

Synopsis
β-thalassemia is a genetic disorder caused by mutations in the β-globin gene and characterized by chronic anemia due to ineffective erythropoiesis, and accompanied by a variety of serious secondary complications such as extramedullary hematopoiesis, splenomegaly, and iron overload. In the past few years, numerous studies have shown that such secondary pathologies have a genetic basis due to the abnormal expression of genes with a role in controlling erythropoiesis and iron metabolism. In this article, the most recent discoveries related to the mechanism(s) responsible for anemia/ineffective erythropoiesis and iron overload will be discussed in detail. Particular attention is paid to the pathway(s) controlling the expression of hepcidin, which is the main regulator of iron metabolism, and the Epo/EpoR/Jak2/Stat5 signaling pathway, which regulates erythropoiesis. Better understanding of how these pathways function and are altered in β-thalassemia has revealed several possibilities for development of new therapeutic approaches to treat of the complications of this disease.
β-thalassemia
Genetic causes, consequences and pleiotropic effects
As discussed in more detail in the Overview by Sankaran and Nathan at the beginning of this issue, β-thalassemia is an inherited disorder characterized by mutations in the gene encoding β-globin that lead to the quantitative reduction or, in the most severe cases, the total absence of β-globin synthesis in human erythroid cells. As a consequence, α-globin chains accumulate in excess, forming aggregates that impair erythroid cell maturation, which ultimately leads to a chronic hemolytic anemia and ineffective erythropoiesis (Fig. 1). The severity of the clinical manifestations in β-thalassemia varies widely, ranging from patients that are almost asymptomatic to individuals who suffer from severe anemia and require regular blood transfusion to sustain life (1-6). In general, the clinical severity of the disease correlates with the size of the free α-chain pool and the degree of imbalance between the production of α- and β-like globin chains.
Fig.1. Consequences of ineffective erythropoiesis and abnormal erythrocyte production.
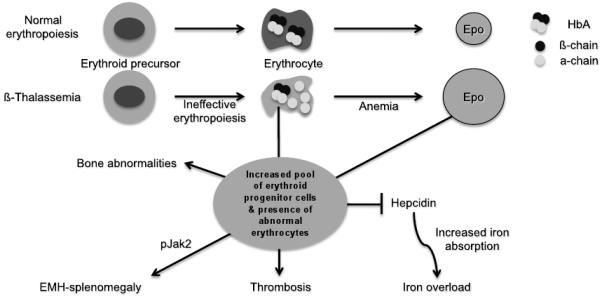
Lines ending with an arrow indicate activation. Lines ending with a line indicate repression.
The α/β globin chain imbalance is responsible for the hemolysis of red blood cells and for the premature death (apoptosis) of erythroid precursors in the bone marrow and at extramedullary sites (Fig. 1). The α-globin chain aggregates form inclusion bodies responsible for oxidative stress and membrane damage within red blood cells and immature developing erythroblasts. These events are followed by the premature death of many late erythroid precursors in the bone marrow and spleen. Together, these phenomena are designated as ineffective erythropoiesis (IE). The anemia and resulting hypoxia lead to a dramatic increase in serum erythropoietin (Epo) levels in an attempt to compensate for the reduced oxygen carrying capacity. The marked increase in Epo stimulation, if it is not inhibited by proper transfusion therapy, can lead to uncontrolled expansion of erythroid precursors in the marrow as well as in other sites, such as the spleen and liver, leading to extramedullary hematopoiesis (EMH) (Fig. 1).
The defect in hemoglobin synthesis that occurs in β-thalassemia ultimately leads to the development of pleiotropic effects on different body compartments. One of the consequences of IE and EMH, for example, is splenomegaly (Fig. 1). The abnormal and damaged red blood cells that are produced in β-thalassemia are sequestered by the reticuloendothelial system in the spleen, causing its enlargement. This enlargement leads to increased sequestration of RBCs, including transfused RBCs, in the spleen, with worsening of the anemia and an increase in transfusion requirement. Increased Epo synthesis is also reflected in marrow expansion, leading to bone marrow hyperplasia, bone deformities and osteopenia (Fig. 1), which contribute to increased morbidity as the disease progresses (7).
Iron overload, however, is the principal and multifaceted complication of β-thalassemia. Physiologically, it is caused by an increased absorption of iron from the gastrointestinal (GI) tract as a consequence of ineffective erythropoiesis., and is greatly aggravated by chronic transfusion therapy. Thus, transfusion-independent individuals with thalassemia intermedia have a slower progression of iron overload and generally develop complications later in life compared to patients with thalassemia major who are chronically transfused (Fig. 1). A remarkable variability of tissue iron distribution has been observed in β-thalassemia with liver, heart and endocrine glands being the organs most severely affected (5). Accumulation of iron at these sites (siderosis) leads to oxidative damage due to the generation of reactive oxygen species (ROS). Death from cardiac failure is the most common clinical consequence of iron excess. Pathological analysis of cardiac tissue has shown that this occurs initially by hypertrophy and dilatation followed by degeneration of myocardial fibers and fibrosis. Endocrine problems are also related to iron overload (Fig. 1). These include hypogonadism, which leads to disturbances of growth and sexual maturation, as well as hypothyroidism, hypoparathyroidism and diabetes mellitus that are seen in variable numbers of patients. Therefore, effective iron chelation therapy is a critical component in the management of thalassemia (8-10). For more than three decades, deferoxamine (DFO, Desferal®) was the chelator of choice (11). While this drug is capable of placing all patients into net negative iron balance, it suffers from the fact that it must be infused subcutaneously over 8 to 12 hours, 5 to 7 days a week for the duration of life. Not surprisingly, adherence to such a regimen is generally poor. More recently, two orally effective iron chelators, Exjade® (DFX, deferasirox) and deferiprone (DFP, Ferriprox®), have been developed to overcome this limitation (11). The topics of iron overload and chelation therapy are discussed in greater detail elsewhere in this issue in the article by Porter and Shah.
Currently, the only definitive cure for thalassemia is the transplant of hematopoietic stem cells (HSC) from cord blood or the bone marrow. This topic is discussed in detail elsewhere in this issue in the article by Gaziev and Lucarelli, as well as that by Kanathezhath and Walters. Splenectomy is another facet in the management of β-thalassemia. However, removal of the spleen can lead to further problems such as an increased risk of infections, pulmonary hypertension and thrombosis (12-14), and is usually considered only in the most severe cases of splenomegaly. New approaches to treat/cure the disease are being developed, such as gene therapy, discussed in the article by Bank elsewhere in this issue, or the use of induced pluripotent stem cells as alternatives to the use of HSC (15,16).
Mouse models of β-thalassemia
The use of mouse models of β-thalassemia has been of fundamental importance in clarifying the molecular mechanisms responsible for disease. The two available mouse models of β-thalassemia intermedia are th1/th1 and th3/+. In the th1/th1 mouse, the deletion removes the βmajor gene in the homozygous state (17), producing hemoglobin levels in the range of 9 to 10 g/dL (17-19). In the th3/+ mouse model, the deletion removes both the βminor and βmajor genes in the heterozygous state (20,21). These mice have hemoglobin levels in the range of 8 to 9 g/dL (19-21). Both models have a degree of disease severity (hepatosplenomegaly, anemia, aberrant erythrocyte morphology) comparable to that of patients affected by transfusion-independent β-thalassemia intermedia. Their ineffective erythropoiesis is characterized by a modest reduction in red blood cells and an increase in reticulocytes (22). Homozygous (th3/th3) mice that completely lack any adult β-globin chain synthesis die late in gestation (20), precluding their use as a model for β-thalassemia major. To overcome this limitation, th3/th3 mice are generated by transplantation of hematopoietic fetal liver cells (HFLCs) into lethally irradiated syngeneic adult recipients, the HFLCs being harvested from th3/th3 embryos at 14.5 days of gestation (22). These mice exhibit severe anemia (3-4 g/dL) 6-8 weeks post-transplant as well as very low red blood cell (RBC) levels and reticulocyte counts, together with massive splenomegaly and extensive EMH (22).
Ineffective erythropoiesis
The Epo/EpoR/Jak2/Stat5 pathway and its potential effect(s) on iron intake in erythroid cells
The hallmark of β-thalassemia is IE that stems from a lack of or reduced synthesis of β-globin, which leads to an excess of α-globin chains that aggregate and precipitate, adhering to the membrane of erythroid precursors. These α-globin aggregates cause cellular and membrane damage, apoptosis of the erythroid precursors in the bone marrow and generation of mature red cells that are abnormal and accumulate in very limited numbers. Moreover, the production of red cells, EMH and the anemia can change significantly over time. Therefore, β-thalassemia serves as a good example of the dynamic balance between expansion of the erythroid pool and production of red cells, and of the many factors that can alter this relationship with critical effects. When pathological levels of damaged erythrocytes are trapped in the spleen they cause splenomegaly, anemia and hypoxia. Anemia and hypoxia, in turn, stimulate Epo synthesis, which increases the number of erythroid precursors and abnormal mature red cells in circulation. This exacerbates the trapping of erythrocytes in the spleen thereby worsening the splenomegaly. Moreover, increased erythropoiesis augments iron absorption. Iron overload can increase the formation of ROS causing damage to many organs and further aggravating the anemia. Blood transfusion is a very effective method for ameliorating the anemia and the consequences of IE. But good adherence to iron chelation therapy is necessary to prevent the detrimental effects of iron overload.
Epo is a 34-kDa renal glycoprotein that functions as the main regulator of erythropoiesis, both under basal and stress conditions. Epo binds to its specific receptor, the erythropoietin receptor (EpoR), at the surface of erythroid cells. EpoR is expressed by the earliest erythroid progenitors at the burst forming unit-erythroid (BFU-E) stage (23-25). The Epo/EpoR signaling pathway begins with dimerization of the receptor and activation of the tyrosine-Janus kinase 2 (Jak2) (26-29), which preassembles at a conserved site in the cytoplasmic domain of the EpoR. Jak2 catalyzes transfer of the gamma-phosphate group of adenosine triphosphate to the hydroxyl groups of specific tyrosine residues in signal transduction molecules and mediates signaling downstream of cytokine receptors after ligand-induced autophosphorylation of itself and of tyrosine residues of the receptor. In particular, Jak2 mediates the phosphorylation of tyrosine residues localized in a conserved cytoplasmic domain of the EpoR. The resulting phosphorylated tyrosine residues of the EpoR function as a ‘scaffold’ or docking sites for the assembly of signal transduction factors containing Src-homology 2 (SH2) domains. The main downstream effectors of Jak2 are a family of transcription factors known as signal transducers and activators of transcription (Stat). In erythroid cells, the main target is Stat5 which, upon phosphorylation, translocates to the nucleus as a dimer to drive expression of target genes. The ultimate effect of Jak2 and Stat5 activation is to induce multiple signaling pathways designed to regulate erythroid proliferation and differentiation, and to protect the cells from apoptosis (28,30,31).
Polycythemia vera (PV), essential thombocytosis (ET) and primary myelofibrosis (PMF) are classified as myeloproliferative disorders (MPD), a subgroup of myeloid malignancies (32). These are clonal stem cell diseases characterized by an expansion of morphologically mature cells of the granulocyte, erythroid, megakaryocyte, or monocyte lineage. Interestingly, several studies have described a close association between an activating JAK2 mutation (Val 617 to Phe; JAK2V617F) and these disorders (33). This mutation is thought to prevent the pseudokinase domain from inhibiting the kinase domain, resulting in a constitutively active state of the protein. Thus, JAK2V617F confers constitutive kinase activation. In erythroid cells this leads to STAT5 phosphorylation and Epo-independent erythroid colony formation (34).
Mice lacking Stat5 expression (Stat5−/−) have shown early lethality associated with microcytic anemia and enhanced apoptosis of early erythroblasts (35-37). While the anemia in these mice correlates with loss of expression of the antiapoptotic Bcl-XL gene and enhanced apoptosis (38), additional analyses indicate that Stat5−/− mice have a significant decrease in expression of the iron transporter transferrin receptor-1 (Tfr1) (38,39). Therefore, it is possible that erythroid cells might express higher levels of Tfr1 under conditions of constitutive activation or up regulation of Jak2. Potential consequences of Jak2 modulation of iron intake in β-thalassemic red cells will be discussed in the next section.
New studies on Jak2 and IE in β-thalassemia
Original ferrokinetic studies and analysis of erythroid precursors in β-thalassemia indicated that many of these cells die in the marrow and extramedullary sites (40-43), and suggested that the relative excess of α chains triggering apoptosis was responsible for IE. However, several recent observations suggest that the balance between proliferation and differentiation in some of the erythroid precursors might be different under normal conditions and those of IE (44). These results and those of new studies in mice (discussed later) suggest that the mechanism(s) and various factors associated with the process known as IE have not yet been completely elucidated.
The splenomegaly exhibited by th3/+ mice(44) is very similar to that observed in patients affected by β-thalassemia intermedia (transfusion independent). Studies on th3/+ mice have shown that their spleen fills with erythroid precursors and sequesters abnormal/damaged erythrocytes, likely contributing to lowering of the hemoglobin level over time (44). Similar observations have been confirmed in splenic specimens from patients with thalassemia intermedia (15,44,45). However, analysis of the erythroid precursors in both the bone marrow and spleen in these animals indicated that a large number of these cells were actively proliferating while the proportion of cells undergoing apoptosis, although higher than in normal mice, was relatively modest (44). Compared to wild-type animals, th3/+ mice have a higher number of erythroid cells associated with the expression of cell cycle-promoting genes such as EpoR, Jak2 (Fig. 1), Cyclin-A, Cdk2 and Ki-67, together with increased levels of the Bcl-XL protein (44). These cells also differentiate less than the corresponding normal cells in vitro. Based on this observation, we can speculate that thalassemic patients may increase the number of erythroid precursors in their spleen and liver over time, this being one factor leading to hepatosplenomegaly. In turn, this would also exacerbate the anemia in β-thalassemia (15,44,45).
We already discussed the increase of Epo levels in response to anemia and hypoxia. Based on the observations above, it is reasonable to expect that increased Epo levels could also activate the EpoR/Jak2 pathway, leading to a “physiological” gain of function of Jak2. Under these circumstances, the persistent phosphorylation of Jak2 might lead to an increased number of erythroid progenitor cells. Therefore, suppression of Jak2 activity might modulate IE. Based on this model, we showed that use of a Jak2 inhibitor has a beneficial effect in limiting IE, splenomegaly and the number of erythroid progenitor cells in β-thalassemia (44). In particular, limiting the number of erythroid progenitors might have a beneficial effect on iron metabolism, since it has been suggested that these cells are the most plausible source of the erythroid factor, whose function is to increase iron absorption by repressing hepcidin expression, as shown in Fig. 1 and 2 and further discussed below. Therefore, future studies will need to address whether the use of Jak2 inhibitors could also limit iron absorption in β-thalassemia.
Fig.2. Potential pathways controlling iron absorption in β-thalassemia.
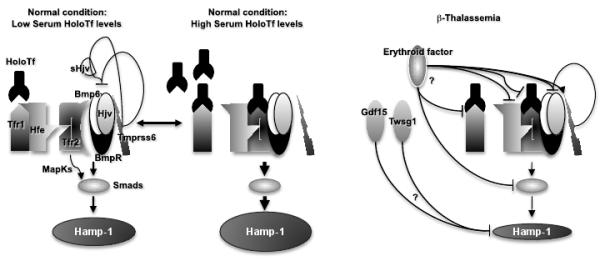
Lines ending with an arrow indicate activation. Lines ending with a line indicate repression.
Activation of the Epo/EpoR/Jak2 pathway is not likely to be the only cause for the limited erythroid differentiation observed in β-thalassemia. For example, in the absence of other erythroid defects, mutations responsible for the constitutive activation of Jak2 lead to the development of polycythemia vera rather than IE (46). Therefore, it is possible to predict that other factors and/or abnormal physiological conditions present in β-thalassemia interfere with erythroid cell differentiation. Among the possible factors acting together with Jak2, iron overload, ROS, the unbalanced synthesis of globin chains and/or heme can be considered (45). Iron is essential for all cells but is toxic in excess. We recently focused on the potential role of iron and heme in modulating IE. Our hypothesis was that thalassemic erythroid cells accumulate an excess of toxic heme associated with free α chains, leading to the formation of ROS, which has been associated with red cell hemolysis and altered differentiation (47,48). Our preliminary data suggest that this is the case and that if the hemoglobin content per cell (MCH) and the overall amount of heme is reduced in thalassemic erythroid cells, both ROS and the accumulation of α-chains on red cell plasma membranes are reduced, ameliorating not only the quality and lifespan of the RBCs, but also the anemia and IE (Gardenghi et al, unpublished data). The amelioration of IE was associated with decreased splenomegaly and a better balance between erythroid precursors and mature RBCs. Based on these preliminary observations, we speculate that ROS play a role in modulating the balance between proliferation and differentiation of the erythroid cells. Additional studies point to the role of ROS in cell differentiation in erythropoiesis, stem cells and cancer, supporting this hypothesis (49,50).
Serum iron is bound to transferrin and enters erythroid cells primarily via receptor-mediated endocytosis of the transferrin/transferrin receptor 1 (Tfr1) complex. Tfr1 is essential for developing erythrocytes. Reduced Tfr1 expression is associated with anemia. Stat5-null mice are severely anemic and die perinatally. Two studies have associated Stat5 with iron homeostasis (38,39), showing that ablation of Stat5 leads to a dramatic reduction in both the mRNA and protein levels of iron regulatory protein 2 (IRP-2) and Tfr1. Both genes have been shown to be direct transcriptional targets of Stat5, establishing a clear link between EpoR/Jak2/Stat5 signaling and iron metabolism. As we proposed previously, reduced iron intake in erythroid cells limits the formation of toxic α-chain/heme aggregates and ROS formation, having a beneficial effect on IE. This has been supported indirectly by the work of Ginzburg and colleagues (51). In their studies, iron delivery to erythroid cells in mice affected by β-thalassemia intermedia was modulated by administration of apo-transferrin. This was associated with reduction of splenomegaly and IE, improvement in hemoglobin levels and an increased number of RBCs. The hemoglobin content per cell (MCH) was also decreased, as was the formation of membrane bound α-chain aggregates. These data reinforce the notion that decreasing iron availability, either by decreasing iron absorption or transferrin saturation, is beneficial to abnormal erythroid cells. Therefore, based on the association between Jak2 and Tfr1, Jak2 inhibitors might also ameliorate IE by decreasing expression of Tfr1, iron intake, formation of toxic α-chain/heme aggregates and ROS. In conclusion, use of Jak2 inhibitors in β-thalassemia might also be beneficial due to their role in controlling iron metabolism in erythroid cells.
Iron metabolism
Hepcidin and regulation of iron absorption
Erythropoiesis and iron metabolism are closely interconnected. The iron utilized by the body is obtained by recycling that present in senescent red blood cells or absorbed from the diet at the level of the proximal intestine. More than two-thirds of the body’s iron content is incorporated into hemoglobin in developing erythroid precursors and mature red blood cells (52). Hepcidin (HAMP/Hamp) (53,54), a cysteine-rich 25-amino acid peptide synthesized in the liver from an 84-amino acid prepropeptide plays a major role in iron homeostasis. Hepcidin was originally isolated from blood ultra filtrate and urine (53,54). Its target is ferroportin, which is the only known iron exporter on enterocytes, hepatocytes and macrophages. Hepcidin binds ferroportin promoting is internalization and degradation (55,56), thereby negatively regulating iron absorption and iron recycling within the body. Hepcidin is up regulated in response to iron overload (57) and inflammation (58-61), and down regulated by erythropoietic stimuli such as anemia, hypoxia or EPO synthesis/administration (58,62,63). In all these scenarios, hepcidin acts primarily on ferroportin controlling iron egress from enterocytes and macrophages, and modulating dietary iron absorption as well as erythropoiesis.
In the last few years, many studies have characterized proteins that contribute to hepcidin regulation. Fig. 2 is a non-exhaustive representation of the proteins and pathways involved in hepcidin regulation. Hepatic cells can take up holo transferrin (holoTf) through Tfr1 by receptor-mediated endocytosis. The same region of Tfr1 that binds holoTf is also recognized by Hfe (Fig. 2) (64,65). HFE is an atypical HLA class I protein. Mutations in HFE cause hemochromatosis, which is a disorder characterized by excess total body iron due to hyper absorption from the diet (66). When the concentration of holoTf in the serum increases, both holoTf and Hfe bind to Tfr2, which is a protein homologous to Tfr1, although the relative affinity of Tfr2 for holoTf and Hfe is reduced compared to that of Tfr1 (67-69). The Hfe/Tfr2 complex then interacts with a second protein complex to signal up-regulation of hepcidin. This second complex of proteins involves the association of bone morphogenetic protein-6 (Bmp6) ligand with a complex of type I and type II serine threonine kinase receptors and the hemojuvelin (Hjv) co-receptor yielding the “hepcidin signaling” or “iron sensor complex” (Fig. 2) (68,70-74). The resulting complex propagates the signal through phosphorylation of cytoplasmic effectors Smad1, Smad5 and Smad8. Once phosphorylated, Smad1/5/8 form heteromeric complexes with the common mediator Smad4 and translocate to the nucleus where they modulate transcription of target genes, including hepcidin (73,75). Moreover, it has been shown that Tfr2 can be activated by its ligand holoTf leading to stimulation of the extracellular signal regulated kinase (Erk)/mitogen activated protein kinase (MapK) pathway, and induction of hepcidin (76,77). Even in this case, Erk activation by holoTf provokes increased levels of phospho-Smad1/5/8.
Mutations in some of these proteins (Hamp, Hfe, Hjv, Tfr2, Bmp6) have clearly been associated with conditions that lead to iron overload due to the fact that they impair hepcidin expression, either directly or indirectly (78,79). In contrast, mutations in TMPRSS6, a type II transmembrane serine protease, are associated with a condition termed iron-refractory iron-deficiency anemia (IRIDA), in which hepcidin expression is increased and both patients and mice suffer from iron deficiency and severe anemia (80,81). Several studies indicate that Tmprss6 targets and degrades Hjv, preventing its assembly in the “iron sensor complex” profoundly impairing the Bmp6/Smad pathway (Fig. 2) (82). A soluble form(s) of Hjv (sHjv) might also act as a decoy for some of the proteins in the complex, limiting its assembly (83). Moreover, it has been shown that mice deficient in Tmprss6 have decreased iron stores and decreased Bmp6 mRNA, but markedly increased mRNA for Id1, another target gene of Bmp6 signaling. Id1, whose promoter is strongly activated by Bmp6 in a Smad-dependent manner, encodes a negative inhibitor of basic helix-loop-helix (bHLH) proteins. Mice deficient in both Tmprss6 and Hjv showed decreased hepatic levels of hepcidin and Id1 mRNA whereas Bmp6 mRNA was markedly increased (84). These mice suffer from systemic iron overload similar to mice deficient in Hjv alone (85). Such findings suggest that regulation of hepcidin expression and maintenance of systemic iron homeostasis by Tmprss6 requires down regulation of Bmp6/Smad signaling and expression of Id1.
As mentioned previously, hepcidin synthesis is down regulated by erythropoietic stimuli such as anemia, hypoxia and Epo synthesis/administration (58,62,63), thereby increasing the GI absorption of iron and its release from stores. It has been postulated that an “erythroid regulator” modulates these responses by acting, directly or indirectly, on the synthesis of hepcidin (Fig. 2). The existence of an erythroid factor is supported by studies in which the serum of patients affected by β-thalassemia or Hfe-related hemochromatosis were compared in terms of their ability to induce the expression of hepcidin and other genes related to iron metabolism in hepatic cells. Sera from β-thalassemia major and intermedia patients down regulated hepcidin expression, while that from those affected by hemochromatosis had no effect on hepcidin (86). While it has been suggested that hypoxia and Epo suppress hepcidin expression, there is considerable evidence indicating that the erythropoietic regulator must involve a soluble factor from the hematopoietic bone marrow. For instance, inhibitors of erythropoiesis can be administered after phlebotomy to disassociate the effects of anemia, hypoxia, and Epo from those of increased erythropoiesis. Phlebotomized mice develop anemia, tissue hypoxia, increased levels of Epo and erythropoiesis and decreased levels of hepatic hepcidin mRNA. When erythropoietic inhibitors were administered, hepcidin mRNA rose dramatically, even though the mice were anemic, hypoxic and exhibited elevated Epo levels (87). If this scenario is correct, it is reasonable to predict that the mediator is a factor secreted by erythroid cells, most likely immature and proliferating erythroid cells. Therefore, we propose that this factor should be more abundant under conditions in which the number of erythroid progenitors is increased, such as after recovery from acute anemia or when IE is present (Fig. 1 and 2). Since erythropoiesis is the most important process utilizing iron in the body, we also speculate that the “erythroid regulator” operates very efficiently in controlling iron metabolism. In other words, the function of the erythroid regulator should be to maintain production of erythrocytes irrespective of the body’s iron balance. However, as we do not know yet how this factor works, we can only speculate that it might operate by targeting one or more components of the “iron sensor complex”, the mediators activated by this complex or the hepcidin promoter, as shown in Fig.2.
The role of hepcidin in β-thalassemia
Studies have shown that the rate of iron absorption from the GI tract in patients affected by β-thalassemia is approximately 3-4 times greater than that in healthy individuals (88). But how does the increased iron absorption affect tissue iron distribution and ultimately erythropoiesis in β-thalassemia? Ferrokinetic studies showed that when donor serum labeled with 59Fe was injected into healthy subjects, 75% to 90% of the iron was incorporated into newly formed red cells within 7 to 10 days. However, when 59Fe was injected into thalassemic patients, only 15% to 20% of it was found in circulating erythrocytes (40). It was hypothesized that the remaining iron was sequestered in those organs where erythroid precursors are subject to premature destruction, such as the bone marrow in humans, and the bone marrow and spleen in mice.
Numerous studies have demonstrated that altered hepcidin expression is responsible for the increased iron absorption observed in β-thalassemia (89-91). In particular, the correlation between IE, iron distribution, and the expression of hepcidin have been investigated in mice affected by thalassemia intermedia and major, models characterized by different degrees of anemia and IE (19,89). The major conclusion is that the pattern of iron distribution in β-thalassemia is dictated by the degree of IE. Where severe anemia exists, hepcidin levels are extremely low, and iron overload occurs rapidly involving predominantly liver parenchymal cells. On the other hand, when the anemia is milder, iron accumulates progressively in splenic macrophages and Kupffer cells in the liver. When thalassemia major mice were transfused, their anemia and IE improved, while iron deposition in the liver was reduced. Analyses using human specimens indicated that urinary hepcidin levels were lower in β-thalassemic patients than those which would be predicted by their iron burden, while transfusion therapy led to an increase in the hepcidin levels(92-96).
In the case of mice affected by β-thalassemia intermedia, it has been observed that they exhibit very low hepcidin levels during the first months of life (16,89,91,97-99). As these animals age, the level of hepcidin increases, resulting in increased organ iron concentrations (16,89,98). This indicates that hepcidin is still partially responsive to iron overload when IE is relatively low. In contrast, the extreme degree of IE in mice affected by thalassemia major limited hepcidin from sensing the iron burden and kept its expression very low. Altogether these observations indicate that the relative levels of IE and iron overload mediate the synthesis of hepcidin in β-thalassemia.
Two erythroid regulators have been proposed called growth differentiation factor 15 (GDF15) and twisted gastrulation protein homolog 1 (TWSG1) (100,101). They are members of the TGF-β super family of proteins known to control proliferation, differentiation, and apoptosis in numerous cell types. Both GDF15 and TWSG1 are elevated in the serum of β-thalassemic patients and suppress hepcidin expression in vitro (101). Comparing normal and thalassemic erythroid cells differentiating in vitro, GDF15 was isolated during the final stages of erythroid differentiation (101). This suggests that GDF15 is secreted by erythroid precursors undergoing cell death. In fact, GDF15 is not elevated after stem cell transplantation in patients with hematopoietic malignancies or in patients with iron deficiency secondary to blood donation, conditions in which apoptosis of erythroid cells is not observed (102,103). These findings confirm that GDF15 is not produced by proliferating erythroid precursors but rather by apoptotic erythroid cells, as in β-thalassemia or refractory anemia with ring-sideroblasts (104). Therefore, GDF15 may limit hepcidin synthesis when erythroid precursors undergo cell death (44). In contrast to GDF15, the highest levels of TWSG1 were detected at early stages of erythroblast differentiation, before hemoglobinization of the cells (100). Future studies will determine whether TWSG1 is an erythroid factor present only in sera of thalassemic patients or whether it is associated with many other conditions characterized by increased erythropoiesis and hepcidin suppression.
Questions and potential novel therapies
Administration of Jak2 inhibitors, and potential effects following reduced iron intake by erythroid cells
Previously, we discussed how Jak2 might influence IE, splenomegaly and anemia in β-thalassemia. One obvious consequence of these observations has been to investigate whether Jak2 inhibitors might have beneficial effects in reducing/preventing splenomegaly and ameliorating the clinical phenotype of this disease. Our preclinical data obtained by using Jak2 inhibitors in mice affected by β-thalassemia intermedia support the notion that patients might benefit from using such compounds (44). In fact, many patients with thalassemia intermedia develop splenomegaly and the need for transfusion therapy, most of them eventually requiring splenectomy. However, because the V617F mutation is localized in a region away from the adenosine triphosphate (ATP)-binding site of JAK2, the ATP-competitive inhibitors of JAK2 kinase (ATP analogues) presently utilized in clinical trials are not likely to discriminate between the wild-type and mutant enzyme. Therefore, their use could decrease RBC synthesis and worsen anemia. With these facts in mind, we tested the effect of administering a Jak2 inhibitor to thalassemic mice in association with blood transfusion. Our preliminary data indicate that the combined use of a Jak2 inhibitor and blood transfusion is superior to either treatment alone in ameliorating splenomegaly and IE (Melchiori et. al. unpublished data). Thus, Jak2 inhibitors might be used temporarily to reduce the spleen size and, in the presence of blood transfusions, to treat or prevent worsening of the anemia. The ultimate goal would be to prevent or delay the need for splenectomy and indirectly to improve the management of the anemia thereby reducing the need for blood transfusions. Moreover, future studies should investigate whether Jak2 inhibitors, through their potential modulation of Tfr1 synthesis, can also modify iron uptake into forming thalassemic erythroid cells, limiting the formation of α-chain/heme aggregates and ameliorating the phenotype of the erythroid cells.
Administration of hepcidin agonists or activators of hepcidin expression
Due to hepcidin deficiency, patients with β-thalassemia intermedia develop iron overload in a manner similar to those with hereditary hemochromatosis. Accordingly, abnormal iron absorption in these patients might be prevented by administration or up regulation of hepcidin (Gardenghi et al., Ann. N.Y. Acad. Sci., 2010, in press; Gardenghi et al. unpublished data). In β-thalassemia major, transfusions rather than dietary iron absorption are the predominant cause of iron overload. In affected individuals, hepcidin levels are higher because IE is suppressed by transfusion therapy. Moreover, hepcidin production is stimulated by the additional iron load derived from the transfusion of red cells. However, hepcidin concentrations decrease in the intervals between transfusions, as the effect of each transfusion is lost (92,93,101). Thus, although intestinal iron absorption contributes less to the total iron load in these patients, hepcidin therapy may be effective when endogenous hepcidin falls and intestinal iron uptake increases. The potential benefits of hepcidin therapy are already supported by a few studies using mouse models of hemochromatosis (65,105). Based on these assumptions and observations, we are further investigating whether or not modulation of hepcidin could be beneficial in β-thalassemia. For this purpose we are utilizing mice affected by β-thalassemia intermedia genetically altered to over-express hepcidin.
Our preliminary data indicate that hepcidin-mediated iron restriction may not only ameliorate iron overload in mice affected by β-thalassemia intermedia, but also improve their erythropoiesis. We hypothesize that amelioration of erythropoiesis in these mice is due to decreased α-chain/heme aggregates and ROS formation. α-chain/heme aggregates can precipitate and lodge in red cell plasma membranes, affecting the properties and lifespan of RBCs. Therefore, reduction of these aggregates might improve the phenotype of the thalassemic red cells and increase their lifespan, with positive feedback on IE (Fig. 3). Moreover, these aggregates are also likely to cause the formation of ROS in erythroid cells. In fact, ROS levels are elevated in red cells derived from patients with β-thalassemia. As a result, glutathione (GSH) levels are lower than in normal red cells (106). ROS likely exacerbate the anemia, further decreasing the already shortened survival of mature RBCs in the circulation (47,107). Excessive ROS formation in maturing erythroid cells might also affect the cell cycle and worsen IE. The Forkhead Box O (FoxO) family of transcription factors plays an essential role in the regulation of oxidative stress in hematopoietic stem and erythroid cells (108). In FoxO3 null mice, red cell survival is reduced and is associated with enhanced mitotic arrest of intermediate erythroid progenitor cells, resulting in a decreased rate of erythroid maturation (48). This raises the interesting possibility that ROS levels regulate not only RBC survival, but also the maturation process of erythroid progenitor cells, modulating IE. Therefore, using hepcidin as a tool to reduced iron absorption might be a novel and exciting approach to controlling IE and managing β-thalassemia.
Fig.3. Potential effects of Hepcidin agonists or activators on iron absorption under normal and β-thalassemic conditions.
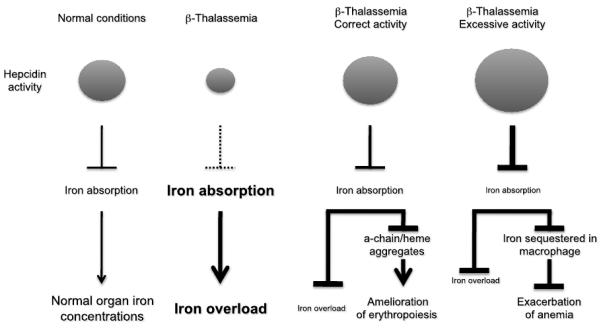
Lines ending with an arrow indicate activation. Lines ending with a line indicate repression.
Thus far, the studies undertaken to evaluate the effect of over expressing hepcidin on iron overload and erythropoiesis have utilized genetic models and did not explore the feasibility of using hepcidin or its agonists as drugs. Development of such compounds will determine their potential to prevent iron overload or reverse its toxic effects based on a dose-response relationships. However, since ferroportin is localized on macrophages as well, the administration or up-regulation of hepcidin in thalassemia may also affect iron recycling and its availability for erythropoiesis, ultimately worsening anemia (Fig. 3). For this reason, preclinical studies must address the effect of these drugs on both iron overload and erythropoiesis. Based on the fact that the levels of hepcidin are low in thalassemia and body iron is in excess, hepcidin therapy may be feasible depending upon the level of hepcidin achieved (Fig. 3). Further study in mouse models of thalassemia followed by rigorous clinical trials in patients will address these issues.
Summary
Patients with β-thalassemia develop secondary effects such as splenomegaly and iron overload.
IE is the hallmark of β-thalassemia, characterized by the premature death of erythroid precursors in the bone marrow and extramedullary sites.
Erythrocytes trapped in the spleen are the cause of splenomegaly, anemia and hypoxia, which leads to increased Epo production.
Splenomegaly eventually contributes to worsening of the anemia, necessitating splenectomy.
Because of increased Epo expression, the Epo/EpoR/Jak2/Stat5 pathway, which regulates erythropoiesis, is over-active in β-thalassemia, contributing to EMH and splenomegaly.
Jak2 inhibitors, used in combination with transfusion therapy, represent an alternative approach aimed at modulating IE and preventing splenomegaly/splenectomy.
Inhibition of Jak2 activity might also modulate the number of erythroid progenitor cells in β-thalassemia. These cells are the most plausible source of the erythroid factor, the proposed function of which is to increase iron absorption by suppressing hepcidin expression. Therefore, reducing Jak2 activity might have a beneficial effect on iron metabolism as well.
Currently, the only effective treatment for iron overload is chelation therapy.
Decreased expression of hepcidin is the cause of increased iron absorption in β-thalassemia.
Alternative therapies to prevent iron overload include up regulation of hepcidin expression and the direct administration of hepcidin agonists.
Acknowledgments
This work was supported by the Cooley’s Anemia Foundation (CAF), the Associazione Veneta Lotta alla Talassemia (AVLT) (S.G.), and by grants from the Carlo and Micol Schejola Foundation, the Children’s Cancer and Blood Foundation and NIH-R21DK065169 (S.R.), R01DK55463 (R.W.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflict of interest.
References
- 1.Cooley TB, Lee P. A series of cases of splenomegaly in children with anemia and peculiar bone changes. Trans. Am. Pediatr. Soc. 1925;37:29. [Google Scholar]
- 2.Weatherall DJ. Single gene disorders or complex traits: lessons from the thalassaemias and other monogenic diseases. BMJ. 2000;321(7269):1117–20. doi: 10.1136/bmj.321.7269.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001;2(4):245–55. doi: 10.1038/35066048. [DOI] [PubMed] [Google Scholar]
- 4.Forget BG. Molecular mechanisms of ß thalassemia. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, editors. Disorders of Hemoglobin: Genetics, Pathophysiology and Clinical Management. Cambridge University Press; Cambridge, UK: 2001. pp. 252–276. [Google Scholar]
- 5.Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. Disorders of Hemoglobin: Genetics, Pathophysiology and Clinical Management. 2nd ed. Cambridge University Press; Cambridge, UK: 2009. pp. 1–826. [Google Scholar]
- 6.Cao A, Pintus L, Lecca U, et al. Control of homozygous beta-thalassemia by carrier screening and antenatal diagnosis in Sardinians. Clin Genet. 1984;26(1):12–22. doi: 10.1111/j.1399-0004.1984.tb00781.x. [DOI] [PubMed] [Google Scholar]
- 7.Vichinsky EP. The morbidity of bone disease in thalassemia. Ann N Y Acad Sci. 1998;850:344–8. doi: 10.1111/j.1749-6632.1998.tb10491.x. [DOI] [PubMed] [Google Scholar]
- 8.Wonke B. Clinical management of beta-thalassemia major. Semin Hematol. 2001;38(4):350–9. doi: 10.1016/s0037-1963(01)90029-0. [DOI] [PubMed] [Google Scholar]
- 9.Cohen AR, Galanello R, Pennell DJ, et al. Thalassemia. Hematology Am Soc Hematol Educ Program. 2004:14–34. doi: 10.1182/asheducation-2004.1.14. [DOI] [PubMed] [Google Scholar]
- 10.Piomelli S. The management of patients with Cooley’s anemia: transfusions and splenectomy. Semin Hematol. 1995;32(4):262–8. [PubMed] [Google Scholar]
- 11.Giardina PJ, Grady RW. Chelation therapy in beta-thalassemia: an optimistic update. Semin Hematol. 2001;38(4):360–6. doi: 10.1016/s0037-1963(01)90030-7. [DOI] [PubMed] [Google Scholar]
- 12.Crary SE, Buchanan GR. Vascular complications after splenectomy for hematologic disorders. Blood. 2009;114(14):2861–8. doi: 10.1182/blood-2009-04-210112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eldor A, Rachmilewitz EA. The hypercoagulable state in thalassemia. Blood. 2002;99(1):36–43. doi: 10.1182/blood.v99.1.36. [DOI] [PubMed] [Google Scholar]
- 14.Taher A, Mehio G, Isma’eel H, et al. Stroke in thalassemia: a dilemma. Am J Hematol. 2008;83(4):343. doi: 10.1002/ajh.21117. [DOI] [PubMed] [Google Scholar]
- 15.Rivella S, Rachmilewitz E. Future alternative therapies for beta-thalassemia. Expert Rev Hematol. 2009;2(6):685. doi: 10.1586/ehm.09.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breda L, Gambari R, Rivella S. Gene therapy in thalassemia and hemoglobinopathies. Mediterranean Journal of Hematology and Infectious Diseases. 2009;1(1) doi: 10.4084/MJHID.2009.008. epub e2009008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skow LC, Burkhart BA, Johnson FM, et al. A mouse model for beta-thalassemia. Cell. 1983;34(3):1043–52. doi: 10.1016/0092-8674(83)90562-7. [DOI] [PubMed] [Google Scholar]
- 18.Curcio MJ, Kantoff P, Schafer MP, et al. Compensatory increase in levels of beta minor globin in murine beta-thalassemia is under translational control. J Biol Chem. 1986;261(34):16126–32. [PubMed] [Google Scholar]
- 19.De Franceschi L, Daraio F, Filippini A, et al. Liver expression of hepcidin and other iron genes in two mouse models of beta-thalassemia. Haematologica. 2006;91(10):1336–42. [PubMed] [Google Scholar]
- 20.Yang B, Kirby S, Lewis J, et al. A mouse model for beta 0-thalassemia. Proc Natl Acad Sci U S A. 1995;92(25):11608–12. doi: 10.1073/pnas.92.25.11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ciavatta DJ, Ryan TM, Farmer SC, et al. Mouse model of human beta zero thalassemia: targeted deletion of the mouse beta maj- and beta min-globin genes in embryonic stem cells. Proc Natl Acad Sci U S A. 1995;92(20):9259–63. doi: 10.1073/pnas.92.20.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rivella S, May C, Chadburn A, et al. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta -globin gene transfer. Blood. 2003;101(8):2932–9. doi: 10.1182/blood-2002-10-3305. [DOI] [PubMed] [Google Scholar]
- 23.Wu H, Liu X, Jaenisch R, et al. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell. 1995;83(1):59–67. doi: 10.1016/0092-8674(95)90234-1. [DOI] [PubMed] [Google Scholar]
- 24.D’Andrea A, Fasman G, Wong G, et al. Erythropoietin receptor: cloning strategy and structural features. Int J Cell Cloning. 1990;8(Suppl 1):173–80. doi: 10.1002/stem.5530080716. [DOI] [PubMed] [Google Scholar]
- 25.D’Andrea AD, Lodish HF, Wong GG. Expression cloning of the murine erythropoietin receptor. Cell. 1989;57(2):277–85. doi: 10.1016/0092-8674(89)90965-3. [DOI] [PubMed] [Google Scholar]
- 26.Li K, Miller C, Hegde S, et al. Roles for an Epo receptor Tyr-343 Stat5 pathway in proliferative co-signaling with kit. J Biol Chem. 2003;278(42):40702–9. doi: 10.1074/jbc.M307182200. [DOI] [PubMed] [Google Scholar]
- 27.Menon MP, Fang J, Wojchowski DM. Core erythropoietin receptor signals for late erythroblast development. Blood. 2006;107(7):2662–72. doi: 10.1182/blood-2005-02-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menon MP, Karur V, Bogacheva O, et al. Signals for stress erythropoiesis are integrated via an erythropoietin receptor-phosphotyrosine-343-Stat5 axis. J Clin Invest. 2006;116(3):683–94. doi: 10.1172/JCI25227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Lindern M, Zauner W, Mellitzer G, et al. The glucocorticoid receptor cooperates with the erythropoietin receptor and c-Kit to enhance and sustain proliferation of erythroid progenitors in vitro. Blood. 1999;94(2):550–9. [PubMed] [Google Scholar]
- 30.Fang J, Menon M, Kapelle W, et al. EPO modulation of cell-cycle regulatory genes, and cell division, in primary bone marrow erythroblasts. Blood. 2007;110(7):2361–70. doi: 10.1182/blood-2006-12-063503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Socolovsky M, Murrell M, Liu Y, et al. Negative Autoregulation by FAS Mediates Robust Fetal Erythropoiesis. PLoS Biol. 2007;5(10):e252. doi: 10.1371/journal.pbio.0050252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.James C. The JAK2V617F mutation in polycythemia vera and other myeloproliferative disorders: one mutation for three diseases? Hematology Am Soc Hematol Educ Program. 2008:69–75. doi: 10.1182/asheducation-2008.1.69. [DOI] [PubMed] [Google Scholar]
- 33.Levine RL. Mechanisms of mutations in myeloproliferative neoplasms. Best Pract Res Clin Haematol. 2009;22(4):489–94. doi: 10.1016/j.beha.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 34.Garcon L, Rivat C, James C, et al. Constitutive activation of STAT5 and Bcl-xL overexpression can induce endogenous erythroid colony formation in human primary cells. Blood. 2006;108(5):1551–4. doi: 10.1182/blood-2005-10-009514. [DOI] [PubMed] [Google Scholar]
- 35.Socolovsky M, Nam H, Fleming MD, et al. Ineffective erythropoiesis in Stat5a(−/−)5b(−/−) mice due to decreased survival of early erythroblasts. Blood. 2001;98(12):3261–73. doi: 10.1182/blood.v98.12.3261. [DOI] [PubMed] [Google Scholar]
- 36.Socolovsky M, Fallon AE, Wang S, et al. Fetal anemia and apoptosis of red cell progenitors in Stat5a−/−5b−/− mice: a direct role for Stat5 in Bcl-X(L) induction. Cell. 1999;98(2):181–91. doi: 10.1016/s0092-8674(00)81013-2. [DOI] [PubMed] [Google Scholar]
- 37.Dolznig H, Grebien F, Deiner EM, et al. Erythroid progenitor renewal versus differentiation: genetic evidence for cell autonomous, essential functions of EpoR, Stat5 and the GR. Oncogene. 2006 doi: 10.1038/sj.onc.1209308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerenyi MA, Grebien F, Gehart H, et al. Stat5 regulates cellular iron uptake of erythroid cells via IRP-2 and TfR-1. Blood. 2008;112(9):3878–88. doi: 10.1182/blood-2008-02-138339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu BM, McLaughlin SK, Na R, et al. Hematopoietic-specific Stat5-null mice display microcytic hypochromic anemia associated with reduced transferrin receptor gene expression. Blood. 2008;112(5):2071–80. doi: 10.1182/blood-2007-12-127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finch CA, Deubelbeiss K, Cook JD, et al. Ferrokinetics in man. Medicine. 1970;49(1):17–53. doi: 10.1097/00005792-197001000-00002. [DOI] [PubMed] [Google Scholar]
- 41.Centis F, Tabellini L, Lucarelli G, et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with beta-thalassemia major. Blood. 2000;96(10):3624–9. [PubMed] [Google Scholar]
- 42.Mathias LA, Fisher TC, Zeng L, et al. Ineffective erythropoiesis in beta-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp Hematol. 2000;28(12):1343–53. doi: 10.1016/s0301-472x(00)00555-5. [DOI] [PubMed] [Google Scholar]
- 43.Yuan J, Angelucci E, Lucarelli G, et al. Accelerated programmed cell death (apoptosis) in erythroid precursors of patients with severe beta-thalassemia (Cooley’s anemia) Blood. 1993;82(2):374–7. [PubMed] [Google Scholar]
- 44.Libani IV, Guy EC, Melchiori L, et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in {beta}-thalassemia. Blood. 2008;112(3):875–85. doi: 10.1182/blood-2007-12-126938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rivella S. Ineffective erythropoiesis and thalassemias. Curr Opin Hematol. 2009;16(3):187–94. doi: 10.1097/MOH.0b013e32832990a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang YL, Vandris K, Jones A, et al. JAK2 Mutations are present in all cases of polycythemia vera. Leukemia. 2008;22(6):1289. doi: 10.1038/sj.leu.2405047. [DOI] [PubMed] [Google Scholar]
- 47.Fibach E, Rachmilewitz E. The role of oxidative stress in hemolytic anemia. Curr Mol Med. 2008;8(7):609–19. doi: 10.2174/156652408786241384. [DOI] [PubMed] [Google Scholar]
- 48.Marinkovic D, Zhang X, Yalcin S, et al. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J Clin Invest. 2007;117(8):2133–44. doi: 10.1172/JCI31807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Callens C, Coulon S, Naudin J, et al. Targeting iron homeostasis induces cellular differentiation and synergizes with differentiating agents in acute myeloid leukemia. J Exp Med. 2010;207(4):731–50. doi: 10.1084/jem.20091488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abdel-Wahab O, Levine RL. Metabolism and the leukemic stem cell. J Exp Med. 2010;207(4):677–80. doi: 10.1084/jem.20100523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li H, Rybicki AC, Suzuka SM, et al. Transferrin therapy ameliorates disease in beta-thalassemic mice. Nat Med. 2010;16(2):177–82. doi: 10.1038/nm.2073. [DOI] [PubMed] [Google Scholar]
- 52.Andrews NC. Disorders of iron metabolism. N Engl J Med. 1999;341(26):1986–95. doi: 10.1056/NEJM199912233412607. [DOI] [PubMed] [Google Scholar]
- 53.Krause A, Neitz S, Magert HJ, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000;480(2-3):147–50. doi: 10.1016/s0014-5793(00)01920-7. [DOI] [PubMed] [Google Scholar]
- 54.Park CH, Valore EV, Waring AJ, et al. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276(11):7806–10. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 55.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–3. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 56.Nemeth E, Preza GC, Jung CL, et al. The N-terminus of hepcidin is essential for its interaction with ferroportin: structure-function study. Blood. 2005 doi: 10.1182/blood-2005-05-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276(11):7811–9. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 58.Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110(7):1037–44. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113(9):1271–6. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nemeth E, Valore EV, Territo M, et al. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101(7):2461–3. doi: 10.1182/blood-2002-10-3235. [DOI] [PubMed] [Google Scholar]
- 61.Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108(9):3204–9. doi: 10.1182/blood-2006-06-027631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nicolas G, Viatte L, Bennoun M, et al. Hepcidin, a new iron regulatory peptide. Blood Cells Mol Dis. 2002;29(3):327–35. doi: 10.1006/bcmd.2002.0573. [DOI] [PubMed] [Google Scholar]
- 63.Vokurka M, Krijt J, Sulc K, et al. Hepcidin mRNA levels in mouse liver respond to inhibition of erythropoiesis. Physiol Res. 2006;55(6):667–74. doi: 10.33549/physiolres.930841. [DOI] [PubMed] [Google Scholar]
- 64.Lebron JA, West AP, Jr., Bjorkman PJ. The hemochromatosis protein HFE competes with transferrin for binding to the transferrin receptor. J Mol Biol. 1999;294(1):239–45. doi: 10.1006/jmbi.1999.3252. [DOI] [PubMed] [Google Scholar]
- 65.Schmidt PJ, Toran PT, Giannetti AM, et al. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. 2008;7(3):205–14. doi: 10.1016/j.cmet.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13(4):399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 67.Kawabata H, Yang R, Hirama T, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem. 1999;274(30):20826–32. doi: 10.1074/jbc.274.30.20826. [DOI] [PubMed] [Google Scholar]
- 68.Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281(39):28494–8. doi: 10.1074/jbc.C600197200. [DOI] [PubMed] [Google Scholar]
- 69.Gao J, Chen J, Kramer M, et al. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 2009;9(3):217–27. doi: 10.1016/j.cmet.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang RH, Li C, Xu X, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2(6):399–409. doi: 10.1016/j.cmet.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 71.Babitt JL, Huang FW, Wrighting DM, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38(5):531–9. doi: 10.1038/ng1777. [DOI] [PubMed] [Google Scholar]
- 72.Babitt JL, Huang FW, Xia Y, et al. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J Clin Invest. 2007;117(7):1933–9. doi: 10.1172/JCI31342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kautz L, Meynard D, Monnier A, et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood. 2008;112(4):1503–9. doi: 10.1182/blood-2008-03-143354. [DOI] [PubMed] [Google Scholar]
- 74.Xia Y, Babitt JL, Sidis Y, et al. Hemojuvelin regulates hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin. Blood. 2008;111(10):5195–204. doi: 10.1182/blood-2007-09-111567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meynard D, Kautz L, Darnaud V, et al. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. 2009;41(4):478–81. doi: 10.1038/ng.320. [DOI] [PubMed] [Google Scholar]
- 76.Ramey G, Deschemin JC, Vaulont S. Cross-talk between the mitogen activated protein kinase and bone morphogenetic protein/hemojuvelin pathways is required for the induction of hepcidin by holotransferrin in primary mouse hepatocytes. Haematologica. 2009;94(6):765–72. doi: 10.3324/haematol.2008.003541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Calzolari A, Raggi C, Deaglio S, et al. TfR2 localizes in lipid raft domains and is released in exosomes to activate signal transduction along the MAPK pathway. J Cell Sci. 2006;119(Pt 21):4486–98. doi: 10.1242/jcs.03228. [DOI] [PubMed] [Google Scholar]
- 78.Andrews NC. Forging a field: the golden age of iron biology. Blood. 2008;112(2):219–30. doi: 10.1182/blood-2007-12-077388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nemeth E, Ganz T. The role of hepcidin in iron metabolism. Acta Haematol. 2009;122(2-3):78–86. doi: 10.1159/000243791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Finberg KE, Heeney MM, Campagna DR, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA) Nat Genet. 2008;40(5):569–71. doi: 10.1038/ng.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Du X, She E, Gelbart T, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science. 2008;320(5879):1088–92. doi: 10.1126/science.1157121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Silvestri L, Pagani A, Nai A, et al. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008;8(6):502–11. doi: 10.1016/j.cmet.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lin L, Goldberg YP, Ganz T. Competitive regulation of hepcidin mRNA by soluble and cell-associated hemojuvelin. Blood. 2005;106(8):2884–9. doi: 10.1182/blood-2005-05-1845. [DOI] [PubMed] [Google Scholar]
- 84.Finberg KE, Whittlesey RL, Fleming MD, et al. Downregulation of Bmp/Smad signaling by Tmprss6 is required for maintenance of systemic iron homeostasis. Blood. 2010 doi: 10.1182/blood-2009-05-224808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang FW, Pinkus JL, Pinkus GS, et al. A mouse model of juvenile hemochromatosis. J Clin Invest. 2005;115(8):2187–91. doi: 10.1172/JCI25049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weizer O, Adamsky K, Breda L, et al. Hepcidin expression in cultured liver cells responds differently to iron overloaded sera derived from patients with thalassemia and hemochromatosis; The American Society of Hematology 46th Annual Meeting; San Diego. [Google Scholar]
- 87.Pak M, Lopez MA, Gabayan V, et al. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006;108(12):3730–5. doi: 10.1182/blood-2006-06-028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fiorelli G, Fargion S, Piperno A, et al. Iron metabolism in thalassemia intermedia. Haematologica. 1990;75(Suppl 5):89–95. [PubMed] [Google Scholar]
- 89.Gardenghi S, Marongiu MF, Ramos P, et al. Ineffective erythropoiesis in {beta}-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood. 2007;109(11):5027–35. doi: 10.1182/blood-2006-09-048868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Weizer-Stern O, Adamsky K, Amariglio N, et al. Downregulation of hepcidin and haemojuvelin expression in the hepatocyte cell-line HepG2 induced by thalassaemic sera. Br J Haematol. 2006;135(1):129–38. doi: 10.1111/j.1365-2141.2006.06258.x. [DOI] [PubMed] [Google Scholar]
- 91.Weizer-Stern O, Adamsky K, Amariglio N, et al. mRNA expression of iron regulatory genes in beta-thalassemia intermedia and beta-thalassemia major mouse models. Am J Hematol. 2006;81(7):479–83. doi: 10.1002/ajh.20549. [DOI] [PubMed] [Google Scholar]
- 92.Origa R, Galanello R, Ganz T, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica. 2007;92(5):583–8. doi: 10.3324/haematol.10842. [DOI] [PubMed] [Google Scholar]
- 93.Kearney SL, Nemeth E, Neufeld EJ, et al. Urinary hepcidin in congenital chronic anemias. Pediatr Blood Cancer. 2007;48(1):57–63. doi: 10.1002/pbc.20616. [DOI] [PubMed] [Google Scholar]
- 94.Papanikolaou G, Tzilianos M, Christakis JI, et al. Hepcidin in iron overload disorders. Blood. 2005;105(10):4103–5. doi: 10.1182/blood-2004-12-4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kattamis A, Papassotiriou I, Palaiologou D, et al. The effects of erythropoetic activity and iron burden on hepcidin expression in patients with thalassemia major. Haematologica. 2006;91(6):809–12. [PubMed] [Google Scholar]
- 96.Jenkins ZA, Hagar W, Bowlus CL, et al. Iron homeostasis during transfusional iron overload in beta-thalassemia and sickle cell disease: changes in iron regulatory protein, hepcidin, and ferritin expression. Pediatr Hematol Oncol. 2007;24(4):237–43. doi: 10.1080/08880010701360700. [DOI] [PubMed] [Google Scholar]
- 97.Adamsky K, Weizer O, Amariglio N, et al. Decreased hepcidin mRNA expression in thalassemic mice. Br J Haematol. 2004;124(1):123–4. doi: 10.1046/j.1365-2141.2003.04734.x. [DOI] [PubMed] [Google Scholar]
- 98.Rechavi G, Rivella S. Regulation of iron absorption in hemoglobinopathies. Curr Mol Med. 2008;8(7):646–62. doi: 10.2174/156652408786241401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.de Franceschi L, Turrini F, Honczarenko M, et al. In vivo reduction of erythrocyte oxidant stress in a murine model of beta-thalassemia. Haematologica. 2004;89(11):1287–98. [PubMed] [Google Scholar]
- 100.Tanno T, Porayette P, Sripichai O, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood. 2009;114(1):181–6. doi: 10.1182/blood-2008-12-195503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007 doi: 10.1038/nm1629. [DOI] [PubMed] [Google Scholar]
- 102.Kanda J, Mizumoto C, Kawabata H, et al. Serum hepcidin level and erythropoietic activity after hematopoietic stem cell transplantation. Haematologica. 2008;93(10):1550–4. doi: 10.3324/haematol.12399. [DOI] [PubMed] [Google Scholar]
- 103.Tanno T, Rabel A, Lee YT, et al. Expression of growth differentiation factor 15 is not elevated in individuals with iron deficiency secondary to volunteer blood donation. Transfusion. 2010 doi: 10.1111/j.1537-2995.2010.02601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ramirez JM, Schaad O, Durual S, et al. Growth differentiation factor 15 production is necessary for normal erythroid differentiation and is increased in refractory anaemia with ring-sideroblasts. Br J Haematol. 2009;144(2):251–62. doi: 10.1111/j.1365-2141.2008.07441.x. [DOI] [PubMed] [Google Scholar]
- 105.Nicolas G, Viatte L, Lou DQ, et al. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat Genet. 2003;34(1):97–101. doi: 10.1038/ng1150. [DOI] [PubMed] [Google Scholar]
- 106.Ghoti H, Amer J, Winder A, et al. Oxidative stress in red blood cells, platelets and polymorphonuclear leukocytes from patients with myelodysplastic syndrome. Eur J Haematol. 2007;79(6):463–7. doi: 10.1111/j.1600-0609.2007.00972.x. [DOI] [PubMed] [Google Scholar]
- 107.Shinar E, Rachmilewitz EA. Oxidative denaturation of red blood cells in thalassemia. Semin Hematol. 1990;27(1):70–82. [PubMed] [Google Scholar]
- 108.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24(50):7410–25. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]