

Abstract
In the two step synthesis of thiolate-monolayer protected clusters (MPCs), the first step of the reaction is a mild reduction of gold(III) by thiols that generates gold(I) thiolate complexes as intermediates. Using tiopronin (Tio) as the thiol reductant, the characterization of the intermediate Au4Tio4 complex was accomplished with various analytical and structural techniques. Nuclear magnetic resonance (NMR), elemental analysis, thermogravimetric analysis (TGA), and matrix-assisted laser desorption/ionization-mass spectrometry (MALDI-MS) were all consistent with a cyclic gold(I)-thiol tetramer structure, and final structural analysis was gathered through the use of powder diffraction and pair distribution functions (PDF). Crystallographic data has proved challenging for almost all previous gold(I)-thiolate complexes. Herein, a novel characterization technique when combined with standard analytical assessment to elucidate structure without crystallographic data proved invaluable to the study of these complexes. This in conjunction with other analytical techniques, in particular mass spectrometry, can elucidate a structure when crystallographic data is unavailable. In addition, luminescent properties provided evidence of aurophilicity within the molecule. The concept of aurophilicity has been introduced to describe a select group of gold-thiolate structures, which possess unique characteristics, mainly red photoluminescence and a distinct Au-Au intramolecular distance indicating a weak metal-metal bond as also evidenced by the structural model of the tetramer. Significant features of both the tetrameric and aurophilic properties of the intermediate gold(I) tiopronin complex are retained after borohydride reduction to form the MPC, including gold(I) tiopronin partial rings as capping motifs, or “staples”, and weak red photoluminescence that extends into the Near Infrared region.
Introduction
Monolayer protected clusters (MPCs) are nanosized materials which show potential as sensors, catalysts, and biological transporters.1, 2 Depending upon their size, MPCs may exhibit molecular or bulk metallic properties.1–3 In all MPC syntheses size control is paramount, and determination of the size and composition of the resulting nanoparticle is essential. MPCs consist of a metallic core and capping ligands which can be subjected to post-synthesis exchange of various peptides or functional ligands.2, 4, 5 The ability to modify the particle with biologically functional ligands makes MPCs very appealing for transport in vivo.6–9
Traditionally, the MPC reaction has been concerned with the characterization of the final products. According to Templeton et al.2, the synthetic pathway of MPCs is shown as a two-step reaction (Scheme 1) in which an intermediate exists between the initial ligand-gold mixture and the reduction step. They further state that the details of the two-step reaction have not been fully dissected.
Scheme 1.

Two-step synthesis in which a precursor molecule is shown as a medium between products and reactants.
More recent work by Jadzinsky et al.10 has provided crystallography data for MPCs, showing gold MPCs consist of a metallic core surrounded by, what they term, “staple structures”, which are primarily composed of gold (I) and sulfur bonds. These structures have re-emphasized the importance of Au(I)-thiolate complexes and their relationship to MPC formation. Much work has been published regarding the characterization and features of Au(I)-thiolate complexes.11–26
Corbierre et al.11 explored preparation of nanoparticles from various Au(I)-thiolate complexes. In their work, they constructed various Au(I)-thiolate complexes, reduced them, and analyzed the resulting particle. Depending upon which complex was used, a variety of nanoparticles resulted with varying size and solubility, confirming that Au(I)-thiolate complexes are rational precursors to nanoparticles. Other work has confirmed changes within the Au(I)-thiolate complex, specifically pH, show size effects to the resulting particle,12 and provide more evidence the Au(I)-thiolate complex is the precursor to nanoparticles. Given these findings, exploration into the structure of these Au(I)-thiolate complexes is needed to enhance future nanoparticle synthesis and applications.
Speculation regarding the composition and structure of these Au(I)-thiolate complexes was shown as early as 1984, when Shaw et al. 13 hypothesized the complexes were composed of either a straight-chain polymer (Figure 1a) or a gold-thiol ring (Figure 1b). Since then some crystallographic studies have been shown for Au[I]-thiolates.15–17, 22–24 Most notably are structures showing 12-membered rings,16 helices,23 and even discrete linear chains.24 The differences noted are presumably dependent on the choice of thiolate ligand and various crystallization parameters. Therefore, crystallographic structure will vary from system to system. Unfortunately, one thing is clear throughout the literature: gold [I]-thiolate complexes are difficult to crystallize and often require months if not years to acquire. This proves problematic for a rapidly growing interest in these structures. For this reason, more novel analytical methods for structural characterization of these complexes are needed.
Figure 1.
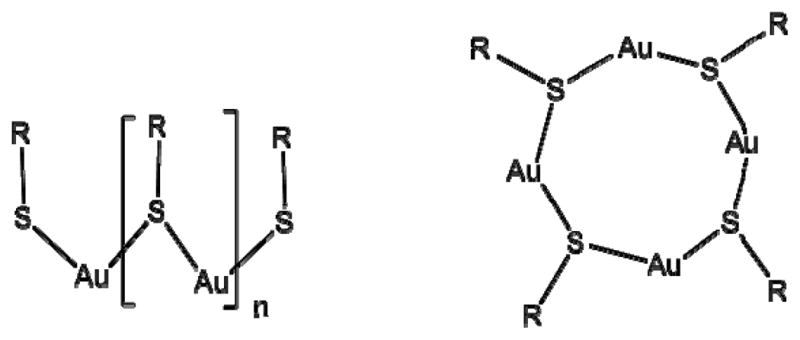
Proposed structures for precursor molecule. A) straight-chain polymer and B) gold-thiol ring compound
There have been many published works regarding luminescent Au(I)-thiolate complexes and their gold-gold interactions.21 Schmidbaur19, 20 has introduced the concept of aurophilicity to describe specific classes of gold compounds. Aurophilicity is characteristic of a certain structural class of gold compounds in which the gold complexes undergo intermolecular aggregation via gold-gold contacts of ca. 3.05Å. If the ligands are sufficiently small, the gold complexes have been shown to form multimolecular structures such as dimers or strings. Some literature report Au(I)-thiolate repeating ring structures.14–18 These structures are probably attributable to strong Au-Au interactions.15 If a given complex is known to exhibit aurophilicity, it may be assumed that the structure correlates to the known structural characteristics previously published for aurophilic species i.e. the complex will be a ring with strong Au-Au interaction, the S-Au bond distance will be ca. 2.5 Å, etc. This could be used in conjunction with other easily obtainable data to predict a structure without crystallography.
Schmidbaur states that luminescence is a strong diagnostic for the aurophilicity phenomena.19–21 Therefore, if a given Au(I)-thiolate complex was luminescent, it could be assumed aurophilicity exists within the structure, and likewise the structure could be comprised of dimers or strings. This could lead to a novel structural interpretation of a given gold-thiolate complex if crystallographic data was unavailable since luminescence measurements are relatively easily obtained. Furthermore, if the ligands are sufficiently small, knowledge of specific bond distances within the complex could lead to further confirmation of aurophilicity and therefore, structural interpretation. However, without crystallographic data, another method for bond length acquisition is needed.
Recently the pair distribution function (PDF) analysis of diffraction data has been shown to give useful information on the structure and composition of molecules on the nanoscale.27, 28 This method allows for the characterization of nanoscale materials that may be very difficult for traditional crystallographic measurements or incapable of forming solid crystals. It provides the same data parameters as crystallography, that is, bond lengths, without having to produce a crystal. Thus, this method would be ideal for assessment of the Au(I)-thiolate compounds given the difficulty in their crystallization. It would further support any luminescent data gathered to deduce if the complex possessed aurophilic properties. Furthermore, the bond length data could be compared to the previous theoretical Auxthiolatex parameters calculated by Gronbeck et al.26 to further elucidate structure.
The previous electrospray ionization mass spectrometry (ESI-MS) work by our group has hinted at the idea that the tiopronin monolayer protected cluster (TMPC) precursor molecule is a Au(I)-thiolate compound.29 These compounds were discovered during ESI-MS runs of a TMPC mixture. These compounds were hypothesized to be remnants of the precursor molecule post-reduction; they were also hypothesized to be cyclic in nature, possessing an Au4thiolate4 predominant composition. However, more analytical scrutiny was needed of the precursor itself to more definitively prove the precursor was cyclic. While the work in Geis et al. supports the theoretical calculations noted in Gronbeck et al.26 for the tetrameric species, subsequent analytical data can further validate this claim. Correlation with the predicted bond distances shown in Hakkinen et al. would further support the argument of a 4:4 structure, and is possible with the aid of PDF analysis. Since crystallization of these complexes has been noted as being difficult and time-consuming, a systematic analysis of the precursor for the parameters associated with aurophilicity would provide extensive structural knowledge. This combined with the prior mass spectrometry could be used to elicit a sound, structural interpretation of this widely scrutinized system without the aid of crystallography.
This work seeks to introduce a new structural method for characterization of Au(I)-thiolates. It describes the analytical assessment of the precursor by conventional MPC characterization tools: NMR, UV-Visible spectroscopy, and thermogravimetric analysis (TGA). The lack of a solid core within the complex made transmission electron microscopy (TEM) invalid for analysis due to resolution limitation. We have also employed the use of matrix assisted laser desorption ionization mass spectrometry (MALDI-MS) for a soft technique to analyze the TMPC precursor.30, 31 Further confirmation was gathered from luminescent and PDF analysis to confirm aurophilicity within the precursor molecule; this combined with MALDI-MS was used to elicit its structure. We have further explored an acidic pH range and initial gold composition of the precursor and observed the changes to the resulting particle.
Experimental Section
Reagents
N-(2-mercaptopropionyl)-glycine (tiopronin) (Sigma), methanol, acetic acid (Fischer Scientific), α-cyano-4-hydroxycinnamic acid (α-cyano) (Sigma), acetonitrile, trifluoroacetic acid, hydrochloric acid, chloroform-d, and DMSO-d6 were all used as received. Tetrachloroauric acid trihydrate was synthesized according to the literature32 and stored in the freezer at −20° C. Briefly gold shot (99.99%) was purchased from precious metal vendors and was initially converted to HAuCl4 × 3H2O by boiling Au0 in HCl/HNO3 solution. A Millipore Nanopure water purification system was used to obtain low conductivity (18 MOmega;) water for analysis.
Synthesis of Precursor
Tiopronin Monolayer Protected Cluster (TMPC) synthesis follows the two-step modified Brust method33 in which tiopronin (1.2 g, 7 mmol) was added to tetrachloroauric acid (1 g, 2.6 mmol) and subsequently further reduced with sodium borohydride (1 g, 26 mmol, 10× excess). Synthesis of the precursor modifies this procedure by elimination of the borohydride reduction step. A three-fold excess of tiopronin reduced the Au (III) to a gold(I)-tiopronin complex which was accompanied by a color change from orange-red to clear. Stirring the solution for 24 hours at room temperature produces a white solid suspended in solution. This solid is presumably an Au(I)-thiolate complex. The solid was then removed via filtration and utilized for analysis after a 24-hour drying period.
Nuclear Magnetic Resonance
1H NMR spectra were recorded of highly concentrated precursor and TMPC samples (c.a. 50 mg/mL) on a Bruker AC400 MHz NMR spectrometer. Spectra were collected in DMSO-d6 and CDCl3.
Thermogravimetric Analysis
TGA was performed on dried precursor and TMPC samples (c.a. 5 – 10 mg) with a TGA 1000 instrument under N2 flow (60 mL min−1), recording data from 25 to 800 °C at a heating rate of 20 °C min−1. The first step of the TGA is the loss of water and is subtracted from the overall mass. The second step is solely due to the loss of organic layer (tiopronin) while the last step is due to the loss of the staple structures. 10
MALDI of precursor
After precipitation from solution using a 50:50 H2O:MeOH mixture with an addition of 30 mM sodium chloride (certified A.C.S., Fisher), the sample was dried. A small quantity of dried precursor complex with a mass of 26.2 mg was dissolved in 400 μL water and combined with 400 μL of saturated α-cyano-4-hydroxycinnamic acid (CHCA, Sigma-Aldrich) in methanol. The sample was sonicated, and a 1 μL aliquot of the solution was immediately deposited on a stainless steel plate using the dried droplet method.34 Positive ion mass spectra were collected using a Voyager DE-STR mass spectrometer in reflectron mode.
Elemental Analysis of precursor
A sample (c.a. 30 mg) of dried precursor was sent to Columbia Analytics for analysis of carbon, sulfur, gold, hydrogen, and nitrogen. Oxygen percentage was calculated by hand.
Powder Diffraction of precursor
A sample of dried precursor (c.a. 50 mg) was crushed into a fine powder for PDF analysis. X-ray powder diffraction experiments were carried out on the precursor at beamline 11-ID-B at the Advanced Photon Source at Argonne National Laboratory, using the rapid acquisition PDF method (RaPDF).35 The x-ray energy used was 58.26 KeV, corresponding to a wavelength of λ =0.2128 Å.
The powdered sample of precursor was sealed in 1.0 mm diameter Kapton tubes. A 2D image plate detector (MAR345) was mounted orthogonal to the path of the beam with a sample to detector distance of 188.592 mm. The measurements were performed at 80 K and 300 K. For each of them, the exposure time was 30 s, repeated five times and summed together for a total collection time of 2.5 minutes per data-set. The 2D data from the image plate were integrated using the program Fit2D36 to obtain a 1D intensity vs. Q function, where Q =4π sin θ/λ is the magnitude of the momentum transfer and θ is the Bragg angle which is half the scattering angle. The data were converted from intensity to the corrected, normalized total scattering structure function, S(Q), according to standard procedures37, 38 using the program PDFgetX2.39 The pair distribution function, G(r), was obtained by Fourier transformation of the reduced total scattering structure F (Q)= Q(S(Q) – 1) according to Eq 1.40
| Eq. 1 |
To minimize the termination effects and noise in the PDF, the optimal Qmax was found to be 16.3 Å−1.
PDF modeling of precursor
Structure refinements were performed using home-written modeling software employing a real-space38 calculation for crystal models and a Debye equation approach40 for isolated molecule models. The software utilizes portions of the ObjCryst++ library.41
The PDF is calculated directly from a given atomic arrangement.38, 39 A variety of initial configurations are tried, taking starting models from the literature and guided by the other characterization measurements. The fitting process iteratively changes the positions of the atoms subject to constraints on bond lengths, angles or atomic positions until the best fit to the data is obtained. The constraints determine the number of modeling parameters of the structure configurations. Intermolecular correlations involving the ligand were excluded from the modeling and are discussed with the results. Atomic vibrations were modeled as isotropic, with one adjustable atomic displacement parameter (ADP) for each atom type and a single atom-pair vibrational-correlation term. A scale factor was also included in the modeling while experimental resolution factors were excluded.
ADF modeling of precursor
All calculations were performed with the Amsterdam Density Functional suite (ADF)42 using the gradient-generalized approximation of Becke43 and Perdew44. Relativistic effects were included using the zeroth order relativistic approximation to the Dirac equation (ZORA).45 All-electron triple zeta double polarized (TZ2P) basis sets included with the ADF suite were employed. The ADF integration accuracy parameter was set to 5.0 for all calculations to ensure sufficient accuracy for numerical integrals.
pH adjustment of precursor
Dried precursor samples were suspended in water (pH 7) and subjected to changes in pH via addition of hydrochloric acid to the desired values of 1, 3, and 5. Samples were then reduced with sodium borohydride and analyzed for subsequent changes to the final TMPC product via UV-Vis and TEM.
Addition of HAuCl4 to precursor
Additional HAuCl4 was added to the precursor in water in 5, 10, 15, and 20 mL quantities of a 5 M solution HAuCl4 and subsequently reduced. Resulting TMPCs were analyzed via TEM.
Transmission Electron Microscopy of TMPCs
The nanoparticles samples were prepared by adding ~1 mg dried particle to 5 mL DI water, sonicating for at least 10 minutes, and then dropped for slow evaporation onto ultrathin carbon grids (400 mesh, Ted Pella, Inc.). Images were obtained with a Phillips CM20T operating at 200 keV with a calibrated magnification of 414 kx. Results are reported as the mean ± the standard deviation obtained from the negatives using ImageJ software (NIH, http://rsb.info.nih.gov/ij/) using sample sizes of at least 50 particles from the TEM grid.
Results and Discussion
1H NMR Spectroscopy
One of the most well-known methods of characterization for TMPCs is NMR.38 MPCs produce many definitive characteristics that provide insight into their structure, specifically line broadening.46, 47 The TMPC precursor was collected and suspended in a solution of DMSO-d6 and CDCl3 for NMR analysis and compared to the tiopronin ligand (Figure S1 in Supporting Information).
TMPC spectra show a loss of a sulfur-hydrogen bond at 2.5 ppm, representative of the ligand-core binding.48 This loss is also present in the TMPC precursor spectrum (asterisk), indicative of a sulfur-gold bond within the molecule. The spectrum itself also shows a coupling change from a quintet to a quartet (arrows), also indicative of the loss of a sulfur-hydrogen bond.
Interesting to note is the sharpness of the peaks within the TMPC precursor spectrum. TMPC spectra show characteristic line-broadening features as a result of T2 relaxation time due to the ligand attachment to the core.46 This is discussed at length elsewhere but is believed to be attributable to the slowing down of the rotational (T2) relaxation times due to the size of the core. It has also been shown that larger clusters produce broader peaks.48 However, broadening is not present within the precursor spectrum, indicating the lack of a gold core within the precursor molecule, which implies the core is not formed before reduction. Rather, the gold-sulfur attachment occurs in the precursor formation step and packs into a core structure during the reduction step.
Composition Analysis
Further understanding of the chemical nature of the precursor was explored through two methods of compositional analysis: elemental analysis and TGA. A sample of approximately 50 mg of dried TMPC precursor sample was given to Columbia Analytical for elemental analysis of gold, sulfur, carbon, hydrogen, and nitrogen. Table 1 displays the results obtained from the duplicate analyses.
Table 1.
Elemental Analysis of TMPC precursor molecule. Run I and II are actual analyses; the third row is theoretical percentages based upon a 1:1 gold:tiopronin structure.
| Run I | Au: 55.80; S: 8.70; C: 17.50; N: 3.59; O: 11.86; H: 2.55 |
| Run II | Au: 55.90; S: N/A; C: 16.90; N: 3.68; O: N/A; H: 2.21 |
| 1:1 Au:Tio | Au: 54.80; S: 8.93; C: 13.40; N: 3.90; O: 8.90; H: 1.97 |
From the analyses, an approximate 55% of the molecule was attributable to gold, whereas the other 45% was attributable to the elemental composition of the tiopronin ligand: hydrogen, carbon, nitrogen, sulfur, and oxygen. While care was taken to completely dry the sample before elemental analysis, it should be noted that methanol and acetic acid were solvents of crystallization. Solvent entrapment has been demonstrated in the isolation of Au-thiolate nanoparticles in previous studies2, 49 using elemental analysis. The elemental analysis data was consistent with a 1:1 gold:tiopronin ratio, (molecular weight = 359.14 g/mol) and predicted values were calculated, producing the third row of the table. For a 1:1 ratio of gold:tiopronin, approximately 55% would be attributable to gold whereas the other 45% would be attributable to the elemental composition of the tiopronin ligand. Further calculations for the individual elements within the tiopronin ligand produced comparable results to the percentages obtained from the sample. It may, therefore, be assumed that the TMPC precursor is comprised of a 1:1 ratio of gold:tiopronin.
Previous characterizations of the tiopronin monolayer protected cluster prove that the final molecular composition is not a 1:1 ratio after reduction.42 In fact, there is always less tiopronin than gold due to the formation of the core and its inner atoms which are internalized and barred from attachment to the ligands.42 The elemental analysis of a 1:1 ratio provides evidence that the core is not present within the precursor molecule, but is rather formed during reduction.
For correlation to elemental analysis, thermogravimetric analysis was applied. TGA provides the percentage of organic within the sample. The results of the TGA analysis of the precursor are shown in Supporting Information (Figure S2 in Supporting Information). Previous work has shown TMPCs decompose thermally by loss of the organic monolayer and subsequently leave behind the gold residue.48 Thus, TGA is commonly used to determine the organic weight of the TMPC. Organic loss percentage has been shown to correlate with core size.48 Application of TGA was used to determine the exact composition of the TMPC precursor for comparison to the elemental analysis.
From the spectrum, an organic loss equivalent to approximately 45% was observed. As was calculated before, for a ratio of 1:1 gold:tiopronin, the percentage distribution would be 55% gold, 45% tiopronin. This approximate percentage is observed from the TGA analysis, providing further evidence that the precursor molecule is composed of a 1:1 ratio of gold:tiopronin.
Mass spectrometry of precursor
Following the work of Gies et al.,29 MALDI-TOF MS was used for the characterization of the precursor complex. The relevant portion of the mass spectrum is shown in Figure 2.
Figure 2.
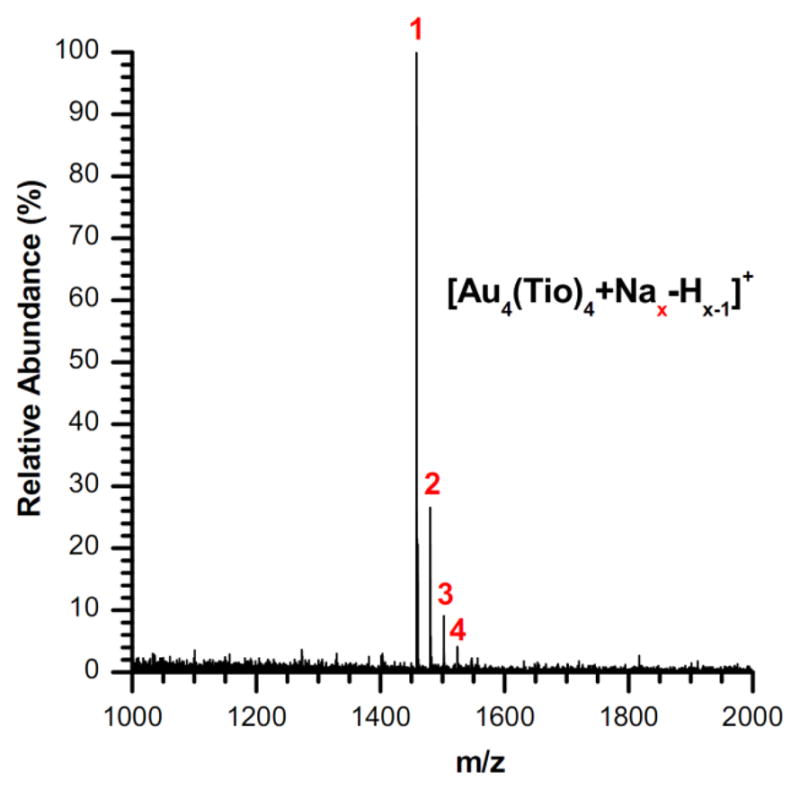
Positive ion MALDI-TOF mass spectrum of tetrameric precursor Au4(Tio)4 after crystallization. The numbers in red signify the number of sodium-coordinations.
Gold(I)-thiolate complexes are known to behave as multimeric compounds with an open chain or ring structure.50 An open chain would be expected to yield an array of regularly spaced peaks as have been observed in mass spectra of polymers. However, in this case the Au4(Tio)4 ion species is exclusively preferred, with no other gold-thiolate ions present above the noise threshold (Figure 2, see Figure S3 in Supporting Information for full spectrum). This result confirms that the precursor complex is a discrete structure with no variation in the number of monomer units.
Aurophilicity measurements of precursor
Previous works19–21 have shown luminescence to be characteristic of the aurophilicity phenomena. These molecules are often organized as rings which further form multimolecular higher order structures like dimers or strings.19 These published structures typically are excited round 350 nm and show emission between 600–700 nm, or red emission. If the precursor molecule is luminescent, it would provide evidence that the structure is most likely a ring conformation and likewise aurophilic.
It is clear that the precursor emits red under long-wave UV light (Figure 3). This was the first confirmation of luminescent properties as well as potential aurophilicity within the molecule.
Figure 3.

Precursor under a) normal and b) long-wave UV light
This is quite close to the observations of Assefa et al.21 in which they observed an emission at 674 nm for compounds at relative room temperature (250 K). It has been previously shown that the emission energy at 700 nm is correlative to the Au-Au bond distance.51 This was demonstrated by two gold tetrameric emission studies, one by Vogler52 [Au4Cl4(piperidine)4] with Au-Au distance of 3.301 Å versus [Au(dta)4] Au-Au distance of 3.01 Å by Forward and co-workers53. The emission energy observed by Tzeng et al.54 which was assigned to the S-Au charge-transfer transition, is also likely the case with the TMPC tetramer. Future work to explore the luminescent properties of the precursor are currently underway as this could provide an explanation of the fluorescent properties noted in their reduced form.
As a final confirmation of a ring composition, pair distribution function (PDF) analysis was applied to the precursor molecule. PDF data measured at 80K and 300K are shown in Figure 4.
Figure 4.

PDF spectra of TMPC precursor at 300K and 80K. Sharp peaks at 2.4 and 3.3 Å are indicative of the gold-sulfur and gold-gold bonds respectively.
The spectra show sharp peaks at low r, signifying rigid intramolecular correlations. The peaks past 7 Å, are relatively strong, which suggests that gold is participating in these correlations. The broadness of these peaks indicates that either the precursor molecule is greater than 10 Å in diameter and is flexible or weakly ordered on this length scale, or that the molecule is smaller and has correlated packing on this length scale. The uniform broadening of the PDF spectrum with respect to temperature suggests that the molecules are not large and flexible.
Refinements were performed using isolated molecule starting models in order to determine the arrangement of atoms that contribute to the first four significant PDF peaks indicated in Figure 4. The starting models are an 8-member ring structure and the 7- and 9-member straight-chain polymer observed by Shaw et al13 (Figure 1). The modeling results are shown in Figure 5. The 9-member straight-chain polymer structure (Figure 5A) was refined by adjusting the nearest-neighbor gold-sulfur distances, sulfur-gold-sulfur angles and isotropic ADPs. The fit spectrum (Figure 5D) fails to reproduce the third peak when modeled over the first four peaks of the PDF (1.8 – 6.25 Å) and introduces spurious peaks over a broader range (1.8 – 9 Å). The misfit of the third peak suggests that the chain structure does not have the atom-pair multiplicity indicated at this distance by the data. The spurious peak indicates that the structure is not compact enough. Increasing the modeling range to include all atom-pair distances from the model does not improve the fit by fitting the peak near 7.5 Å, but rather results in a poorer fit overall. The results are qualitatively similar when considering a 7-member chain, indicating that the chain model is inconsistent with the PDF data.
Figure 5.

PDF molecular models of TMPC precursor. A) Refined 9-member straight-chain polymer model. B) Refined 8-member closed-chain model. C) Refined 8-member ring model. Model bond length values are shown. D, E, F) PDF fits corresponding to A, B, C, respectively. In the PDF fits the blue circles are the 80 K data, the solid red line is the calculated PDF of the best-fitting model over the range of r from 1.8 – 6.25 Å, and the dashed red line is the PDF from the refined model extended out to 9 Å. The green line is the difference between the data and fit over the fit range and is offset for clarity.
By removing an atom from the 9-member chain structure and bending it into a closed-chain (Figure 5B), the spurious peaks at large distances are eliminated but the multi-component peak that should lie between 5 and 6 Å is displaced (Figure 5E). The planar 8-member ring structure (Figure 5C), which relates to Shaw’s ring structure, is modeled by constraining the closed chain structure such that all gold-sulfur bonds are the same length, and allowing three unique bond angles to enforce the symmetry shown in the figure. This structure reproduces all the relevant PDF peaks without introducing spurious ones (Figure 5F). Together with the results from mass-spectrometry the PDF results strongly suggest that the molecular size and configuration are correct in this model. Allowing this structure to refine out of plane does not improve the model agreement with the PDF.
We considered two non-planar clusters to further test the planar 8-member ring model. The butterfly cluster reported by Fackler and coworkers55 has an 8-member metal-sulfur core that resembles a buckled 8-member ring. The core sulfur-metal distances are comparable to the first peak position in the PDF, but the metal-metal distances are in the range 2.7 to 3.0 Å, where there are no PDF peaks. To produce a better fit to the PDF, it was necessary to increase the metal-metal distances while preserving the sulfur-metal distances, which bent the core back towards the planar 8-member ring, recovering the planar 8-ring model again. Similarly, another non-planar structure candidate was tried based on a 9-atom cluster56 but it gave less satisfactory results than the 8-fold ring and was not supported by the mass spectrometry evidence.
For completeness, and to gain insight on the packing of the molecular units within the precursor, the PDF data were also modeled using several crystalline structures suggested in the literature as candidate structures. The fits of these crystalline models were extended out to 9 Å in order to include intermolecular correlations and are shown in Figure 6. The first model is the 12-ring crystal suggested by LeBlanc and Lock16. As seen in Figure 6, this model fits poorly over nearly the entire spectrum. In particular, the atom-pair distance at 4.66 Å suggested by PDF the does not occur in the 12-ring structure and refinement of the ring compensates for this by distorting other regions of the calculated spectrum. The second model from Heaven et al.22 is an array of 43-atom clusters with a suggestive staple-like structure on the surface. As expected based on the mass-spectrometry results, this structure also performs poorly, especially at low-r where it completely fails to reproduce the intramolecular correlations. This confirms the mass-spectrometry result that only the precursor ligated species and not gold clusters are present in these samples. Furthermore, it highlights the fact that the PDF data can distinguish these cases.
Figure 6.
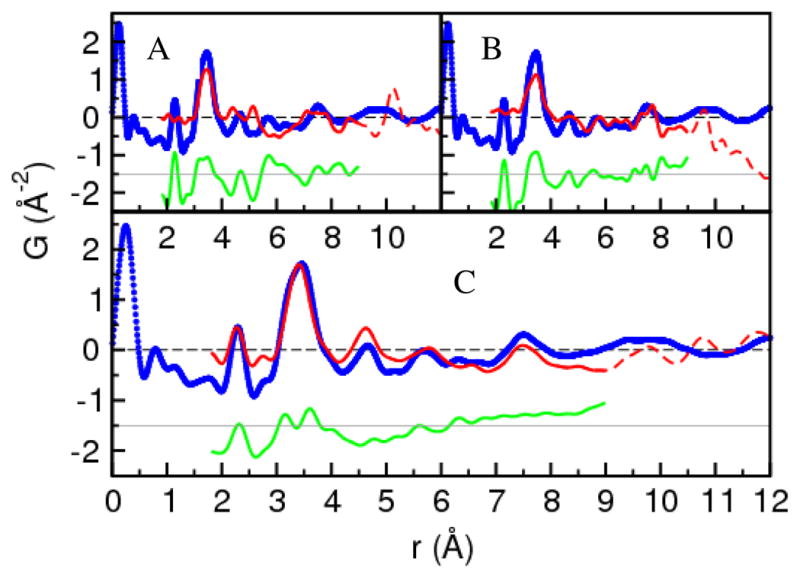
PDF crystal model fits of TMPC precursor. A) Fit from crystallized 12-member ring structure of LeBlanc and Lock16. B) Fit from crystallized cluster model of Heaven et al. 22 C) Fit from double-helix structure of Bau23. The blue circles are the 80 K PDF data, the solid red line is the calculated PDF of the best-fitting model over the range of r from 1.8 – 9 Å, and the dashed red line is the PDF from the refined model extended out to 12 Å. The green line is the difference between the data and fit over the fit range and is offset for clarity.
The third model is a double-helix suggested by Bau.23 This model is closest to the 8-member ring structure modeled in Figure 5, since the double-helix can be viewed a stack of broken rings. From Figure 6 it is apparent that this model fits nearly as well as the 8-member ring structure up to 6.25 Å, though it does not fully reproduce the nearest-neighbor gold-sulfur correlations. The model captures the intermolecular correlations near 7.5 Å very well. The quality of this fit strongly suggests that the 8-member ring molecular units are stacked one on top of another. From Figure 4, the correlations in the PDF disappear by 15 Å, which implies that the packing is not perfect and becomes uncorrelated already by this distance.
The low-frequency oscillatory misfit from the 8-member ring and double-helix models that is apparent in Figures 5F and 6C can be explained by the density of the soft and disordered ligand molecules that were excluded from the structure models. They produce broad, low peaks in the PDF due to the weak ordering. This broad intensity does not obscure the strong peaks from the gold and sulfur precursor clusters. It contributes to the calculated model error but does not significantly affect the refined atomic positions within the ring. In other words, the misfit shown in Figures 5F and 6C is an artifact of the modeling method and is not structurally significant.
A second modeling effort was made using another program, ADF, for energy minimization calculations. The results of the ADF modeling are shown in Figure 7.
Figure 7.

ADF molecular model of TMPC precursor. Model bond length values are shown in bold.
From the model, a variation of the previously published staple structure and the octahedral structure proposed is observed. The structure is not perfectly symmetrical in which two “staples” would perfectly align. It, in fact, has a slight distortion in which the gold atoms “bow” from the center. The gold-sulfur bond distances are well within the given range of 2.2 – 2.6 Å in the previous crystallographic data. 10, 26 The gold-gold distances are comparable to the gold-gold distances observed on the outside of the core in Jadzinsky et al,.10 in which the range is said to be up to 5.5 Å. Further, the extreme similarities of the precursor structure to the “staple” structures found on the outside of the particle are of particular interest. While some bond distances are not equivalent between the same atoms, in comparison to the PDF modeling, the bond distances are quite close, most within 0.05 Å. The Au-S bond length for PDF analysis was given as approximately 2.33 Angstroms vs. ADF at 2.325 Angstroms. Likewise Au-Au distance from PDF was 3.37 (diagonal) vs. ADF at 3.351 Angstroms. Finally Au-Au distance (across ring) PDF = 5.69 Angstroms vs. 4.746 Angstroms ADF. It should also be recognized that the modeling program assumes molecules within the gas phase; solid phase packing could lend itself to shorter observed distances, which would be much closer to the PDF values.
Given the molecular modeling bond length data and the PDF analysis, the precursor is most likely a ring compound with strong aurophilicity. The gold-gold bond distance is extremely close to the energy minimum described in previous works,19, 26 which were noted at 3.05 Å and 3.24 Å respectively. While the value of 3.37 Å is not at absolute minimum, it is well within the range of lowest energy minima, as noted in Schmidbaur. This further proves the precursor molecule exhibits aurophilicity. This value is also in strong correlation with previous theoretical calculations for the Au4thiolate4 in which the predicted Au-Au bond was noted as 3.24 Å. The slight difference between this value and the theoretical is attributable to the choice in ligand. The theoretical calculations were made with a similar, but slightly larger ligand, glutathione. Within the work, it is explicitly stated that bond length will vary due to ligand choice; presumably there should be differences between their observations for glutathione and those noted for tiopronin. Nevertheless, the two values are extremely close, and provide further evidence that the TMPC precursor is tetrameric in structure. Both glutathione and tiopronin have some steric bulk to them which influence the stability of the resulting structures. The higher-r peaks within the PDF suggest that the precursor molecules are stacked though proof of this would require more intricate modeling and computational calculations.
Modifications to the precursor molecule
Many modifications to MPCs post-synthesis have been shown to affect particle size and composition. 11, 12 Specifically, a change in pH conditions after reduction of glutathione MPCs has been shown to produce larger particles at low pH (pH = 5) and smaller particles at high pH (pH= 8).12 Given the synthesis of the TMPC precursor and TMPCs themselves are synthesized in acidic conditions (pH=1), we felt it was necessary to expand on previous studies to include the extreme acidic pH range. To our knowledge, no work has been published showing the effects of acidic pH on gold-thiolate precursors and the subsequent effects on the resulting particle.
The UV-Vis results for the pH modifications to the precursor are shown in Figure S6 in the Supporting Information. The UV-Vis spectrum shows an SPR band present for the particles synthesized from the precursor at pH 1 and pH 3. The presence of the SPR band signifies greater size dispersity and the presence of larger particles. The spectrum does not show an SPR band for the particles synthesized at pH 5, denoting more mono disperse, smaller particles at the higher pH value. The precursor with no modification did not show an SPR band, signifying the sample particles were originally small and monodisperse. This provides evidence that lowering the pH creates more poly dispersity and larger particles.
Further confirmation of this trend was explored with TEM images of the different pH value particles. Results from the TEM analysis are shown in Figure 8.
Figure 8.

TEM images and average sizes for each pH value. A reverse correlation is noted with precursor pH and size of the resulting particle after reduction. Between pH = 3 and pH = 5 the appearance of the SPR band is eliminated; particles at high precursor pH are smaller in size.
The TEM averages calculated show a descending trend in size with increasing pH. Also noted from the standard deviation bars is a decreasing trend in dispersity. This phenomenon could best be explained by repulsive forces between the tiopronin ligands. At lower pH values, all sites are protonated, therefore the ligands are neutral. This neutrality creates minimal repulsion between ligands, allowing for close association. When the sample is reduced, there will be areas of large aggregation of precursor as well as areas of low aggregation. These disperse areas of aggregation create a dispersity of particles upon reduction. The areas of large aggregation will create large particles while areas of small aggregation will create small particles.
Conversely, at pH 5, some sites may become deprotonated, decreasing the repulsive forces between the tiopronin ligands. This slight repulsion causes the ligands to spread apart, evenly throughout the sample, creating a uniform distribution. Once reduced, the particles will be of uniform size since no aggregates were formed to create larger particles. The data confirms that reducing the precursor at a high pH value leads to mono disperse samples; likewise if larger dispersity and particle size is warranted, lowering the pH of the precursor before reduction will produce the desired results. This result proves extensively interesting given the current method for TMPC synthesis is performed in acidic conditions. As a result of these findings, it may be advantageous to raise the pH of the solution prior to reduction to elicit more uniform, smaller particles, as is often sought for biological applications.
A second study to elicit changes within the particle as a result of modifying the precursor was also implemented. Work presented in this paper has suggests that the gold core is formed during reduction as a result of the breaking of bonds within the precursor structure. Therefore, the composition of the TMPC should be limited to the metals and ligands only within the precursor molecule. To test this hypothesis, excess gold was added to the solution containing the precursor molecule. Assuming the structure of the precursor is not the only determinant in the composition of the resulting particle, the addition of more gold after the precursor formation should yield a product with a larger core. If, however, the components of the precursor are the limiting factor in the resulting particle composition, then the addition of excess gold should not affect core size once reduced.
The results of the UV-Vis traces for the addition of HAuCl4 after precursor formation clearly show the absence of a surface plasmon band not only within the precursor alone with no gold addition but also after gold addition. From the spectra, one may ascertain that there was no core growth as a result of the addition of gold to the precursor solution prior to reduction. Also noticed was the accumulation of solid gold aggregation post-reduction within the TMPC solution. If the HAuCl4 did not interact with the precursor prior or during reduction, the product solution should contain solid gold aggregates due to reduction of pure HAuCl4, and this was in fact observed. Samples synthesized with additional HAuCl4 contained many gold aggregates, thus saturating the grid making TEM imaging impossible. Dialysis of the solutions was somewhat ineffective since the molecular weight cut-off (MWCO) used in TMPC synthesis enabled the gold aggregates to remain in solution. Nevertheless, additional Au(III) was not able to be successfully added to the intermediate Au4Tio4 complex create larger MPCs that were protected from agglomeration.
Conclusion
This work seeks to show alternative structural methods to crystallization for gold-thiolate complexes through the characterization of the tiopronin monolayer protected gold cluster precursor. The analytical results show the precursor is formed in a 1:1 ratio of gold: tiopronin and is in fact a 4:4 structure with ring configuration as has been hypothesized previously by our group.29 The PDF modeling strongly suggests this by showing that 7- and 9-member straight-chain polymer structures and previously reported crystal structures cannot account for the observed correlations, whereas the refined 8-member ring structure does. We further correlate our structural data to those observed in aurophilicity accounts as well as those shown for theoretical Au4thiolate4 molecules. Further study of the precursor molecule showed changing the pH prior to reduction can significantly affect the size and dispersity of the resulting particle sample (high pH yields smaller and more uniform sample), and may prove useful in synthesis. We have also shown preliminary work that the gold core size is predetermined by the precursor, not by the amount of gold present in solution at the time of reduction. Further exploration is currently underway of the precursor formation and its role in particle synthesis.
Supplementary Material
Acknowledgments
This work was made possible by the National Institutes of Health (R01 GM076479), a Chemical Biology Interface Training Grant from the National Institutes of Health (T32 GM065086) (K.M.H.), and the Vanderbilt Institute of Nanoscale Science and Engineering. We would like to thank Stephen Chmely and Dr. Timothy Hanusa for their help with the molecular modeling. We would also like to acknowledge Emil Bozin, Pavol Juhas and Timur Dykhne for help with PDF data collection. The PDF work was supported by the US National Science Foundation through Grant DMR-0703940. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Footnotes
Supporting Information Available: NMR, TGA, full MALDI-TOF MS spectrum, Refined 6-atom and 7- member chain models and their PDF fits, and UV-Visible data of the gold nanoparticles. This material is available free of charge via the internet at http://pubs.acs.org
References
- 1.Templeton AC, Chen S, Gross SM, Murray RW. Langmuir. 1999;15:66–76. [Google Scholar]
- 2.Templeton AC, Wuelfing WP, Murray RW. Acc Chem Res. 2000;33:27–36. doi: 10.1021/ar9602664. [DOI] [PubMed] [Google Scholar]
- 3.Shon YS, Mazzitelli C, Murray RW. Langmuir. 2001;17:7735–7741. [Google Scholar]
- 4.Guo R, Song Y, Wang G, Murray RW. J Am Chem Soc. 2005;127:2752–2757. doi: 10.1021/ja044638c. [DOI] [PubMed] [Google Scholar]
- 5.Hostetler MJ, Templeton AC, Murray RW. Langmuir. 1999;15:3782–3789. [Google Scholar]
- 6.Gerdon AE, Wright DW, Cliffel DE. Biomacromolecules. 2005;6:3419–3424. doi: 10.1021/bm050475o. [DOI] [PubMed] [Google Scholar]
- 7.Gerdon AE, Wright DW, Cliffel DE. Angew Chem, Int Ed Engl. 2006;45:594–598. doi: 10.1002/anie.200503328. [DOI] [PubMed] [Google Scholar]
- 8.Herold DM, Das IJ, Stobbe CC, Iyer RV, Chapman JD. Int J Radiat Biol. 2000;76:1357–1364. doi: 10.1080/09553000050151637. [DOI] [PubMed] [Google Scholar]
- 9.Farrell N. Transition metal complexes as drugs and chemotherapeutic agents. Vol. 11. Kluwer Academic Publishers; Boston: 1989. pp. 243–256. [Google Scholar]
- 10.Jadzinsky PD, Calero G, Ackerson CJ, Bushnell DA, Kornberg RD. Science. 2007;318:430–433. doi: 10.1126/science.1148624. [DOI] [PubMed] [Google Scholar]
- 11.Corbierre MK, Lennox RB. Chem Mater. 2005;17:5691–5696. [Google Scholar]
- 12.Brinas RP, Hu M, Qian L, Lymar ES, Hainfeld JF. J Am Chem Soc. 2008;130:975–982. doi: 10.1021/ja076333e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw CF, Schaeffer NA, Elder RC, Eidsness MK, Trooster JM, Callis GHM. J Am Chem Soc. 1984;106:3511–3521. [Google Scholar]
- 14.Schroter I, Strahle J. Chem Ber. 1991;124:2161–2164. [Google Scholar]
- 15.Wiseman MR, Marsh PA, Bishop PT, Brisdon BJ, Mahon MF. J Am Chem Soc. 2000;122:12598–12599. [Google Scholar]
- 16.LeBlanc D, Lock CJL. Acta Crystallogr. 1997;C53:1765–1768. [Google Scholar]
- 17.Bonasia PJ, Gindelberger DE, Arnold J. Inorg Chem. 1993;32:5126–5131. [Google Scholar]
- 18.Wojnowski W, Becker B, Sabmannshausen J, Peters EM, Peters K, von Schnering HGZ. Anorg Allg Chem. 1994;620:1417. [Google Scholar]
- 19.Schmidbaur H. Chem Soc Rev. 1995;24:391–400. [Google Scholar]
- 20.Schmidbaur H. Gold Bull. 1990;23:11. [Google Scholar]
- 21.Assefa Z, McBurnett BG, Staples RJ, Fackler JP, Assmann B, Angermaier B, Schmidbaur H. Inorg Chem. 1995;34:75–83. [Google Scholar]
- 22.Heaven MW, Dass A, White PS, Holt KM, Murray RW. J Am Chem Soc. 2008;130:3754–3755. doi: 10.1021/ja800561b. [DOI] [PubMed] [Google Scholar]
- 23.Bau R. J Am Chem Soc. 1998;120:9380–9381. [Google Scholar]
- 24.Bates PA, Waters JM. Acta Crystallogr Section C. 1985;41:862–865. [Google Scholar]
- 25.Akola J, Walter M, Whetten RL, Hakkinen H, Gronbeck H. J Am Chem Soc. 2008;130:3756–3757. doi: 10.1021/ja800594p. [DOI] [PubMed] [Google Scholar]
- 26.Gronbeck H, Walter M, Hakkinen H. J Am Chem Soc. 2006;128:10268–10275. doi: 10.1021/ja062584w. [DOI] [PubMed] [Google Scholar]
- 27.Billinge SJL, Levin I. Science. 2007;316:561–565. doi: 10.1126/science.1135080. [DOI] [PubMed] [Google Scholar]
- 28.Juhas P, Cherba DM, Duxbury PM, Punch WF, Billinge SJL. Nature. 2006;440:1095–1104. doi: 10.1038/nature04556. [DOI] [PubMed] [Google Scholar]
- 29.Gies AP, Hercules DM, Gerdon AE, Cliffel DE. J Am Chem Soc. 2007;129:1095–1104. doi: 10.1021/ja0639057. [DOI] [PubMed] [Google Scholar]
- 30.Dreisewerd K. Chem Rev. 2003;103:395–425. doi: 10.1021/cr010375i. [DOI] [PubMed] [Google Scholar]
- 31.Teng CH, Ho KC, Lin YS, Chen YC. Anal Chem. 2004;76:4337–4342. doi: 10.1021/ac049963x. [DOI] [PubMed] [Google Scholar]
- 32.Brauer G. Handbook of Preparative Inorganic Chemistry. 1. Academic Press; New York: 1965. p. 1054. [Google Scholar]
- 33.Brust M, Fink J, Bethel D, Schiffrin DJ, Whyman R. J Chem Soc Chem Comm. 1994:801. [Google Scholar]
- 34.Karas M, Hillenkamp F. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 35.Chupas PJ, Qui X, Hanson JC, Lee PL, Grey CP, Billinge SJL. J Appl Crystallogr. 2003;36:1342–1347. [Google Scholar]
- 36.Hammersley AP, Svenson SO, Hanfland M, Hauserman D. High Pressure Res. 1996;14:235–248. [Google Scholar]
- 37.Billinge SJL. Powder diffraction: theory and practice. 1. Royal Society of Chemistry; London: 2008. Local Structure from total scattering and atomic pair distribution fuction (pdf) analysis; pp. 464–493. [Google Scholar]
- 38.Egami T, Billinge SJL. Underneath the Bragg peaks: Structural analysis of complex materials. Elsevier; Oxford: 2004. [Google Scholar]
- 39.Qui X, Thompson JW, Billinge SJL. J Appl Crystallogr. 2004;37:678. [Google Scholar]
- 40.Farrow CL, Billinge SJL. Acta Crystallogr. 2009;A65:232. doi: 10.1107/S0108767309009714. [DOI] [PubMed] [Google Scholar]
- 41.Favre-Nicolin V, Cerny R. J Appl Crystallogr. 2002;13:734–743. [Google Scholar]
- 42.Theoretical Chemistry. Vrije Universiteit; Amsterdam, The Netherlands: http://www.scm.comADF2008.01. [Google Scholar]
- 43.Becke AD. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 44.Perdew JP. Phys Rev B. 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 45.van Lenthe E, Ehlers A, Baerends EJ. J Chem Phys. 1999;110:8943–8953. [Google Scholar]
- 46.Kohlman O, Steinmetz WE, Mao XA, Wuelfing WP, Templeton AC, Murray RW, Johnson CS. J Phys Chem B. 2001;105:8801–8809. [Google Scholar]
- 47.Badia A, Demers L, Dickinson L, Morin FG, Lennox RB, Reven L. J Am Chem Soc. 1997;119:11104–11105. [Google Scholar]
- 48.Hostetler MJ, Wingate JE, Zhong CJ, Harris JE, Vachet RW, Clark MR, Londono JD, Green SJ, Stokes JJ, Wignall GD, Glish GL, Porter MD, Evans ND, Murray RW. Langmuir. 1998;14:17–30. [Google Scholar]
- 49.Terrill R, Postlethwaite TA, Chen CH, Poon CD, Terzis A, Chen A, Hutchison JE, Clark MR, Wignall G. J Am Chem Soc. 1995;117:12537–12548. [Google Scholar]
- 50.Shaw CF. Chem Rev. 1999;99:2589–2600. doi: 10.1021/cr980431o. [DOI] [PubMed] [Google Scholar]
- 51.Yam VWW, Lo KKW. Chem Soc Rev. 1999;28:323–334. [Google Scholar]
- 52.Vogler A, Kunkely H. Chem Phys Lett. 1988;150:135–137. [Google Scholar]
- 53.Forward JM, Bohmann D, Fackler J, JP, Staples RJ. Inorg Chem. 1995;34:6330. [Google Scholar]
- 54.Tzeng BC, Yeh HT, Huang YC, Chao HY, Lee GH, Peng SM. Inorg Chem. 2003;42:6008–6014. doi: 10.1021/ic034041i. [DOI] [PubMed] [Google Scholar]
- 55.Fackler JP, Jr, Lopez CA, Staples RJ, Wang S, Winpenny REP, Lattimer RP. J Chem Soc Chem Comm. 1992:146–148. [Google Scholar]
- 56.Slovokhotov YL, Struchkov YT. J Organomet Chem. 1984;277:143–146. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.