

Abstract
Drug discovery often begins with the screening of large compound libraries to identify lead compounds. Recently, the enzymes involved in the biosynthesis of natural products have been investigated for their potential to generate new, diverse compound libraries. There have been several approaches toward this end, including altering the substrate specificities of the enzymes involved in natural product biosynthesis and engineering functional communication between enzymes from different biosynthetic pathways. While there exist assays to assess substrate specificity of enzymes involved in these pathways, there is no simple method for determining whether enzymes from different synthases will function cooperatively to generate the desired product(s). Herein we report a method which provides insight into both substrate specificity and compatibility of protein-protein interactions between the acyl carrier protein (ACP) and ketosynthase (KS) domains involved in fatty acid and polyketide biosynthesis. Our technique uses a one-pot chemoenzymatic method to generate post-translationally modified ACPs capable of covalently interacting with KS domains from different biosynthetic systems. The extent of interaction between ACPs and KSs from different systems is easily detected and quantified by a gel-based method. Our results are consistent with previous studies of substrate specificity and ACP-KS binding interactions and provide new insight into unnatural substrate and protein interactions.
Introduction
Fatty acid and polyketide biosynthesis are evolutionarily related pathways that use similar mechanisms to synthesize their products.[1,2] In both pathways small acyl carrier proteins (ACPs) serve as a central covalent template to which biosynthetic intermediates are tethered while iterative cycles of loading, condensation, and processing are performed by accessory enzymes such as acyltransferse (AT), ketosynthase (KS), and ketoreductase (KR) domains.[3-5] In type I fatty acid synthase (FAS) and polyketide synthase (PKS) systems these enzyme activities are all housed on a single multidomain “megasynthase,” while in type II FAS and PKS systems each accessory enzyme is a discrete polypeptide which must interact in trans with the growing ACP-tethered intermediate.[1,2, 4-8]
ACP-bound acyl intermediates are tethered through a thioester bond formed by the terminal thiol of a post-translationally introduced 4’-phosphopantetheine arm. This 4’-phosphopantetheine prosthetic group is introduced by the action of a phosphopantetheinyltransferase (PPTase) enzyme, which converts apo-ACP to holo-ACP by the transfer of the 4’-phosphopantetheine from coenzyme A (CoA) to a conserved serine residue of ACP.[9] Recently our group[10-12] and others[13,14] have shown that promiscuous PPTases, most notably the surfactin PPTase Sfp,[15] are highly tolerant of modification to the terminal portion of CoA and will transfer a variety of 4’-phosphopantetheine analogues from CoA analogues to produce crypto-ACPs. Crypto-ACP refers to an ACP in which the terminal thiol moiety is replaced or “hidden” by an unnatural modification.[11] This ability to modify ACPs with virtually any substitution at the thiol terminus of the 4’-phosphopantetheine group has been used for the proteomic identification of PKS and FAS systems in crude cell lysate[16, unpublished results] and to provide essential insights into the substrate selectivity of these enzymes.[17]
One further area in which this technology has proven useful is in probing the structural interactions of carrier protein mediated biosynthesis. In type II FAS and PKS biosynthesis, tight yet transient interactions between the loaded ACP and each of its partner enzymes are essential for efficient biosynthetic productivity. The transient and non-covalent nature of these protein-protein interactions has hindered attempts to study the individual steps of these biosynthetic processes using traditional structural methods. To help alleviate this problem we recently developed a method to trap ACPs during their interaction with KS domains.[18] In natural type II FAS and PKS systems, KS domains are responsible for the condensation reaction in which the growing acyl-chain of the FAS or PKS intermediate is first transferred to the nucleophilic cysteine of the KS, followed by decarboxylation and Claisen condensation of a malonyl-ACP intermediate with the KS-bound acyl-enzyme intermediate.[19-21] To study this transient interaction we used a chemoenzymatic synthesis of CoA analogues combined with posttranslational modification by the PPTase Sfp to produce crypto-ACPs in which non-hydrolyzable electrophilic probes are tethered in place of the natural thiol terminus of holo-ACP. This reactive-moiety can then interact with the nucleophilic active site cysteine of a KS domain, resulting in the formation of a covalent crosslink. This interaction can be detected by running the one-pot reaction on SDS-PAGE and observing a gel shift. Tighter interactions between the ACP and KS yield a greater signal, while a diminished or negligible signal is an indication that either the two proteins are failing to associate or that the reactive CoA analog is effectively competing with the crosslinking reaction for modification of the KS active site.
In addition to its utility in isolating covalently crosslinked ACP-KS complexes for crystallographic studies,[22] this crosslinking procedure presents a novel method for studying the step-wise compatibility of ACP and partner enzymes that originate from alternate biosynthetic systems. Currently there is no simple procedure for determining whether an ACP and KS from two different biosynthetic systems will possess the requisite protein-protein interactions to insure tight-binding and efficient processing of biosynthetic intermediates, a shortcoming which has presented an obstacle in the rational manipulation of modular synthases for the purposes of generating novel combinatorial compound libraries.[23-25] By comparing the extent of crosslinking between an ACP and KS we can demonstrate how well the KS will accept alternate carrier proteins from other organisms and/or biosynthetic pathways.
Here we present a full study of ACP-KS crosslinking in which we apply nine new electrophilic 4’-phosphopantetheine analogs towards crosslinking studies of three systems: KASI-ACP and KASII-ACP from E. coil type II FAS, and EncAB-EncC, a KS-ACP pair from the S. maritimus type II enterocin PKS (Figure 1).[26-28] A gel-based assay is used to probe and quantify the interactions between both natural and unnatural ACP-KS patners using our panel of reactive pantetheine analogs, with results reflecting the specificity of a KS for its natural ACP partner. In addition we verify the active-site specificity of our crosslinking experiments by performing MALDI and LC-MS/MS analyses on in-gel digests of crosslinked proteins. Finally we use our panel of electrophilic pantetheine analogs to correlate substrate specificity with structure of the electrophilic 4’-phosphopantetheine arm of the crypto-ACP (Figure 2). This study represents the first application of this technology to a medicinally relevant type II PKS biosynthetic system, and our findings accurately reflect the known differences in both binding affinities and substrate specificity among KS domains and provide new insight into the interaction between ACP and KS domains in type II FAS and PKS systems.
Figure 1.
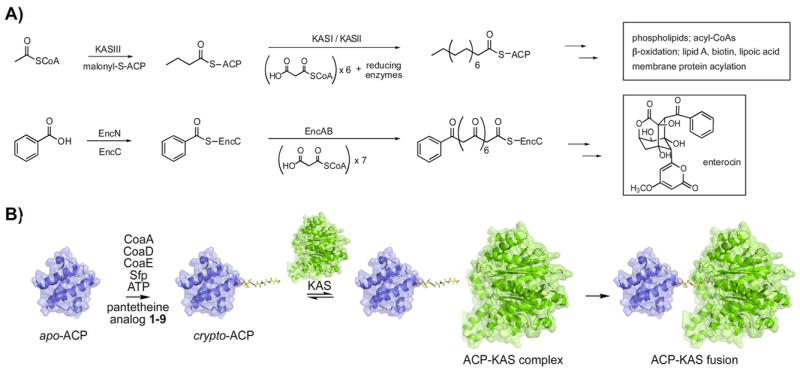
A method to probe interactions between modified CP’s and KS’s of type II FAS and PKS pathways. A) A comparison of type II biosyntheses between FAS in E. coli and enterocin biosyntheis in S. maritimus. B) Apo-ACP is incubated with CoaA, CoaD, CoaE, and Sfp in the presence of ATP and pantetheine analog 1-9 to generate crypto-ACP. Crypto-ACP reacts with the active site cysteine of the ketosynthase to form ACP-KAS fusion product.
Figure 2.
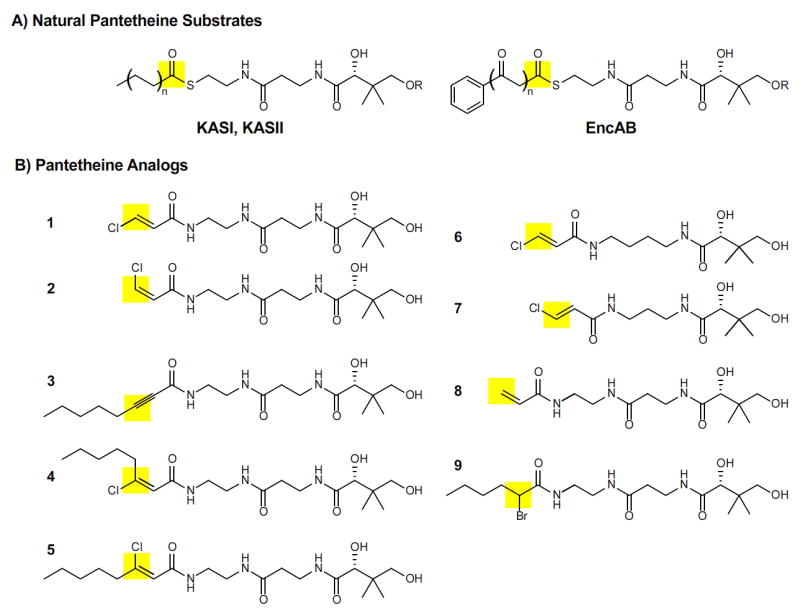
Pantetheine analogs as probes for substrate specificity of KS domains in FAS and PKS. The site of attack by the active site cysteine of KS is highlighted in yellow. A) Natural pantetheine substrates for KS domains KASI and KASII of E. coli FAS and EncAB of S. maritimus PKS. Substrates of length n= 0-6 are bound to the ACP at R. B) Pantetheine analogs 1-9 used to mimic substrates of varying lengths and degrees of oxidation.
Results and Discussion
Design and Synthesis of Reactive Pantetheine Analogs for Crosslinking
This study utilizes a previously reported chemoenzymatic method in which CoaA, CoaD, and CoaE are used to convert pantetheine analogues 1-9 into CoA analogues in one-pot, before transfer of the 4’-phosphopantetheine group to the ACP by the PPTase Sfp (Figure 1B).[10,18] In our initial report, we noticed different reactivities of our crosslinking pantetheine analogs with regards to stereochemistry of the leaving group.[18] These differences are likely a reflection of binding pocket residue interactions. The enzymes responsible for the biosynthesis of the S. maritimus polyketide enterocin belong to the type II polyketide synthase (PKS) family.[27,28] The ketosynthase-chain length factor (KS-CLF) EncAB must recognize not only benzoate and malonate bound to the carrier protein EncC, but also the growing product bound to EncC; this growing product is transacylated to the active site cysteine prior to condensation with malonyl-EncC. E. coli FAS ketosynthases KASI and KASII also recognize their growing fatty acyl chains in addition to recognizing malonate bound to ACP (Figure 1A). While they have overlapping substrate specificities, KASI and KASII have shown slightly different activities towards a panel of fatty acid substrates. KASII shows greater specific activity with all substrates from hexanoyl-ACP (C6) to hexadecanoyl-ACP (C16). Most notably, KASII shows significantly more activity than KASI with myristoyl- (14:0) and palmitoleoyl- (16:1)ACP as substrates.[29] The binding pocket of KASII is slightly larger and thus more receptive to the longer substrates of the latter stages of fatty acid synthesis; structural and modeling studies have shown the binding pocket of KASI is too shallow to hold the C18:1 product of the elongation of C16:1.[30] Other differences in substrate specificity between the two ketosynthases are thought to be due to the identities of residues lining the substrate binding pockets.[31]
Pantetheine analogues 1-9 can be thought of as incorporating two important components for the purpose of this study: 1) an affinity tag of varying stereochemistry and electrophilicity and 2) an alkyl chain designed to mimic a variety of substrate sizes (Figure 2). As terminal chloroacrylamides[32] proved the most efficient ACP-KS crosslinkers in our original study, we first examined the subtle differences of this reactive electrophile in depth. In order to examine the influence of electrophile stereochemistry on crosslinking we synthesized both terminal (1-2), and long chain (4-5) chloracrylamides of varying cis/trans stereochemistry. We also investigated the effect of varying the placement of the Michael accepting double bond of the chloroacrylamide by synthesizing compound 6, which places it at the same chain length as the naturally occurring 4’-phosphopantetheine thioester, and compound 7, which places the electrophilic carbon one carbon shorter than the naturally occurring thioester. To understand the effect of the chlorine group on crosslinking better we synthesized 8, a non-chloro containing terminal acrylamide. Finally we examined the reactivity of two other long-chain electrophile containing pantetheine analogues, α/β unsaturated alkyne 3 and racemic α-bromoacetamide 9.[33] In terms of acyl-chain length these analogues can be grouped into three subgroups: those with acyl chains shorter than acylated 4’-phosphopantetheine (6-7), those with acyl-chains roughly corresponding to a short acyl chain (1-2, 8), and those incorporating a longer acyl-chain (3-5, 9).
After overproduction and purification of the apo-forms of the carrier proteins E. coli AcpP (from type II FAS) and S. maritimus EncC (from type II enterocin PKS) the one-pot chemoenzymatic method was applied with analogues 1-9 to post-translationally modify each ACP and produce crypto-ACPs. Each crypto-ACP then was reacted with its natural partner KS. Two of the more potent crypto-ACP forms, derived from compounds 1 and 3, were also incubated with a KS from a different organism and biosynthetic system. In almost all cases tested there was some interaction between crypto-ACP and KS (Figure 3). This interaction was detected as a gel shift on SDS-PAGE; the degree of crosslinking was quantified using the gel analyzing function of the program ImageJ (Table 1).[34] For all natural systems, crosslinking was confirmed by MALDI MS/MS analyses (Figure 4).[18]
Figure 3.
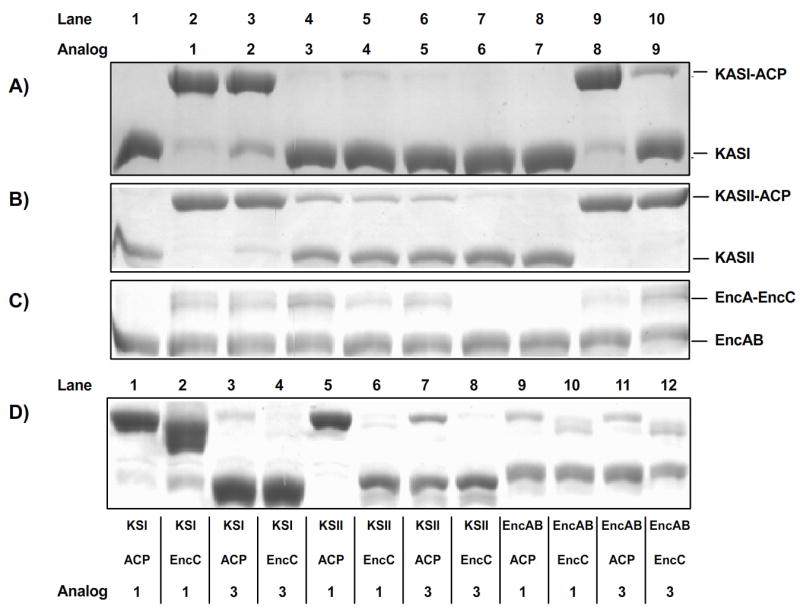
Application of the method for KS-CP formation to E. coli FAS and S. maritimus PKS proteins, using 9 reactive pantetheine analogs. SDS-Page analysis of one-pot reactions to form KS-CP complexes using the panel of analogs and 3 combinations of KS, CP proteins: A) E. coli KASI and ACP B) E. coli KASII and ACP C) S. maritimus EncAB and EncC. D) SDS-Page analysis of one-pot reactions to form KS-CP complexes using the shorter analog 1 and longer analog 3 to compare substrate specificity and binding compatibility between the KSs of type II biosynthesis and both natural and unnatural ACP partners.
Table 1.
Quantification of the degree of crosslinking between KS and ACP using the program ImageJ. A) Comparison of crosslinking using the panel of analogs with natural KS-ACP partners. B) Comparison of natural versus unnatural KS-ACP partners using analogs 1 and 3. Pantetheine analog used in each experiment is designated in bold.
| A) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| KASI-ACP | 87 | 74 | 4 | 5 | 3 | 0 | 0 | 92 | 19 |
| KASII-ACP | 99 | 92 | 27 | 20 | 17 | 4 | 2 | 100 | 100 |
| EncAB-EncC | 42 | 43 | 56 | 23 | 35 | 0 | 0 | 24 | 59 |
| B) | ||
| 1 | 3 | |
| KASI-ACP | 95 | 7 |
| KASI-EncC | 89 | 1 |
| KASII-ACP | 99 | 26 |
| KASII-EncC | 1 | 1 |
| EncAB-ACP | 34 | 28 |
| EncAB-EncC | 32 | 49 |
Figure 4.

Compiled MS-MS data of gel-excised EncA-EncC complex, digested with pepsin, trypsin, or chymotrypsin. Residues highlighted in yellow and blue represent regions of the proteins identified by their peptide fragments. Peptides containing the active site of EncC (S62, highlighted in green) and the active site of EncA (C191, highlighted in purple) were not isolated.
Functional Characteristics of Linker Region and Length of 4’-Phosphopantetheine Arms for ACP-KS Crosslinking
Inspection of the results of the gel-shift assay (Figure 3) indicate several important crosslinking characteristics for the pantetheine analogs in our panel. An initial observation is that our results reaffirm the importance of the second amide bond of pantetheine for enzyme recognition.[35] Analogs 6 and 7, which lack this important recognition element, allowed only minimal interaction with a ketosynthase domain in the KASII-ACP system and showed no detectable interaction in other systems, despite the optimal placement of the electrophilic chloroacrylamide in 6. This is not due simply to a lack of ACP loading by 6 and 7, as MALDI-MS analysis of the one-pot chemoenzymatic reactions before addition of KS shows the expected mass change for formation of crypto-ACPs in both cases (Supporting Figure 1). Among these two poor crosslinkers placement of the reactive electrophile does appear to play a role in ACP-KS crosslinking activity, as evidenced by the slight preference for compound 6 (same length as the natural substrate) over compound 7 (one carbon shorter than the natural substrate) in the KASII-ACP system. Overall placement of the electrophilic trap at a distance greater than that of the position of the natural thioester from the ACP was most effective for crosslinking, an indication of the importance of the length of the 4’-phosphopantetheine arm in modulating carrier protein interactions with accessory enzymes. The identity of the electrophilic functionality at the site of attack of the pantetheine analog also proved very important for ACP-KS crosslinking efficiency, with the most reactive electrophile, α-bromoacetamide 9, showing extreme reactivity with KASII and EncAB systems.
Correlation of 4’-Phosphopantetheine Structure with ACP-KS Crosslinking Efficiency and Substrate Specificity
Crosslinking between a crypto-ACP and KS domain is dependent upon the ability of the KS to both recognize the surface residues of the carrier protein and accommodate and react with the reactive substrate analogue of the modified 4’-phosphopantetheine arm. A greater crosslinking signal, detected as a gel shift on SDS-PAGE, indicates a tighter interaction between the ACP and KS. A weaker signal is indicative of either a lack of favorable binding interactions between the partner ACP and KS, a lack of binding interactions between the 4’-phosphopantetheine arm of the crypto-ACP and the active-site of the KS, or an extremely reactive electrophilic pantetheine analogue which blocks the KS active site before transfer to the ACP. For the purposes of this study we chose electrophilic warheads of tempered reactivity (acrylamides and acetylenamides being some of the least reactive Michael acceptors, haloacetamides being the least reactive α -halocarbonyl compounds) to avoid the latter problem. Furthermore, since the surface residues of our crypto-ACPs are invariable through the full spectrum of our pantetheine analogues, crosslinking efficiency of an ACP and KS from the same system (i.e. E. coli type II FAS) should be mostly a function of the ability of the KS active-site to interact favorably with the structure of the crypto-ACP’s unnatural 4’-phosphopantetheine arm.
For the functional assessment of these interactions we performed thirty one-pot ACP-KS crosslinking reactions, applying each pantetheine analogue 1-9 along with a DMSO control to each of the three systems tested (Figure 3A-C). Our results correlate well with previous studies of substrate specificity in ACP-KS systems and also draw new conclusions yet to be verified. For example, previous studies of ACP-KS dissociation constants in E. coli type II FAS have estimated the Kd as 70 nM between KASII and C14-ACP, versus a weaker interaction of 180 nM between KASI and C14-ACP. Both interactions are stronger than that of the ACP-KS responsible for the biosynthesis of the type II PKS product actinorhodin, which is estimated to lie in the 1-10 uM range.[36] The dissociation constant between EncAB and EncC is expected to be similar to that of the actinorhodin system, as both biosyntheses are carried out by related Streptomyces bacteria. This overall weaker interaction of ACP and KS partners in type II PK biosynthesis is reflected in the relative crosslinking efficiencies of EncC-EncAB when compared to the ACP and KS from E. coli type II FAS. Of the five most active compounds, 1-3, 8, and 9, only two crosslink EncAB with an efficiency of greater than 50%, while three of five crosslink AcpP-KASI to a degree over 70% and four of five crosslink AcpP-KASII to a degree greater than 90% - a direct correlation of crosslinking efficacy to binding of ACP and KS (Table 1A).
In addition to such broad intersystem insights, careful analysis of the crosslinking results provides knowledge of the active sites of the KS enzymes themselves. For example, comparing the results of testing our panel of analogs on the natural KASI-ACP and KASII-ACP systems shows that in addition to its tighter binding, KASII acts more efficiently than KASI on larger, longer substrate mimics. The data generated from the alkyl chain incorporating substrate mimics (compounds 3, 4, 5, and 9) indicate a clear preference (greater than 4-fold) by KASII over KASI for larger substrates. This supports the previously noted study by Edwards et al., which showed KASII acts on larger substrates C14:0 and C16:1 at least two-fold more efficiently than KASI.[37] In comparison, the two KS domains had similar activity (> 70% crosslinking) towards the smaller substrates, compounds 1, 2 and 8. All three of these compounds are similar in size to natural FAS substrates at the beginning of elongation, while compounds 3-5 and 9 are more similar to intermediates in FAS elongation. The slight preference for compound 8 might be a result of a narrow active-site channel which has trouble accepting the bulky halide incorporated by 1 and 2; this hypothesis is supported by the preference for compound 3 over compounds 4 and 5 in the KASII-ACP system.
The substrate binding pocket of the ketosynthase EncAB needs to be larger and bulkier than that of either E. coli FAS ketosynthase. The active site of EncAB holds substrates containing up to seven unreduced ketide units followed by the benzoate starter unit and produces a final product of the condensation reactions which is larger than those produced by either KASI or KASII. The results of our crosslinking studies with compounds 3-5 and 9 reflect this difference. Of the three systems tested in our panel, EncAB showed the greatest activity towards octyl-chain incorporating compounds 3-5. EncAB did not have the greatest reactivity of the three systems with compound 9 (KASII was extremely reactive with this analogue), but hexanoyl bromoacetamide 9 show the greatest crosslinking of EncC-EncAB compared with the rest of the panel. These results are likely due to the inherent volume of the substrate binding pocket. In addition, EncAB does not interact as well with EncC-bound compound 8 as it does with EncC-bound compounds 1 or 2. Even among these starter unit analogues, having a large, bulky leaving group on the pantetheine analog seems to render the analog more effective at binding in the expansive KS active site. However, unlike in the smaller active sites of the E. coli ketosynthases, there is no preference for the trans isomer 1 over the cis isomer 2 in these less bulky crosslinking agents. This lack of a clear stereochemical preference for EncAB crosslinking with 1 and 2, along with the complete crosslinking observed in KASII, suggest that the lack of complete crosslinking by racemic bromoacetamide 9 in the Enc system is due more to the weak protein-protein interactions of EncC-EncAB than to an intrinsic preference for one stereoisomer of 9. To verify this will require the synthesis of enantiopure pantetheine derivatives. Alternatively, the lack of complete crosslinking in the Enc system could be due to competition for the active-site between non-carrier protein-bound pantetheine analogues and crypto-ACP, a phenomenon not observed in the tight-binding type II FAS systems.
Verification of ACP Modification and ACP-KS Crosslinking Site Selectivity
To verify the compatibility of analogues 1-9 with our one-pot chemoenzymatic carrier protein modification protocol we used MALDI-MS to monitor the mass increase upon posttranslational modification of apo to crypto ACPs (Supporting Figure 1). In our initial report the site-specific nature of our crosslinking technique on E. coli AcpP and both KASI and KASII was demonstrated using data from MALDI MS/MS analyses of in gel digested complexes.[18] We extended this approach to the S. maritimus ACP EncC and KS EncAB, which are involved in the biosynthesis of the polyketide enterocin (Figure 4).[26,28] In this experiment, EncC was post-translationally modified by a CoA entity derived from compound 3. Crypto-EncC was then allowed to react with EncAB. The one-pot reaction was run on multiple lanes of SDS-PAGE; the subunits EncA (KS) and EncB (CLF) of the EncAB were dissociated under these denaturing conditions. Bands corresponding to crosslinked EncA-EncC were then excised and digested with either trypsin, chymotrypsin, or pepsin. MALDI MS/MS analyses resulted in the identification of peptides corresponding to 71% sequence coverage of EncC and 96% of EncA. Importantly, the active site residues, Ser72 of our EncC construct and Cys191 of our EncA construct, were not identified in the analysis. This is consistent with post-translational modification of Ser72 and subsequent reaction of crypto-EncC with Cys191, resulting in the site-directed crosslink between EncAB and EncC. While it would have been desirable to obtain peptide fragments representative of the active site regions in the absence of crosslinking conditions, this would have required considerably more experimentation. Instead we chose to address this issue using protein crystallographic studies, which are currently underway. In addition, all of these active-site specific crosslinking reactions failed when the KS was subjected to heat denaturation prior to reaction (unpublished results). Thus, from these experiments and those of our initial report, the site-specific nature of crosslinking has been verified for all ACP and KS species used to date.
Investigation of Intersystem ACP-KS Partner Protein Interactions by Crosslinking Analyses
With each of the compounds tested, KASII from E. coli FAS crosslinks its cognate carrier protein AcpP to a greater degree than KASI. As stated above, this suggests that the AcpP-KASII interaction is stronger than that of AcpP-KASI, a finding supported by previous kinetic analyses of these enzymes.[29] What we intended to examine next was whether the strength of these interactions were mainly functions of the ACP or KS component, and to what extent these protein-protein interactions were compatible across different biosynthetic systems. To study this, we performed combinatorial crosslinking experiments using two compounds: chloroacrylamide 1, our model “starter unit” electrophilic 4’-phosphopantetheine analogue, and α/β unsaturated acetylene amide 3, a long chain 4’-phosphopantetheine precursor which had shown good crosslinking of EncC-EncAB enzymes. Using our one-pot chemoenzymatic method each analogue was loaded onto the carrier proteins AcpP or EncC before incubation with one of the three ketosynthase domains, KASI, KASII, or EncAB (Figure 3D).
Analysis of the crosslinking reactions by gel-shift assay showed that, as expected, each KS domain showed a preference for interacting with its natural carrier protein (Table 1B). In addition, KASII showed negligible crosslinking to crypto- EncC whether modified with analogue 1 or 3. Correlating crosslinking efficiency with compatibility of ACP-KS binding surfaces, this suggests that the binding interactions of KASII are mainly dependent on tight surface residue interaction rather than interactions resulting from the loading of substrate on the 4’-phosphopantetheine arm. In contrast KASI showed a high degree of crosslinking (89%) with EncC modified with starter unit analogue 1 but negligible reactivity (1%) with crypto-EncC loaded with 3, indicating a less permissive active-site coupled to more permissive, less specific binding residues at the protein surface. Together these results suggest KASI and KASII utilize subtly different yet complementary binding modes in E. coli type II FAS (Figure 5).
Figure 5.
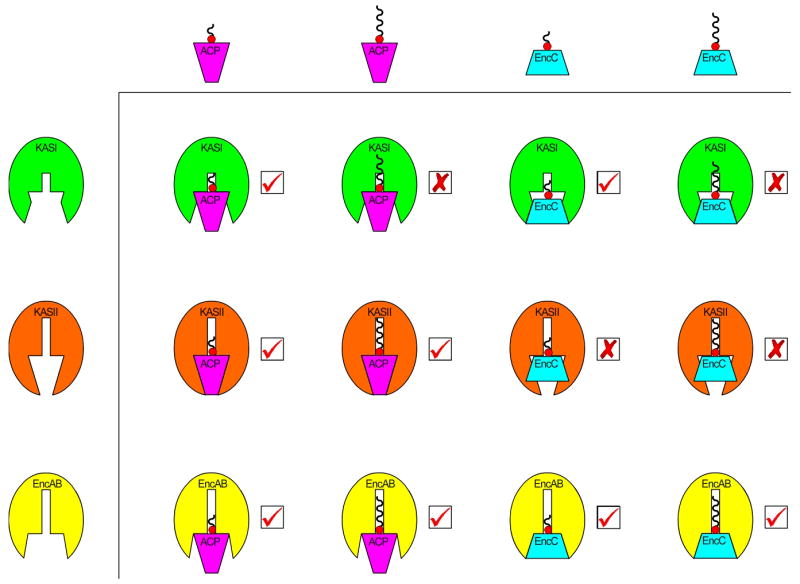
Summary of results from the experiment designed to compare substrate specificity and binding compatibility between the KSs of type II biosynthesis and both natural and unnatural ACP partners. Wavy lines attached to the ACP cartoons correspond to phosphopantetheine analog 1 (shorter line) or 3 (longer line). Significant binding interactions were detected as a gel-shift on SDS-Page in the reactions marked with a “√”, while negligible interactions occurred in the reactions marked with an “X”.
The type II PKS ketosynthase EncAB showed less preference for the carrier protein with which it interacted. Crypto-EncC primed with compounds 1 and 3 crosslinked EncAB reasonably well at values 32% and 49%, respectively. When the unnatural carrier protein AcpP was similarly prepared with 1 and 3 these values were 34% and 28%, respectively. Notably, EncAB showed a significant preference for its natural carrier protein partner when using α/β unsaturated alkyne 3, a compound more similar in size to the natural substrate of EncAB than compound 1. This experiment was comparable to the study by Izumikawa et al, which demonstrated that the benzoate:CoA ligase EncN, responsible for loading benzoate onto EncC, can efficiently load ACPs from type II PKS systems other than enterocin.[27] Together with the observed crosslinking results, this apparent promiscuity of the carrier protein used in crucial steps of enterocin biosynthesis may be an indication that the enterocin system is capable of utilizing the carrier protein from its type II FAS system for enterocin biosynthesis during times of stress. Alternatively, while EncC is located in the enterocin biosynthetic gene cluster, we cannot discount the possibility that this organism expresses only a single ACP that is used for both type II FAS and PKS biosynthesis. To date, only 21 proteins from this organism have been sequenced, all but one of which are associated with the enterocin biosynthetic pathway. Based upon the observed results, one could conjecture that when the S. maritimus FAS gene cluster is eventually deciphered, the sequence of its associated carrier protein may very likely be strikingly similar to EncC.
Conclusions
In the current study we have developed the ACP-KS crosslinking methods from our preliminary reports, synthesizing a variety of 4’-phosphopantetheine analogue precursors in order to identify key structural characteristics necessary for ACP-KS crosslinking. In addition we have tested the compatibility of three new electrophilic functionalities with this approach and expanded the structure of our crosslinking reagents to incorporate fatty alkyl substrate mimics. The utilization of this panel allowed the first crosslinking of an ACP-KS pair from a type II PKS system. Such an approach may allow structural analysis of this single step in the biosynthesis of the medicinally relevant enterocin and aid future efforts at rational design of combinatorial biosynthetic enzymes. We demonstrated the site-specificity of our crosslinking agents in the natural KASI-ACP, KASII-ACP, and EncAB-EncC systems using MS analyses. The site-specificity of crosslinking between unnatural partner proteins was not evaluated in this study. Future studies exploring the functional aspects of these unnatural interactions will address this issue.
Perhaps most interestingly, this study has demonstrated a clear correlation between structure of the electrophilic 4’-phosphopantetheine arm and active-site structure, substrate specificity, and crosslinking efficiency. In particular, our results agree with a variety of studies that have investigated binding affinity and substrate specificity of KS domains in type II FAS and PKS systems. This finding points to the information-rich nature of thoroughly studied crosslinking analyses using our system. Finally, we have used our techniques to investigate the dominant interactions responsible for ACP-KS binding in both fatty acid and enterocin biosynthetic systems. Although the results of these intersystem crosslinking studies must be verified by analysis of substrate turnover, we hope to use this approach in the future to aid in the development of unnatural biosynthetic systems by investigating a wide-range of ACP-KS pairs to help establish a foundational knowledge of ACP-KS interactions. This approach could be further aided by mutagenic studies in which we use crosslinking studies to determine the amino-acid residues crucial for protein surface and enzyme-substrate interactions in these systems.
Methods
Synthetic protocols, compound characterization, and additional experimental procedures may be found in the Supporting Information.
Crosslinking of Recombinant ACP and Ketosynthase
To compare the reactivities of our ketosynthases towards our panel of pantetheine analogs (1-9) and to compare the interaction between ketosynthase and natural vs. unnatural carrier protein by SDS-PAGE, we used the following procedure. To a 50 mM potassium phosphate, pH 7.0, buffer with 12.5 mM Mg2Cl2 and 8 mM ATP, were added carrier protein (4 μg, ≥ 5-fold excess), CoaA (0.1 μg), CoaD (0.5 μg), CoaE (1 μg), and B. subtilis Sfp (0.5 μg). To the mixture, we added pantetheine analog (0.1 mM), and the mixture was incubated at 37°C for 5 minutes. We added ketosynthase (2-3.5 μg), and the reaction was allowed to proceed at 37°C for 1 hour. Samples were run on a 9% SDS-PAGE, where crosslinked product was detected by staining with Coomassie. Gels were photographed with a BioRad Fluor-S MultiImager, and bands were quantified using the ImageJ software provided by NIH.[33]
Mass Spectrometry Analyses
To verify the post-translational modification of carrier protein using matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS), we used the following procedure. To a 50 mM potassium phosphate, pH 7.0, buffer with 12.5 mM Mg2Cl2 and 8 mM ATP, were added carrier protein (4 μg), CoaA (0.1 μg), CoaD (0.5 μg), CoaE (1 μg), and B. subtilis Sfp (0.5 μg). To the mixture, we added pantetheine analog (1 or 3, 0.1 mM), and the mixture was incubated at 37°C for 1 hour. The reaction mix was analyzed by MALDI-MS without further purification.
MALDI MS/MS analyses of in gel digested complexes were performed as previously described.[18]
Supplementary Material
{kind=link}
Acknowledgments
Funding was provided by ACS-PRF 42158-G4, NIH R01GM075797, NIH R01AI47818, and ACS RSG-01-011-01-CDD. We thank H. Mori from the Japanese E. coli consortium for providing genes from the ASKA plasmid ORF library.[38]
References
- 1.Hopwood DA, Sherman DH. Annu Rev Genet. 1990;24:37–66. doi: 10.1146/annurev.ge.24.120190.000345. [DOI] [PubMed] [Google Scholar]
- 2.Smith S, Tsai SC. Nat Prod Rep. 2007;24:1041–1072. doi: 10.1039/b603600g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White SW, Zheng J, Zhang YM, Rock CO. Annu Rev Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 4.Hertweck C, Luzhetskyy A, Rebets Y, Bechthold A. Nat Prod Rep. 2007;24:162–190. doi: 10.1039/b507395m. [DOI] [PubMed] [Google Scholar]
- 5.Mercer AC, Burkart MD. Nat Prod Rep. 2007;24:750–773. doi: 10.1039/b603921a. [DOI] [PubMed] [Google Scholar]
- 6.Maier T, Jenni S, Ban N. Science. 2006;311:1258–1267. doi: 10.1126/science.1123248. [DOI] [PubMed] [Google Scholar]
- 7.Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE. Annu Rev Biochem. 2007;76:195–221. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- 8.Shen B. Curr Opin Chem Biol. 2003;7:285–295. doi: 10.1016/s1367-5931(03)00020-6. [DOI] [PubMed] [Google Scholar]
- 9.Lambalot RH, Gehring AM, Flugel RS, Zuber P, LaCelle M, Marahiel MA, Reid R, Khosla C, Walsh CT. Chem Biol. 1996;3:923–936. doi: 10.1016/s1074-5521(96)90181-7. [DOI] [PubMed] [Google Scholar]
- 10.Worthington AS, Burkart MD. Org Biomol Chem. 2006;4:44–46. doi: 10.1039/b512735a. [DOI] [PubMed] [Google Scholar]
- 11.La Clair JJ, Foley TL, Schegg TR, Regan CM, Burkart MD. Chem Biol. 2004;11:195–201. doi: 10.1016/j.chembiol.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 12.Mercer AC, La Clair JJ, Burkart MD. Chembiochem. 2005;6:1335–1337. doi: 10.1002/cbic.200500051. [DOI] [PubMed] [Google Scholar]
- 13.Yin J, Lin AJ, Golan DE, Walsh CT. Nat Protoc. 2006;1:280–285. doi: 10.1038/nprot.2006.43. [DOI] [PubMed] [Google Scholar]
- 14.Sieber SA, Walsh CT, Marahiel MA. J Am Chem Soc. 2003;125:10862–10866. doi: 10.1021/ja0361852. [DOI] [PubMed] [Google Scholar]
- 15.Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT. Biochemistry. 1998;37:1585–1595. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 16.Meier JL, Mercer AC, Rivera H, Jr, Burkart MD. J Am Chem Soc. 2006;128:12174–12184. doi: 10.1021/ja063217n. [DOI] [PubMed] [Google Scholar]
- 17.Dorrestein PC, Blackhall J, Straight PD, Fischbach MA, Garneau-Tsodikova S, Edwards DJ, McLaughlin S, Lin M, Gerwick WH, Kolter R, Walsh CT, Kelleher NL. Biochemistry. 2006;45:1537–1546. doi: 10.1021/bi052333k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Worthington AS, Rivera H, Torpey JW, Alexander MD, Burkart MD. ACS Chem Biol. 2006;1:687–691. doi: 10.1021/cb6003965. [DOI] [PubMed] [Google Scholar]
- 19.D’Agnolo G, Rosenfeld IS, Vagelos PR. J Biol Chem. 1975;250:5283–5288. [PubMed] [Google Scholar]
- 20.Olsen JG, Kadziola A, von Wettstein-Knowles P, Siggaard-Andersen M, Larsen S. Structure. 2001;9:233–243. doi: 10.1016/s0969-2126(01)00583-4. [DOI] [PubMed] [Google Scholar]
- 21.Drier J, Khosla C. Biochemistry. 2000;39:2088–2095. doi: 10.1021/bi992121l. [DOI] [PubMed] [Google Scholar]
- 22.Haushalter RW, Worthington AS, Hur GH, Burkart MD. Bioorg Med Chem Lett. 2008 doi: 10.1016/j.bmcl.2008.01.026. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu N, Cane DE, Khosla C. Biochemistry. 2002;41:5056–5066. doi: 10.1021/bi012086u. [DOI] [PubMed] [Google Scholar]
- 24.Menzella HG, Reid R, Carney JR, Chandran SS, Reisinger SJ, Patel KG, Hopwood DA, Santi DV. Nat Biotechnol. 2005;23:1171–1176. doi: 10.1038/nbt1128. [DOI] [PubMed] [Google Scholar]
- 25.Weissman KJ, Leadlay PF. Nat Rev Microbiol. 2005;3:925–936. doi: 10.1038/nrmicro1287. [DOI] [PubMed] [Google Scholar]
- 26.Piel J, Hertweck C, Shipley PR, Hunt DM, Newman MS, Moore BS. Chem Biol. 2000;7:943–955. doi: 10.1016/s1074-5521(00)00044-2. [DOI] [PubMed] [Google Scholar]
- 27.Izumikawa M, Cheng Q, Moore BS. J Am Chem Soc. 2006;128:1428–1429. doi: 10.1021/ja0559707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng Q, Xiang L, Izumikawa M, Meluzzi D, Moore BS. Nat Chem Biol. 2007;3:557–558. doi: 10.1038/nchembio.2007.22. [DOI] [PubMed] [Google Scholar]
- 29.McGuire KA, McGuire JN, von Wettstein-Knowles P. Biochem Soc Trans. 2000;28:607–610. doi: 10.1042/0300-5127:0280607. [DOI] [PubMed] [Google Scholar]
- 30.Olsen JG, Kadziola A, von Wettstein-Knowles P, Siggaard-Andersen M, Lindquist Y, Larsen S. FEBS Lett. 1999;460:46–52. doi: 10.1016/s0014-5793(99)01303-4. [DOI] [PubMed] [Google Scholar]
- 31.Moche M, Dehesh K, Edwards P, Lindqvist Y. J Mol Biol. 2001;305:491–503. doi: 10.1006/jmbi.2000.4272. [DOI] [PubMed] [Google Scholar]
- 32.Govardhan CP, Abeles RH. Arch Biochem Biophys. 1996;330:110–114. doi: 10.1006/abbi.1996.0231. [DOI] [PubMed] [Google Scholar]
- 33.Plapp BV, Chen WS. Methods Enzymol. 1981;72:587–591. doi: 10.1016/s0076-6879(81)72048-2. [DOI] [PubMed] [Google Scholar]
- 34.Abramoff MD, Magelhaes PJ, Ram SJ. Biophoton Int. 2004;11:36–42. [Google Scholar]
- 35.Xun J, Huang H, Vogel KW, Drueckhammer DG. Bioorg Chem. 2005;33:90–107. doi: 10.1016/j.bioorg.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 36.Dreier J, Shah AN, Khosla C. J Biol Chem. 1999;274:25108–25112. doi: 10.1074/jbc.274.35.25108. [DOI] [PubMed] [Google Scholar]
- 37.Edwards P, Nelsen JS, Metz JG, Dehesh K. FEBS Lett. 1997;402:62–66. doi: 10.1016/s0014-5793(96)01437-8. [DOI] [PubMed] [Google Scholar]
- 38.Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. DNA Res. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.