

Abstract
The initial, rapid increase in skin blood flow in response to direct application of heat is thought to be mediated by an axon reflex, which is dependent on intact cutaneous sensory nerves. We tested the hypothesis that inhibition of transient receptor potential vanilloid type 1 (TRPV-1) channels, which are putative channels located on sensory nerves, would attenuate the skin blood flow response to local heating in humans. Ten subjects were equipped with four microdialysis fibres which were randomly assigned one of four treatments: (1) vehicle control (90% propylene glycol + 10% lactated Ringer solution); (2) 20 mm capsazepine to inhibit TRPV-1 channels; (3) 10 mm l-NAME to inhibit NO synthase; and (4) combined 20 mm capsazepine + 10 mm l-NAME. Following baseline measurements, the temperature of skin heaters was increased from 33°C to 42°C at a rate of 1.0°C every 10 s and local temperature was held at 42°C for 20–30 min until a stable plateau in skin blood flow was achieved. An index of skin blood flow was measured directly over each microdialysis site via laser-Doppler flowmetry (LDF). Beat-by-beat blood pressure was measured via photoplethysmography and verified via automated brachial auscultation. At the end of the local heating protocol, temperature of the heaters was increased to 43°C and 28 mm nitroprusside was infused to achieve maximal vasodilatation. Cutaneous vascular conductance (CVC) was calculated as LDF/mean arterial pressure and normalized to maximal values (%CVCmax). Initial peak in capsazepine (44 ± 4%CVCmax), l-NAME (56 ± 4%CVCmax) and capsazepine +l-NAME (32 ± 6%CVCmax) sites was significantly attenuated compared to control (87 ± 5%CVCmax; P < 0.001 for all conditions). The plateau phase of thermal hyperaemia was significantly attenuated in capsazepine (73 ± 6%CVCmax), l-NAME (47 ± 5%CVCmax) and capsazepine +l-NAME (31 ± 7%CVCmax) sites compared to control (92 ± 5%CVCmax; P < 0.001 for all conditions). These data suggest TRPV-1 channels contribute substantially to the initial peak and modestly to the plateau phases of thermal hyperaemia. These data further suggest a portion of the NO component of thermal hyperaemia may be due to activation of TRPV-1 channels.
Introduction
Local application of heat to human skin results in a large increase in skin blood flow that is biphasic and characterised by a rapid initial peak and nadir followed by a more prolonged pleateau (Kellogg et al. 1999; Minson et al. 2001). The precise mechanisms underlying this robust cutaneous thermal hyperaemia remain unresolved; however, it is generally believed the initial peak and nadir are mediated by an axon reflex mechanism while the prolonged plateau is a predominantly nitric oxide (NO)-dependent mechanism (Magerl & Treede, 1996; Kellogg et al. 1999; Minson et al. 2001). Mechanisms of cutaneous thermal hyperaemia have been shown to be altered by the rate at which heat is applied to the skin (Magerl & Treede, 1996; Hodges et al. 2009a) and in instances where there is a conscious perception of pain during the local heating (Kellogg et al. 1999), suggesting the skin blood flow response to local heating involves a complex and, most probably, redundant series of mechanisms and vasodilator pathways. Cutaneous thermal hyperaemia is frequently used to assess microvascular function and reactivity and, as such, it is important to delineate mechanisms of this response.
The initial peak and nadir of cutaneous thermal hyperaemia has been shown to be primarily dependent on cutaneous axon reflexes and, to a lesser extent, NO (Kellogg et al. 1999; Minson et al. 2001). It has further been suggested that calcitonin gene-related peptide (CGRP), possibly co-released with substance P, is ultimately responsible for the initial peak and nadir components of thermal hyperaemia (Wallengren et al. 1987; Schmelz et al. 1997); however, to date there has been no experimental evidence to support this claim. In an elegant series of studies, Minson and colleagues (2001) demonstrated that inhibition of NO and local axon reflexes (via local application of EMLA cream, which is a topical anaesthetic that blocks the axon reflexes) independently attenuated both the initial peak and nadir. These investigators also found that EMLA cream attenuated the initial peak and nadir to a greater extent than inhibition of NO synthase; however, EMLA did not abolish the initial peak response. Taken together, Minson et al. (2001) suggested that both axon reflexes and NO contribute to the initial peak and nadir component of thermal hyperaemia, and the majority of this response is mediated by an axon reflex mechanism.
Since EMLA cream inhibits sensory nerve function, the question as to which mechanisms are involved in depolarising the sensory nerves in response to application of heat to the skin remains unresolved. Transient receptor potential vanilloid (TRPV) channels are a group of ion channels characterised by their activation to a host of stimuli, including temperature (Caterina, 2007; Vriens et al. 2009). The TRPV-1 channel is activated by heat (threshold ∼42°C) and by capsaicin, the active ingredient in hot chili peppers, and is located primarily on afferent sensory nerves of the skin and other tissues (Caterina, 2007; Lin et al. 2007; Wu et al. 2007; Vriens et al. 2009). TRPV-1 channels have also been shown to be expressed in human endothelial cells and have been suggested to be involved in regulation of vascular tone, endothelial-dependent vasodilatation and temperature regulation in animals (Yao & Garland, 2005). If sensory nerves and axon reflexes contribute to the initial peak and nadir of thermal hyperaemia, activation of TRPV-1 channels may constitute one mechanism by which the cutaneous sensory nerves are depolarised, which subsequently results in axon reflex-mediated vasodilatation. Recent data further suggest that TRPV channels, specifically TRPV-1, can be activated by NO (Yao & Garland, 2005; Yoshida et al. 2006; Miyamoto et al. 2009) and may enhance the production of NO (Yao & Garland, 2005). Taken together, the above information supports a potential role for TRPV-1 channels to cutaneous thermal hyperaemia.
The purpose of the present study was to test the following hypotheses: (1) inhibition of TRPV-1 channels would substantially attenuate the initial peak and nadir (axon reflex) components of thermal hyperaemia; (2) inhibition of TRPV-1 channels would result in a modest attenuation of the plateau phase of thermal hyperaemia, which would support an interaction between TRPV-1 channel activation and NO; and (3) combined inhibition of TRPV-1 channels and NO synthase would attenuate all phases of thermal hyperaemia to a greater extent than TRPV-1 channel or NO synthase inhibition alone.
Methods
Ethical approval
The Institutional Review Board of Kansas State University approved this study. Prior to participation, the details of the protocol were verbally explained to each subject and written informed consent was obtained. All protocols conformed to guidelines as set forth by the Declaration of Helsinki.
Subjects
Ten subjects (7 men, 3 women) between the ages of 21 and 26 participated in this study. All subjects were healthy, non-smokers, free of cardiovascular disease and diabetes, and were not taking any medications aside from oral contraceptives. All subjects were asked to refrain from caffeine, alcohol and exercise for 12 h prior to the study. Phase of menstrual cycle or oral contraceptive use was noted for female subjects but not controlled for in these experiments. Due to the robust nature of the thermal hyperaemic response and the low number of female subjects, there were no discernable differences in data due to phase of menstrual cycle or oral contraceptive use. Thus, data from men and women were grouped.
Instrumentation
Skin blood flow measurements were made from the ventral aspect of the left forearm and subjects rested supine with the experimental arm at heart level for the entire protocol. Blood pressure was monitored beat by beat via photoplethysmography (NexfinHD; BMEYE, Amsterdam, The Netherlands) and verified via automated brachial auscultation (S/5 Light Monitor; Datex-Ohmeda, GE Healthcare; Madison, WI, USA) every 10 min.
Four microdialysis fibres, approximately 3–5 cm apart, were placed in the dermal layer of the skin on the ventral aspect of the forearm in the absence of anaesthaesia. Ice was used to numb the skin prior to placement of the microdialysis fibres (Hodges et al. 2009a). To place the microdialysis fibres, a 23-gauge needle was placed in the dermal layer of the skin, a fibre was threaded through the lumen of the needle, and the needle was removed, leaving the membrane in place. The needle used in this study (23-guage; ∼1 cm long (1/2 inch) needle) was slightly larger than what has been reported previously (25-guage; 1.5 inch long needle) by our laboratory and others (for example, see: Minson et al. 2001; Wong et al. 2003, 2004, 2006; Holowatz et al. 2005; Hodges et al. 2009a, 2009b; Fieger & Wong, 2010); however, the extent of the trauma response induced by placement of the slightly larger needle was not visibly different than what we have experienced with 25-guage needles and the time to resolution of the trauma response was similar to what has been previously reported. The membranes of the microdialysis fibres were 10 mm in length with a 55 kDa molecular mass cut-off (CMA 31 Linear Probe; CMA Microdialysis, Sweden). Approximately 45–90 min were allowed for resolution of the trauma response induced by microdialysis fibre placement in the skin before the start of the protocol. During this time, all fibres were perfused with lactated Ringer solution at a rate of 4 μl min−1.
Red blood cell (RBC) flux, measured by laser-Doppler flowmetry (PeriFlux 5010 laser-Doppler perfusion monitor; Perimed; Jarfalla, Sweden), served as an estimate of skin blood flow. Local heating units (PF5020 local heating units and PeriFlux 5020 Temperature Unit; Perimed) were placed on the skin directly over each microdialysis membrane, and an integrated laser-Doppler probe with seven receiving and seven emitting fibres (Probe 413; Perimed) was placed in the centre of each local heating unit to measure RBC flux directly over each microdialysis site.
Protocol
Data collection began when skin blood flow was observed to stabilize over the areas of microdialysis insertion via laser Doppler flowmetry, indicating resolution of the trauma response. Pre-infusion baseline skin blood flow measurements were recorded during a 5 min period with local heaters set at 33°C.
Following baseline measurements, drug infusion through each microdialysis fibre was initiated. Each microdialysis fibre was randomly assigned one of four treatments: (1) 90% propylene glycol (USP grade; Sigma; St Louis, MO, USA) in 10% lactated Ringer solution (Baxter Healthcare; Deerfield, IL, USA) to serve as a vehicle control; (2) 20 mm capsazepine (Tocris Biosciences; Ellisville, MO, USA) was used to selectively inhibit TRPV-1 channels; (3) 10 mm of the l-arginine analogue NG-nitro-l-arginine methyl ester (l-NAME; EMD Biosciences; San Diego, CA, USA) to inhibit NO synthase; and (4) combined 20 mm capsazepine and 10 mm l-NAME, to simultaneously inhibit TRPV-1 channels and NO synthase.
Capsazepine was used to selectively inhibit TRPV-1 channels. Capsazepine has a half-maximal inhibitory concentration (IC50) of 10−7–10−6m (Caterina et al. 1997) and dissociation constant, Ki, of 3.2 μm (Tocris) for the TRPV-1 channel. Lin et al. (2007) has demonstrated that intra-arterial infusions of capsazepine nearly abolished capsaicin-induced inflammation in anaesthetized rats and this reduction was more than that achieved with a CGRP or NK1 antagonist. Movahed et al. (2005) have shown that 10 mm capsazepine abolishes cutaneous vasodilatation induced by three TRPV-1-specific agonists, including capsaicin. These two studies, in conjunction with the IC50 and Ki values, provide evidence to suggest capsazepine is a TRPV-1-specific antagonist. The low solubility of capsazepine in aqueous solution required the use of a 90% propylene glycol in 10% lactated Ringer solution vehicle. Propylene glycol is a solvent found in many household and personal hygiene products, such as deodorant and toothpaste, and is frequently used to solubilize prescription medications for intravenous use, such as diazepam (i.e. valium; Baldessarini, 2001; Klaassen, 2001). During initial vehicle control pilot work it was determined that a 90% propylene glycol in 10% lactated Ringer solution and a 20 mm concentration of capsazepine was the optimal vehicle and dose of drug, respectively, inasmuch as higher concentrations of capsazepine required the addition of dimethyl sulfoxide (DMSO). The addition of DMSO was found to significantly increase baseline skin blood flow. To minimize the potentially confounding effects of increased baseline skin blood flow, we chose to use a 20 mm concentration of capsazepine dissolved in 90% propylene glycol and 10% lactated Ringer solution. We, and others, have previously shown that a 10 mm concentration of l-NAME adequately inhibits NO synthase in human skin (Minson et al. 2001; Wong et al. 2003, 2006; Holowatz et al. 2005; McCord et al. 2006; Fieger & Wong, 2010). All drug treatments were dissolved in 90% propylene glycol and 10% lactated Ringer solution and were perfused at a constant rate of 2 μl min−1 with a microinfusion pump (Bee Hive controller and Baby Bee Syringe Pumps; Bioanalytical Systems, West Lafayette, IN, USA). Pre-infusion baseline was taken as the last minute prior to drug infusion. Post-infusion baseline was taken as the last 1–2 min of infusion prior to local heating.
All drugs were infused for at least 60 min prior to commencement of the local heating protocol. Extensive pilot work indicated that at least 60 min of infusion of capsazepine was required for maximal inhibition of TRPV-1 channels and previous studies have demonstrated this duration of infusion adequately inhibits NO synthase (Minson et al. 2001; Wong et al. 2003, 2006; Holowatz et al. 2005; McCord et al. 2006; Fieger & Wong, 2010). Following at least 60 min of drug infusion, the temperature of the local heaters was increased to 42°C at a rate of 1°C every 10 s (equivalent to 0.5°C every 5 s; Minson et al. 2001; Wong et al. 2006; Fieger & Wong, 2010). The temperature of the local heating units was maintained at 42°C until a plateau in skin blood flow was observed and maintained for 20–30 min. Once this stable plateau was established, a maximal skin blood flow response was elicited via infusion of sodium nitroprusside (SNP) at a rate of 4 μl min−1 and a simultaneous temperature increase to 43°C. This temperature increase and dose of SNP have been previously determined effective in eliciting a maximal skin blood flow response (Minson et al. 2001; Holowatz et al. 2005; McCord et al. 2006; Wong et al. 2006; Fieger & Wong, 2010).
Data collection and analysis
Data were digitized and stored at 100 Hz on a personal computer and were subsequently analysed off-line using signal-processing software (Windaq; Dataq Instruments, Akron, OH, USA). Cutaneous vascular conductance (CVC) was calculated as RBC flux divided by MAP and normalized to maximal vasodilatation (%CVCmax) via SNP infusion and local heating to 43°C.
The initial peak and nadir are rapid and transient, and last, on average, 90–150 s. Therefore, a stable 30–90 s period of skin blood flow was used for analysis. For the secondary plateau and maximal skin blood flow responses, a stable 3–5 min period of skin blood flow was used for analysis.
Due to the low number of female subjects who participated in this study (n = 3), any differences due to phase of menstrual cycle or oral contraceptive use would not be expected to significantly alter the data. Therefore, data from all subjects were averaged for statistical analysis. For each experimental site, a paired t test was used to compare pre-drug infusion and post-drug infusion (before heating) baseline values. A one-way repeated measure ANOVA was used to compare the effect of drug treatment between experimental sites. The relative contribution of TRPV-1 channels and NO as well as the interaction between TRPV-1 channels and NO was determined using one-way repeated measure ANOVA. Maximal CVC values for each site were compared using a one-way ANOVA. The per cent contribution of TRPV-1 channels, NO, and combined TRPV-1 channels + NO was calculated using the following equation:
where CVCcon is CVC in the control site and CVCexp is CVC in the respective experimental site (capsazepine, l-NAME or combined capsazepine +l-NAME). This calculation was performed for the initial peak, nadir and plateau for each treatment site, and the data were compared using a one-way ANOVA with repeated measures. For all ANOVAs, Student–Newman–Keuls post hoc analysis was used to determine where significance differences occurred. All statistical analyses were performed using SigmaStat 3.5 (Systat Software; Point Richmond, CA, USA). P values < 0.05 were considered to be significant and all data presented are mean ±s.e.m.
Results
The data depicted in Fig. 1A–D are from a single subject and are representative of the thermal hyperaemic response in each treatment site.
Figure 1. Representative tracing of the skin blood flow response in the four treatment sites.
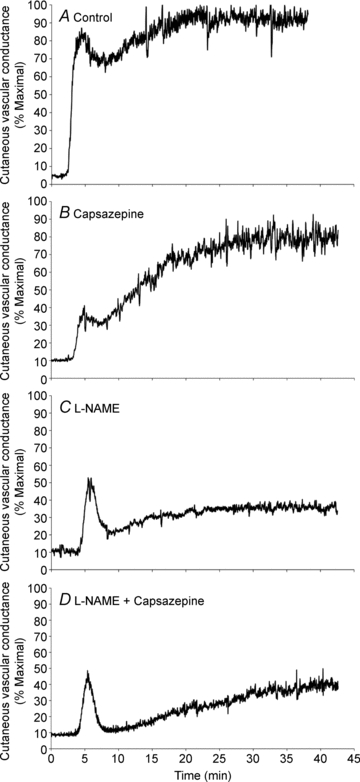
A, vehicle control (90% propylene glycol + 10% lactated Ringer solution); B, capsazepine (TRPV-1 inhibition); C, l-NAME (NO synthase inhibition); D, combined capsazepine +l-NAME. Data are from one subject.
Baseline and maximal skin blood flow
There was no significant difference between pre-infusion and post-infusion baseline values within any of the treatment sites (Table 1) and there was no statistical difference in baseline skin blood flow between any of the treatment sites (Fig. 2). Similarly, there was no significant difference in maximal CVC values between treatment sites (Table 2).
Table 1.
Pre- and post-drug infusion baseline CVC values
| Treatment site | Pre-infusion CVC | Post-infusion CVC |
|---|---|---|
| Control | 13 ± 3 | 11 ± 2 |
| Capsazepine | 15 ± 3 | 14 ± 5 |
| l-NAME | 11 ± 2 | 11 ± 3 |
| Capsazepine +l-NAME | 12 ± 4 | 15 ± 6 |
Values are mean ±s.e.m. There was no statistical difference between pre- and post-drug infusion baseline CVC values. CVC, cutaneous vascular conductance (RBC flux/MAP); l-NAME, NO synthase inhibition.
Figure 2. Comparison of baseline CVC between treatment sites.
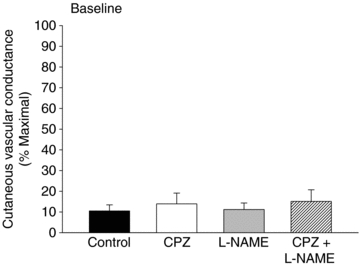
Post-drug infusion baseline CVC values were not significantly different between treatment sites. Values are mean ±s.e.m.
Table 2.
Absolute maximal CVC values
| Treatment site | Maximal CVC |
|---|---|
| Control | 1.99 ± 0.30 |
| Capsazepine | 2.03 ± 0.56 |
| l-NAME | 2.10 ± 0.22 |
| Capsazepine +l-NAME | 1.97 ± 0.41 |
Values are mean ±s.e.m. There was no statistical difference in maximal CVC between treatment sites. CVC, cutaneous vascular conductance (RBC flux/MAP); l-NAME, NO sunthase inhibition.
The initial peak in control sites averaged 87 ± 5%CVCmax. The initial peak was significantly reduced in capsazepine (44 ± 4%CVCmax), l-NAME (56 ± 4%CVCmax) and capsazepine +l-NAME (32 ± 6%CVCmax) sites compared to control (P < 0.001 for all conditions). Capsazepine significantly attenuated the initial peak compared to l-NAME (P < 0.001) and the combination of capsazepine +l-NAME attenuated the initial peak compared to both capsazepine and l-NAME alone (P < 0.001 for both conditions). These data are shown in Fig. 3. These data suggest the contribution of TRPV-1 channels is greater than the contribution of NO to the initial peak. Since the reduction in capsazepine +l-NAME sites was greater than the reduction in l-NAME only sites, the data further suggest that a portion of the NO component of the initial peak may be due to TRPV-1 channel activation.
Figure 3. Effect of TRPV-1 channel and NO synthase inhibiton on the initial peak response to local heating.
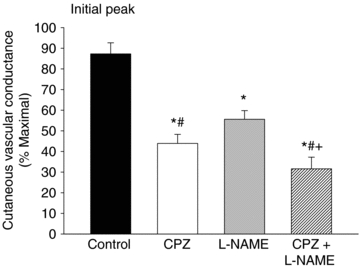
Inhibition of TRPV-1 channels and NO synthase significantly attenuated the initial peak compared to control. The effect of TRPV-1 inhibition was greater than the effect of NO synthase inhibition. Combined inhibition of TRPV-1 channels and NO synthase further attenuated the initial peak. *P < 0.05 vs. control; #P < 0.05 vs.l-NAME; +P < 0.05 vs. capsazepine. CPZ, capsazepine (TRPV-1 inhibition); l-NAME, NO synthase inhibition.
Figure 4 shows the results for the nadir. The nadir in control sites averaged 64 ± 5%CVCmax. The nadir was significantly attenuated in capsazepine (35 ± 4%CVCmax), l-NAME (22 ± 3%CVCmax) and capsazepine +l-NAME (23 ± 3%CVCmax) sites compared to control (P < 0.001 for all conditions). The nadir in l-NAME and combined capsazepine +l-NAME sites was significantly attenuated compared to capsazepine (P < 0.001) but there was no difference between l-NAME and combined capsazepine +l-NAME sites. These data suggest the contribution of NO to the nadir is greater than the contribution of TRPV-1 channels. As there was no further reduction of the nadir in capsazepine +l-NAME sites compared to l-NAME only sites, this suggests TRPV-1 channels do not contribute to the nadir when NO synthase is inhibited and the entirety of the nadir can be explained by NO.
Figure 4. Effect of TRPV-1 channel and NO synthase inhibiton on the nadir response to local heating.
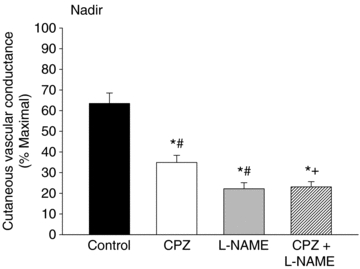
Inhibition of TRPV-1 channels and NO synthase significantly attenuated the nadir compared to control. The effect of NO synthase inhibition was greater than the effect of TRPV-1 inhibition. Combined inhibition of TRPV-1 channels and NO synthase further attenuated the initial peak. *P < 0.05 vs. control; #P < 0.05 vs.l-NAME; +P < 0.05 vs. capsazepine. CPZ, capsazepine (TRPV-1 inhibition); l-NAME, NO synthase inhibition.
The plateau phase of thermal hyperaemia averaged 92 ± 5%CVCmax in control sites, 73 ± 6%CVCmax in capsazepine sites, 47 ± 5%CVCmax in l-NAME sites and 31 ± 7%CVCmax in combined capsazepine +l-NAME sites. The plateau was significantly attenuated in capsazepine, l-NAME, and combined capsazepine +l-NAME sites compared to the control (P < 0.001 for all conditions). In addition, the plateau was significantly attenuated in l-NAME and combined capsazepine +l-NAME sites compared to capsazepine (P < 0.001). The plateau in capsazepine +l-NAME sites was attenuated compared to l-NAME only sites (P < 0.001). These data are shown in Fig. 5. These results suggest the contribution of NO is greater than the contribution of TRPV-1 channels to the plateau phase of thermal hyperaemia. As the attenuation in combined capsazepine +l-NAME sites was greater than either the capsazepine or l-NAME only sites, this suggests a portion of the NO component may be due to an interaction with TRPV-1 channels; however, a large portion of the NO component is from sources other than TRPV-1 channels.
Figure 5. Effect of TRPV-1 channel and NO synthase inhibiton on the plateau response to local heating.

Inhibition of TRPV-1 channels and NO synthase significantly attenuated the plateau compared to control. The effect of NO synthase inhibition was greater than the effect of TRPV-1 inhibition. Combined inhibition of TRPV-1 channels and NO synthase further attenuated the initial peak. *P < 0.05 vs. control; #P < 0.05 vs.l-NAME; +P < 0.05 vs. capsazepine. CPZ, capsazepine (TRPV-1 inhibition); l-NAME, NO synthase inhibition.
Per cent contribution of NO and TRPV-1 channels to thermal hyperaemia
Figure 6A–C depicts the per cent contribution of NO, TRPV-1 channels and combined NO + TRPV-1 channels to the initial peak, nadir and plateau of the thermal hyperaemic response. For the initial peak (Fig. 6A), the contribution of NO (36 ± 4%CVCmax) was significantly less than the contribution of TRPV-1 channels (50 ± 6%CVCmax) and combined NO + TRPV-1 channels (64%± 7%CVCmax; P < 0.001 for all conditions). Similarly, the per cent contribution from TRPV-1 channels was significantly less than the contribution from combined NO + TRPV-1 channels. For the nadir (Fig. 6B), there was no significant difference between the contribution of NO (65 ± 6%CVCmax) and the contribution of combined NO + TRPV-1 channels (63 ± 5%CVCmax); however, these contributions were significantly greater than the contribution of TRPV-1 channels (45 ± 9%CVCmax) to the nadir. For the plateau (Fig. 6C), the contribution from combined NO + TRPV-1 channels (68 ± 5%CVCmax) was significantly greater than the contribution from NO (49 ± 6%CVCmax) or from TRPV-1 channels (20 ± 4%CVCmax; P < 0.001 for all conditions). The contribution of TRPV-1 channels to the plateau was modest and significantly less than the contribution of NO (P < 0.001).
Figure 6. Per cent contribution of TRPV-1 channels, NO, and combined TRPV-1 channels + NO to cutaneous thermal hyperaemia.
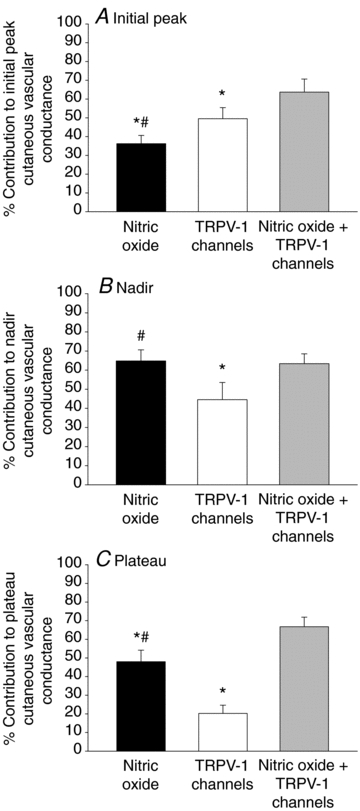
A, per cent contrtibution of TRPV-1 channels and NO to the initial peak; B, per cent contribution of TRPV-1 channels and NO to the nadir; C, per cent contribution of TRPV-1 channels and NO to the plateau. *P < 0.05 vs. nitric oxide + TRPV-1 channels; #P < 0.05 vs. TRPV-1 channels.
Discussion
The major new finding from this study is that TRPV-1 channels contribute to the thermal hyperaemic response in human skin. We observed a substantial role for TRPV-1 channels to the axon reflex component of cutaneous thermal hyperaemia, as evidenced by the attenuated initial peak (Fig. 3) and nadir (Fig. 4) components in sites treated with the TRPV-1-specific inhibitor capsazepine. Inhibition of TRPV-1 channels also attenuated the plateau component (Fig. 5); however, this attenuation was minimal when compared to the attenuation observed in l-NAME and combined capsazepine +l-NAME sites (Fig. 5). The observation that combined capszaepine +l-NAME attenuated the entire thermal hyperaemic response more than individual drug treatments further suggests there is an interaction between TRPV-1 channels and NO.
Axon reflex component of cutaneous thermal hyperaemia
The TRPV-1 channels have been shown to be activated by temperature (heat), localized on sensory afferent nerves and endothelial cells, and to be activated by, or enhance the production of, NO (Yao & Garland, 2005; Yoshida et al. 2006; Caterina, 2007; Lin et al. 2007; Wu et al. 2007; Miyamoto et al. 2009; Vriens et al. 2009). These characteristics of TRPV-1 channels make them an ideal candidate for initiating the sequence of events that ultimately leads to axon reflex-induced vasodilatation.
Consistent with our hypothesis, we observed a significant reduction in the initial peak and nadir in skin sites treated with the TRPV-1-specific antagonist capsazepine, suggesting that TRPV-1 channels make a significant contribution to the axon reflex component of thermal hyperaemia. Approximately 50% of the initial peak can be explained by TRPV-1 channels whereas only ∼35% of the initial peak can be explained by NO (Fig. 6A), which suggests the initial peak appears to rely more on a TRPV-1 channel-dependent mechanism while the nadir appears to rely more on a NO-dependent mechanism.
When TRPV-1 channels and NO synthase were simultaneously inhibited we observed a further reduction in the initial peak (∼65% contribution of combined TRPV-1 channels and NO; Fig. 6A); however, the reduction of the nadir in the combined TRPV-1 and NO synthase-inhibited sites was similar to the NO synthase-inhibited sites alone (∼65% contribution of NO alone and combined TRPV-1 + NO), which suggests TRPV-1 channels do not contribute to the nadir when NO synthase is inhibited. Results from combined TRPV-1 and NO synthase inhibition in the present study provide support for the hypothesis of an interaction between TRPV-1 channel activation and NO to the initial peak component of thermal hyperaemia. Whether the interaction between TRPV-1 channel activation and NO during the axon reflex component of cutaneous thermal hyperaemia is a function of NO-induced TRPV-1 channel activation or TRPV-1-induced enhancement of NO production remains to be determined.
Recent data from Hodges et al. (2008, 2009b) and Houghton et al. (2006) suggest the axon reflex response to local heating involves a complex series of mechanisms that involves not only sensory nerves and vasodilator substances but also vasoconstrictor nerves and substances. To what degree the TRPV-1 component of the axon reflex component of thermal hyperaemia interacts with, or depends on, cutaneous vasoconstrictor nerves remains unknown. It also remains unknown how inhibition of TRPV-1 channels may affect the temperature threshold for the axon reflex. Houghton et al. (2006) first reported that the axon reflex occurs at a local heater temperature of ∼37°C. Studies utilizing a slow local heating protocol will be required to determine how TRPV-1 channels influence the temperature threshold for the axon reflex.
A characteristic of TRPV-1 channels is they are activated by heat with a temperature threshold of ∼42°C (Yao & Garland, 2005; Caterina, 2007; Vriens et al. 2009). Minson et al. (2001) reported skin temperatures between 39 and 40°C when the local heater temperatures were clamped at 42°C. In the present study, skin temperature would not be expected to reach the ∼42°C threshold at which TRPV-1 channels are activated. Recent evidence suggests there is ‘cross talk’ between neurokinin-1 (NK1) receptors, the preferential receptor for substance P, and TRPV-1 channels, where NK1 activation increases the sensitivity of TRPV-1 channels to heat and lowers the threshold of TRPV-1 channels to ∼35–37°C (Zhang et al. 2007), a skin temperature range that could be achieved with the local heating protocol employed in the present study. This idea of an interaction between TRPV-1 channels and NK1 receptors is in accord with the hypothesis that CGRP and substance P are co-released from cutaneous sensory nerve terminals and are the neuropeptides ultimately responsible for the axon reflex-mediated vasodilatation (Wallengren et al. 1987; Schmelz et al. 1997). Future research aimed at investigating a potential interaction between TRPV-1 channels and NK1 receptors will be important in delineating mechanisms of cutaneous thermal hyperaemia.
Secondary plateau component of cutaneous thermal hyperaemia
In contrast to the initial peak and nadir, the plateau phase of cutaneous thermal hyperaemia is largely dependent on NO, where inhibition of NO synthase during the plateau reduces skin blood flow by ∼70% (Minson et al. 2001). In the present study, we observed a role for TRPV-1 channel activation directly contributing to the plateau phase of thermal hyperaemia (Fig. 5). Although modest, we observed an attenuated plateau in sites treated with a TRPV-1 antagonist (capsazepine; Fig. 5) and the present data suggest TRPV-1 channel activation directly contributes ∼20% to the plateau phase (Fig. 6C). The contributions of TRPV-1 channels and NO also appear to be additive, where attenuation of the plateau in combined capsazepine +l-NAME sites appears to be equal to the sum of the attenuation in the independent capsazepine and l-NAME sites. This suggests TRPV-1 channel activation may account for a small portion of the NO component; however, a substantial portion of the NO component is independent of TRPV-1 channel activation.
In the study by Minson et al. (2001), acute inhibition of axon reflexes with EMLA cream did not appear to attenuate the plateau phase of cutaneous thermal hyperaemia. This observation by Minson et al. is in contrast to the present investigation where inhibition of TRPV-1 channels modestly attenuated the plateau component of thermal hyperaemia. Although TRPV-1 channels are predominantly located on afferent sensory nerves in the skin, there is evidence to suggest TRPV-1 channels may also be expressed in endothelial cells (Yao & Garland, 2005; Caterina, 2007; Lin et al. 2007; Vriens et al. 2009; Wu et al. 2009). Thus, the data from the present study may suggest the involvement of neural TRPV-1 channels in the axon reflex component and endothelial TRPV-1 channels in the plateau phase of cutaneous thermal hyperaemia.
Limitations
It is possible the 20 mm concentration of capsazepine did not elicit maximal inhibition of the TRPV-1 channels; however, use of higher concentrations of capsazepine was problematic. Capsazepine is minimally soluble in aqueous solution such that a vehicle of 90% propylene glycol and 10% lactated Ringer solution had to be employed to obtain a solution of capsazepine. Propylene glycol was used to improve solubility because it did not increase baseline skin blood flow; however, higher concentrations of capsazepine were not possible with propylene glycol and lactated Ringer solution. Addition of DMSO improved solubility but resulted in non-specific vasodilatation and an increase in baseline skin blood flow. These and other potential untoward side-effects of DMSO have been described previously and shown in other skin blood flow studies (Shastry & Joyner, 2002; Santos et al. 2003; Kellogg et al. 2008). To minimize the confounding effects of an increased baseline, we chose to use a 20 mm concentration of capsazsepine dissolved in propylene glycol and lactated Ringer solution and, as such, it is possible our data underestimate the contribution of TRPV-1 channels to the thermal hyperaemic response. As our data may underestimate the role of TRPV-1 channels, the data in Fig. 6A–C should be viewed with this in mind. Nevertheless, our data support a substantial role for TRPV-1 channels to the initial peak and a modest role to the plateau component of cutaneous thermal hyperaemia.
Perspectives and conclusion
Mechanisms underlying cutaneous thermal hyperaemia are highly complex and highly redundant. Our current understanding of the thermal hyperaemic response suggests the initial peak and nadir are mediated by an axon reflex mechanism that requires functional sensory afferent nerves and TRPV-1 channels, and the sustained plateau phase is predominantly an NO-dependent mechanism but also involves TRPV-1 channels. Whether substance P and CGRP are released from afferent sensory nerve terminals and are involved in axon reflex vasodilatation remains unknown. Recent data from Kellogg et al. (2009) suggest that endothelial NO synthase (eNOS) is the NOS isoform that is activated in response to local heating of the skin; however, Stewart et al. (2007) have provided evidence to suggest that neuronal NOS (nNOS) is responsible for the increase in NO production during local heating of the skin. Whether eNOS or nNOS is involved in cutaneous thermal hyperaemia remains equivocal. It is highly probable a number of substances act as direct vasodilators while also increasing the production of secondary vasodilator substances, such as NO. For example, a role for adenosine A1/A2 receptor activation, as both a direct vasodilator and as a source of NO, in the thermal hyperaemic response has been demonstrated (Fieger & Wong, 2010). It also appears that an intact adrenergic vasoconstrictor system is required for full expression of cutaneous thermal hyperaemia (Houghton et al. 2006; Hodges et al. 2008, 2009b). There is also evidence to suggest interactions between sensory nerves, TRPV-1 channels and purinergic recpetors, primarily P2X and P2Y receptors, which may constitute another mechanism by which not only are sensory nerves activated during local heating but also may directly induce a portion of the cutaneous vasodilatation (Moriyama et al. 2003; Burnstock, 2006, 2009). Clearly, more research will be required to fully unravel mechanisms attending cutaneous thermal hyperaemia.
In conclusion, we have demonstrated that TRPV-1 channel activation makes a significant contribution to the axon reflex component of cutaneous thermal hyperaemia. Specifically, we observe an approximately 50% attenuation of the initial peak (Fig. 6A) and an approximately 45% reduction of the nadir (Fig. 6B). We also demonstrated TRPV-1 channel activation directly contributes to the plateau phase, although this contribution of TRPV-1 channels is modest (∼20%; Fig. 6C) when compared to NO. These data suggest activation of TRPV-1 channels, which are putative channels primarily located on cutaneous sensory nerves, may constitute a mechanisms by which cutaneous sensory nerves are depolarized during local heating of the skin.
Acknowledgments
The authors wish to thank Matthew Turner for assistance with data collection. We also appreciate the time of the subjects who participated in this study. Funding for this study was provided by Kansas State University Small Research Grants nos 2305 and 2477 and Kansas State University Mentoring Fellowship for Women and Minorities in the Sciences and Engineering to Brett Wong.
Glossary
Abbreviations
- CGRP
calcitonin gene-related peptide
- CPZ
capsazepine
- CVC
cutaneous vascular conductance
- %CVC max
percentage of maximal cutaneous vascular conductance
- DMSO
dimethyl sulfoxide
- LDF
laser-Doppler flowmetry
- l-NAME
NG-nitro-l-arginine-methyl ester
- MAP
mean arterial pressure
- NK1 receptors
neurokinin-1 receptors
- NO
nitric oxide
- SNP
sodium nitroprusside
- TPRV-1 channels
transient receptor potential vanilloid type 1 channels
Author contributions
This study was conducted in the Cardiovascular and Thermal Physiology Laboratory in the Department of Kinesiology at Kansas State University, Manhattan, KS, USA. B.J.W. contributed to conception and design of the experiments; collection, analysis, and interpretation of data; and drafting and revising the manuscript for intellectual content. S.M.F. contributed to design of experiments; collection, analysis, and interpretation of data; and revising the manuscript for intellectual content. All authors approved the final version of the manuscript. The authors have no conflict of interest.
References
- Baldessarini RJ. Drugs and the treatment of psychiatric disorders: psychosis and anxiety. In: Hardman JG, Limbird LE, editors. Goodman and Gilman's The Pharmacological Basis of Therapeutics. 10th edn. New York: McGraw-Hill; 2001. pp. 399–430. [Google Scholar]
- Burnstock G. Purinergic P2 receptors as targets for novel analgesics. Pharmacol Ther. 2006;110:433–454. doi: 10.1016/j.pharmthera.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Purinergic regulation of vascular tone and remodeling. Auton Autacoid Pharmacol. 2009;29:63–72. doi: 10.1111/j.1474-8673.2009.00435.x. [DOI] [PubMed] [Google Scholar]
- Caterina MJ. Transient receptor potential ion channels as participants in thermosensation and thermoregulation. Am J Physiol Regul Integr Comp Physiol. 2007;292:R64–R76. doi: 10.1152/ajpregu.00446.2006. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Fieger SM, Wong BJ. Adenosine receptor inhibition with theophylline attenuates the skin blood flow response to local heating in humans. Exp Physiol. 2010;95:946–954. doi: 10.1113/expphysiol.2010.053538. [DOI] [PubMed] [Google Scholar]
- Hodges GJ, Chiu C, Kosiba WA, Zhao K, Johnson JM. The effect of microdialysis needle trauma on cutaneous vascular responses in humans. J Appl Physiol. 2009a;106:1112–1118. doi: 10.1152/japplphysiol.91508.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges GJ, Kosiba WA, Zhao K, Johnson JM. The involvement of norepinephrine, neuropeptide Y, and nitric oxide in the cutaneous vasodilator response to local heating in humans. J Appl Physiol. 2008;105:233–240. doi: 10.1152/japplphysiol.90412.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges GJ, Kosiba WA, Zhao K, Johnson JM. The involvement of heating rate and vasoconstrictor nerves in the cutaneous vasodilator response to skin warming. Am J Physiol Heart Circ Physiol. 2009b;296:H51–H56. doi: 10.1152/ajpheart.00919.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowatz LA, Thompson CS, Minson CT, Kenney WL. Mechanisms of acetylcholine-mediated vasodilatation in young and aged human skin. J Physiol. 2005;563:965–973. doi: 10.1113/jphysiol.2004.080952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton BL, Meendering JR, Wong BJ, Minson CT. Nitric oxide and noradrenaline contribute to the temperature threshold of the axon reflex response to gradual local heating in human skin. J Physiol. 2006;572:811–820. doi: 10.1113/jphysiol.2005.104067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg DL, Jr, Liu Y, Kosiba IF, O’Donnell D. Role of nitric oxide in the vascular effects of local warming of the skin in humans. J Appl Physiol. 1999;86:185–1190. doi: 10.1152/jappl.1999.86.4.1185. [DOI] [PubMed] [Google Scholar]
- Kellogg DL, Jr, Zhao JL, Wu Y. Neuronal nitric oxide synthase mechanisms in the cutaneous vasculature of humans in vivo. J Physiol. 2008;586:847–857. doi: 10.1113/jphysiol.2007.144642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg DL, Jr, Zhao JL, Wu Y. Endothelial nitric oxide synthase control mechanisms in the cutaneous vasculature of humans in vivo. Am J Physiol Heart Circ Physiol. 2009;295:H123–H129. doi: 10.1152/ajpheart.00082.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen CD. Nonmetallic environmental toxicants: air pollutants, solvents and vapors, and pesticides. In: Hardman JG, Limbird LE, editors. Goodman and Gilman's The Pharmacological Basis of Therapeutics. 10th edn. New York: McGraw-Hill; 2001. pp. 399–430. [Google Scholar]
- Lin Q, Li D, Xu X, Zou X, Fang L. Roles of TRPV1 and neuropeptidergic receptors in dorsal root reflex-mediated neurogenic inflammation induced by intradermal injection of capsaicin. Mol Pain. 2007;3:30–45. doi: 10.1186/1744-8069-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord GR, Cracowski J-L, Minson CT. Prostanoids contribute to cutaneous active vasodilatation in humans. Am J Physiol Regul Integr Comp Physiol. 2006;291:R596–R602. doi: 10.1152/ajpregu.00710.2005. [DOI] [PubMed] [Google Scholar]
- Magerl W, Treede RD. Heat-evoked vasodilatation in human hairy skin: axon reflexes due to low-level activity of nociceptive afferents. J Physiol. 1996;497:837–848. doi: 10.1113/jphysiol.1996.sp021814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minson CT, Berry LT, Joyner MJ. Nitric oxide and neurally mediated regulation of skin blood flow during local heating. J Appl Physiol. 2001;91:1619–1626. doi: 10.1152/jappl.2001.91.4.1619. [DOI] [PubMed] [Google Scholar]
- Moriyama T, Iida T, Kobayashi K, Higashi T, Fukuoka T, Tsumura H, Leon C, Suzuki N, Inoue K, Gachet C, Noguchi K, Tominaga M. Possible involvement of P2Y2 metabotropic receptors in ATP-induced transient receptor potential vanilloid receptor 1-mediated thermal hypersensitivity. J Neurosci. 2003;23:6058–6062. doi: 10.1523/JNEUROSCI.23-14-06058.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Dubin AE, Petrus MJ, Patapoutian A. TRPV1 and TRPA1 mediate peripheral nitric oxide-induced nociception in mice. PLoS ONE. 2009;4:1–11. doi: 10.1371/journal.pone.0007596. e7596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movahed P, Evilevitch V, Andersson TLG, Jonsson BAG, Wollmer P, Zygmunt PM, Hogestatt ED. Vascular effects of anandamide and N-acylvanillylamines in the human forearm and skin microcirculation. Br J Pharmacol. 2005;146:171–179. doi: 10.1038/sj.bjp.0706313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos NC, Figueira-Coelho J, Martins-Silva J, Saldanha C. Multidisciplinary utilization of dimethyl sulfoxide: pharmacological, cellular, and molecular aspects. Biochem Pharmacol. 2003;65:1035–1041. doi: 10.1016/s0006-2952(03)00002-9. [DOI] [PubMed] [Google Scholar]
- Schmelz M, Luz O, Averback B, Bickel A. Plasma extravasation and neuropeptide release in human skin as measured by intradermal microdialysis. Neurosci Lett. 1997;230:117–120. doi: 10.1016/s0304-3940(97)00494-1. [DOI] [PubMed] [Google Scholar]
- Shastry S, Joyner MJ. Geldanamycin attenuates NO-mediated dilation in human skin. Am J Physiol Heart Circ Physiol. 2002;282:H232–H236. doi: 10.1152/ajpheart.2002.282.1.H232. [DOI] [PubMed] [Google Scholar]
- Stewart JM, Medow MS, Minson CT, Taneja I. Cutaneous neuronal nitric oxide synthase is specifically decreased in postural tachycardia syndrome. Am J Physiol Heart Circ Physiol. 2007;293:H2161–H2167. doi: 10.1152/ajpheart.00600.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriens J, Appendino G, Nilius B. Pharmacology of vanilloid transient receptor potential cation channels. Mol Pharmacol. 2009;75:1262–1279. doi: 10.1124/mol.109.055624. [DOI] [PubMed] [Google Scholar]
- Wallengren J, Ekman R, Sundler F. Occurrence and distribution of neuropeptides in the human skin. An immunochemical and immunocytochemical study on normal human skin and blister fluid from inflamed skin. Acta Derm Venereol. 1987;66:185–192. [PubMed] [Google Scholar]
- Wong BJ, Wilkins BW, Holowatz LA, Minson CT. Nitric oxide synthase inhibition does not alter the reactive hyperemic response in the cutaneous circulation. J Appl Physiol. 2003;95:504–510. doi: 10.1152/japplphysiol.00254.2003. [DOI] [PubMed] [Google Scholar]
- Wong BJ, Wilkins BW, Minson CT. H1 but not H2 histamine receptors contribute to the rise in skin blood flow during whole body heating in humans. J Physiol. 2004;560:941–948. doi: 10.1113/jphysiol.2004.071779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong BJ, Williams SJ, Minson CT. Minimal role for H1 and H2 histamine receptors in cutaneous thermal hyperaemia to local heating in humans. J Appl Physiol. 2006;100:535–540. doi: 10.1152/japplphysiol.00902.2005. [DOI] [PubMed] [Google Scholar]
- Wu M, Komori N, Qin C, Farber JP, Linderoth B, Foreman RD. Roles of peripheral terminals of transient receptor potential vailloid-1 containing sensory fibers in spinal cord stimulation-induced peripheral vasodilation. Brain Res. 2007;1156:80–92. doi: 10.1016/j.brainres.2007.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, Garland CJ. Recent developments in vascular endothelial cell transient receptor potential channels. Circ Res. 2005;97:853–863. doi: 10.1161/01.RES.0000187473.85419.3e. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y, Mori Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cang C-L, Kawasaki Y, Liang L-L, Zhang Y-Q, Ji R-R, Zhao Z-Q. Neurokinin-1 receptor enhances TRPV1 activity in primary sensory neurons via PKCɛ: a novel pathway for heat hyperalgesia. J Neurosci. 2007;237:12067–12077. doi: 10.1523/JNEUROSCI.0496-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]