 μM stock concentration. From this solution, 4 μL were incubated with 8 μL of Bfl-1 working solution (7.4 nM GST-Bfl-1 fusion protein in 25 mM Bis-Tris pH 7.0, 1 mM TCEP, 0.005% Tween 20) at room temperature away from direct sunlight in 384-well black plates (Greiner Bio-One, Monroe, NC). After 1 hour, FITC-Bid BH3 peptide (8 μL of 5.6 nM in 25 mM Bis-Tris pH 7.0, 1 mM TCEP, 0.005% Tween 20) was added, and plates were incubated at room temperature away from direct sunlight. Fluorescence polarization was measured using an Analyst GT plate reader (Molecular Devices, Inc., Sunnyvale, CA) using fluorescein filters (excitation filter-485 nm, emission filter 530 nm, dichroic mirror # 505 nm). Data analysis was done using CBIS software (ChemInnovations, Inc., San Diego, CA). Dose-response confirmation: Chemical inhibitors were serially diluted in DMSO then in water so final concentration was in 10% DMSO. Ten concentrations of each compound were used. Each inhibitor (4 μL) was transferred to 384-well black plates (Greiner Bio-One, Monroe, NC) followed by GST-Bfl-1 solution described above (8 μL). Plates were incubated for 1 h at 4 °C. FITC-Bid BH3 peptide solution described above (8 μL) was added, and plates were incubated for 4 h at room temperature. Fluorescence polarization was measured at described above. Data analysis was performed using sigmoidal dose-response equation through non-linear regression.
- 18.Peptide Synthesis: FITC-Bid BH3 peptide was synthesized on the ACT 350 Multiple Peptide Synthesizer using Fmoc chemistry on Wang resin. FITC was coupled to the N-terminus while the peptide was still on the resin. Peptide was cleaved from the resin using 94% TFA/2.5% H2O/2.5% EDT/1% TIS for two hours at room temperature. Crude peptide was purified on a Gilson HPLC with 0.1% TFA water/acetonitrile and characterized by mass spectroscopy on an Applied Biosystems Voyager System 6264 using MALDI-TOF ionization. TR-FRET Assay: Dose-response curves contained 10 concentrations of compounds obtained using 2-fold serial dilution. Compounds were serially diluted in 100% DMSO, and then diluted with water to 10% final DMSO concentration. Compounds in 10% DMSO (4 μL) were transferred into columns 3-22 of Greiner 384-well white small-volume plates (784075). Each compound concentration was assayed in duplicate wells. Columns 1-2 and 23-24 contained 4 μL of 10% DMSO. Assay buffer (8 μL of 25 mM Bis-Tris, pH 7.0, 1 mM TCEP, 0.005% Tween 20) was added to columns 1-2, which were reserved for positive controls, using a WellMate bulk dispenser (Matrix). Bfl-1 working solution (8 μL of 7.4 nM GST-Bfl-1 in assay buffer) was added to columns 3-24 using WellMate bulk dispenser (Matrix). Columns 23-24 represent negative control wells. Plates were incubated for 1h at +4 °C. Freshly prepared FITC-Bid/Tb-Ab working solution (8 μL of 5.6 nM FITC-Bid and 2.5 nM Tb-Ab in assay buffer) was added to the whole plate using WellMate bulk dispenser (Matrix). Plates were incubated for 4 h at room temperature protected from direct light. Fluorescence was measured on an M5 plate reader, Molecular Devices (excitation: 340 nm, emission: 490 and 520 nm, cutoff: 475 and 515 nm, respectively) in Time Resolved (TR) mode with signal integrated for 1 ms after initial delay 0.1 ms and averaged from 5 readings. The TR-FRET signal was calculated as the ratio of TR-Fluorescence at 520 nm to TR-Fluorescence at 490 nm. Data analysis was done using a sigmoidal dose-response equation through non-linear regression. Statistical Analysis: Analysis of the inhibition kinetics was done with Graphpad Prism (San Diego, CA) software. A website explaining the meaning of SEM (reported as ± in the IC50 data in the Tables) can be found at: http://graphpad.com/help/prism5/prism5help.html?usingstatistical_analyses_step_by_s.htm.
- 19.Zhai D, Jin C, Shiau C.-w., Kitada S, Satterthwait AC, Reed JC. Mol Cancer Ther. 2008;7:1639. doi: 10.1158/1535-7163.MCT-07-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanaineh-Abdelnour L, Bayyuk S, Theodorie R. Tetrahedron. 1999;55:11859. [Google Scholar]
- 21. http://www.ncbi.nlm.nih.gov/pccompound.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.