

Abstract
Loss of gap junctional intercellular communication (GJIC) between cancer cells is a common characteristic of malignant transformation. This communication is mediated by connexin proteins that make up the functional units of gap junctions. Connexins are highly regulated at the protein level and phosphorylation events play a key role in their trafficking and degradation. The metastasis suppressor BRMS1 up-regulates GJIC and decreases PI3K signaling. Based on these observations we set out to determine if there was a link between PI3K and GJIC in tumorigenic and metastatic cell lines. Treatment of cells with the well-known PI3K inhibitor LY294002, and its structural analog LY303511 which does not inhibit PI3K, increased homotypic GJIC; however, we found the effect to be independent of PI3K/AKT inhibition. Additionally, while levels of the connexin Cx43 remained unchanged, Cx43 relocalization from the cytosol to the plasma membrane was observed. Both LY294002 and LY303511 increased the activity of protein kinase A (PKA). Moreover, PKA blockade by the small molecule inhibitor H89 decreased the LY294002/LY303511 mediated increase in GJIC. Collectively, our findings demonstrate a connection between PKA activity and GJIC mediated by PI3K-independent mechanisms of LY294002 and LY303511. Manipulation of these signaling pathways could prove useful for anti-metastatic therapy.
Keywords: connexin 43, gap junction, protein kinase A, LY294002, LY303511
Introduction
The deadliest attribute of cancer cells is their ability to disseminate and colonize other tissues (1). The process of metastasis involves genetic changes which result in dysregulation of tumor cell interactions with other tumor cells and the host (2, 3). Breast cancer metastasis suppressor 1 (BRMS1) blocks the ability of breast (4–7), melanoma (8), ovarian (9), and non-small cell lung (10) cancer cells to metastasize, but does not block the formation of orthotopic tumors following injection. BRMS1 is a component of multiple SIN3:HDAC complexes that alter the expression of numerous genes and proteins (11–13). Among the first identified phenotypic changes reported for BRMS1-expressing cells was the restoration of gap junctional intercellular communication (14).
GJIC defines a process in which small molecules (e.g. IP3, ATP, Ca2+) can be transferred between adjoining cells through physical interaction of highly regulated channels located in the plasma membrane. These functional channels are comprised of hexameric structures (connexons) made up of individual connexin proteins that interact with numerous other components within the plasma membrane (15). GJIC is associated with normal cellular homeostasis, but dysregulation is common in neoplastic progression and even further loss of communication is often associated with acquisition of a metastatic phenotype (16–19). However, exceptions to this trend exist, e.g., Nicolson et al. showed that cells transfected with p21ras exhibited higher metastatic potential, but did not always lose GJIC (20). Nevertheless, restoration of GJIC by expression of connexins in some models decreases proliferation and inhibits metastasis (21–25). Contradictory reports show a rise of migratory and metastatic potential when GJIC is increased (reviewed in (26, 27)), highlighting cell- and context-specific components of the process.
Since some connexins function in a plasma membrane-independent manner (28), and since post-translational modifications and assembly of connexins into connexons can be regulated by altered signaling pathways (29, 30), we hypothesized that the selective alteration of phosphoinositide levels by BRMS1 (31) could be, at least partially, involved in the regulation of GJIC. Specifically, BRMS1 expression results in a dramatic >95% reduction in PtdIns(4,5)P2 levels (31), leading to a decrease in phosphorylation of AKT at Ser 473. Since PtdIns(4,5)P2 is a major substrate for the oncogenic phosphoinositide-3-kinase (PI3K) and is a major component of lipid rafts in which connexons form (32, 33), we tested whether inhibition of PI3K by the commonly used PI3K inhibitor LY294002, would itself mimic BRMS1 in BRMS1 null cancer cells by restoring or enhancing GJIC. While LY294002 indeed enhanced GJIC in numerous cell lines of different origin, we report that the mechanism of action is not likely via its regulation of PI3K, but rather PI3K independent mechanisms related to PKA.
Materials and Methods
Cell lines
MDA-MB-231, MDA-MB-435, MDA-MB-436, MDA-MB-468, T47D and C8161.9 were grown in Dulbecco’s-modified Eagle’s medium mixed 1:1 (v:v) with Ham’s F-12 medium (DMEM/F12; Invitrogen, Carlsbad, CA #11330) supplemented with 2 mM L-glutamine, 0.2 mM non-essential amino acids with 5% fetal bovine serum. MDA-MB-231, MDA-MB-435, MDA-MB-436, MDA-MB-468 and T47D are human breast carcinoma-derived cell lines, while C8161.9 is a clone derived from the C8161 human melanoma. The origin of the MDA-MB-435 has been questioned (34); however recent literature strongly confirms its use as a breast carcinoma cell line (35–37). S2VP10 pancreatic cancer cell line (38) was maintained in RPMI-1640 medium (Invitrogen #11875) supplemented with 5% fetal bovine serum. All cell lines were tested and found to be free of Mycoplasma spp. contamination using a PCR-based kit (Aligent Technologies, Santa Clara, CA #302108).
Chemicals
All chemicals were prepared as stock solutions and stored at −20°C in aliquots; working solutions were diluted fresh at time of experiment. Calcein-AM (#C1430) and CM-DiI (#C7001) were obtained from Invitrogen and dissolved in DMSO at 1 mM stock. LY294002 (Invitrogen #PHZ1144), LY303511 (Santa Cruz Biotechnology #sc-202215) and H-89 (Sigma, St. Louis, MO #B1427) were dissolved in DMSO at 10 mM stock. AKT Inhibitor VII (EMD Chemicals, Gibbstown, NJ #124014) was dissolved in purified water at 10 mM stock. 8-bromo cAMP (8-BR-cAMP) (Sigma #B5386) was dissolved in DMSO at 1 M stock. 2′5′-dideoxyadenosine (Sigma #D7408) and SQ 22,536 (Sigma #S153) were dissolved in DMSO at 10 mM stock. Vehicle controls (DMSO, purified water) had no effect on assays performed in this study. All chemicals were used at concentrations that exhibited no toxicity or morphological changes in the cells.
GJIC assay
Calcein-AM (acetoxymethylester) passively diffuses into cells where the acetyl methoxy group is cleaved by internal esterases generating a green fluorescent dye, calcein, that does not diffuse from the cell, but remains small enough to pass through gap junctions. ‘Donor’ cells were loaded with 10 μM calcein-AM for 10 min at 37°C/5%CO2, washed 3X with Dulbecco’s phosphate buffered saline (DPBS) and plated with non-labeled ‘acceptor’ cells for 6 hr. Donor cells were also labeled with 5 μM CM-DiI, a red fluorescent lipophilic dye that does not transfer between cells and was used to mark donor cells. Calcein spread from donor to acceptor cells was indicative of GJIC. Acceptor cells became calcein positive but remained CM-DiI negative. All experiments reported herein were performed in serum-free media, although addition of serum did not alter the observed trends (Supplementary Figure 1). Flow cytometry with a BD LSRII Cell analytical flow cytometer using BD FACSDiva software was used to calculate the average number of cells which received calcein per donor cell and represented as fold change.
Immunofluorescence
Cells were grown on glass cover slips with indicated treatments followed by performing the GJIC assay described above. Cells were fixed with 3.7% neutral buffered formalin in DPBS and permeabilized with ice-cold methanol for 30 min at −20°C. Cover slips were blocked with 5% BSA in DPBS and primary antibodies over night at 4°C. Anti-Cx43 (Cell Signaling #3512) was used at 1:100 diluted in 2% bovine serum albumin (BSA) in DPBS, washed 3X in DPBS, followed by addition of secondary anti-rabbit IgG FITC conjugated antibody (Sigma #ab27478) at 1:160 in 2% BSA in DPBS for 1 hr at room temperature. Cover slips were then washed 3X with DPBS and mounted using VECTASHIELD with DAPI (Vector Labs #H-1200). For phospho-CREB Ser 133 analysis, Alexa Fluor 488 conjugate antibody (Cell Signaling #9187) was used. Rabbit polyclonal IgG (AbCam #ab27478) was used as an isotype control. Images were obtained on a Nikon Eclipse TE2000-U microscope using Q Imaging QICAM software for image analysis.
Immunoblot analysis
Cells were lysed on ice in 25 mM TRIS-HCl, 1% Triton X-100, 5% glycerol, 0.5 mM EDTA, 50 mM β-glycerolphosphate supplemented with 1.0 mM NaVa, 0.5 mM PMSF and 1× HALT Protease Inhibitor Cocktail (Thermo Scientific #78430). For connexin protein analysis, lysates were sonicated on ice, all other samples were boiled at 95°C for 5 min. Proteins were resolved using 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) before transfer to polyvinylidene fluoride (PVDF) membranes and blocked for 1 hr in 5% non-fat dry milk in TRIS buffered saline with Tween-20 (TBST). For Cx43 immunoblots, proteins were transferred to nitrocellulose membranes. Primary antibodies were diluted at 1:1000 in 5% BSA in TBST.
The following primary antibodies were purchased from Cell Signaling Technology: Cx43 (#3512), phospho-AKT Ser473 (#9271), pan-AKT(#4685), α-tubulin (#2125), CREB (#9104), phospho-CREB Ser 133 (#9198). Secondary antibodies were diluted at 1:2500 in 5% non-fat dry milk in TBST. Secondary HRP-conjugated antibodies were purchased from GE Healthcare: anti-rabbit (#NA934V), anti-mouse (#NA931V). Membranes were developed by chemiluminescence (ECL, Thermo Scientific #32106). ImageJ (NIH) processing and analysis was used for quantification of Cx43 blots and results represented as fold change compared to non-treated cells.
Statistical analysis
Data for GJIC are presented as mean ± SEM and represented as fold-change. Student’s t-test was used for statistical analysis between two groups.
Results
Treatment of Cells with LY294002 Increases Calcein Dye Transfer
Donor cells were labeled with calcein-AM and CM-DiI before plating with non-labeled acceptor cells. We observed that treatment with 10 μM LY294002 exhibited significantly higher rates of dye transfer from donor to acceptor cells (Figure 1A). We extended these results to a total of five breast cancer cell lines (MDA-MB-231, MDA-MB-435, MDA-MB-436, MDA-MB-468, T47D), a metastatic melanoma cell line (C8161.9) and a metastatic pancreatic cell line (S2VP10) (Figure 1B). All of the cell lines exhibited varying levels of dye spread in non-treated conditions (NT), but all cells displayed a significant and consistent increase in GJIC when incubated in LY294002. Cell lines that exhibited the lowest baseline rates of dye spread (MDA-MB-231, S2VP10) experienced the greatest fold change. Calcein could be seen spreading between cells within an hour of addition of donor cells and increased throughout the duration of the experiment with readily noticeable differences at 6 hr.
Figure 1.

LY294002 increases GJIC. (A) Cell lines treated with LY294002 (10 μM) exhibit higher levels of calcein dye transfer from donor to acceptor cells in a fluorescence dye transfer assay. (B) Seven cancer cell lines exhibited a significant increase in calcein spread. Data shown are represented as fold increase in dye transfer compared to non-treated (NT) cells. Arrows highlight donor cells. [* = p<0.05, ** = p<0.01]
Treatment with Wortmannin and Direct Inhibition of AKT do not Increase GJIC Compared to LY294002
To determine if inhibition of AKT phosphorylation by LY294002 was mediating the effects on GJIC, MDA-MB-231, MDA-MB-435 and C8161.9 cells were treated with increasing concentrations of Wortmannin (50, 100, 500 nM), another PI3K inhibitor. Phosphorylation of AKT at Ser 473 was potently inhibited via immunoblot analysis (Figure 2A). However, in comparison to LY294002, no increase in GJIC was noted compared to non-treated cells in MDA-MB-231, MDA-MB-435 and C8161.9 (Figure 2B), while slight decreases were observed, signifying that inhibition of phosphorylation of AKT was not responsible for the LY294002-mediated increase in GJIC. To further confirm that signaling factors downstream of AKT do not increase GJIC in these cell lines, we used a direct inhibitor of AKT, AKT Inhibitor VII (AKTinbVII) which binds to the pleckstrin homology domain of AKT. Treatment of MDA-MB-231 and MDA-MB-435 with AKTinbVII reduced phosphorylation of AKT in MDA-MB-231 cells and completely abolished phosphorylation in MDA-MB-435 (Figure 3A). However, despite these decreases, no significant increase was observed in GJIC (Figure 3B). Similar to treatment with Wortmannin, the cells exhibited slight decreases in GJIC upon direct AKT inhibition. These results demonstrate that directly decreasing AKT activity does not cause an increase in GJIC.
Figure 2.
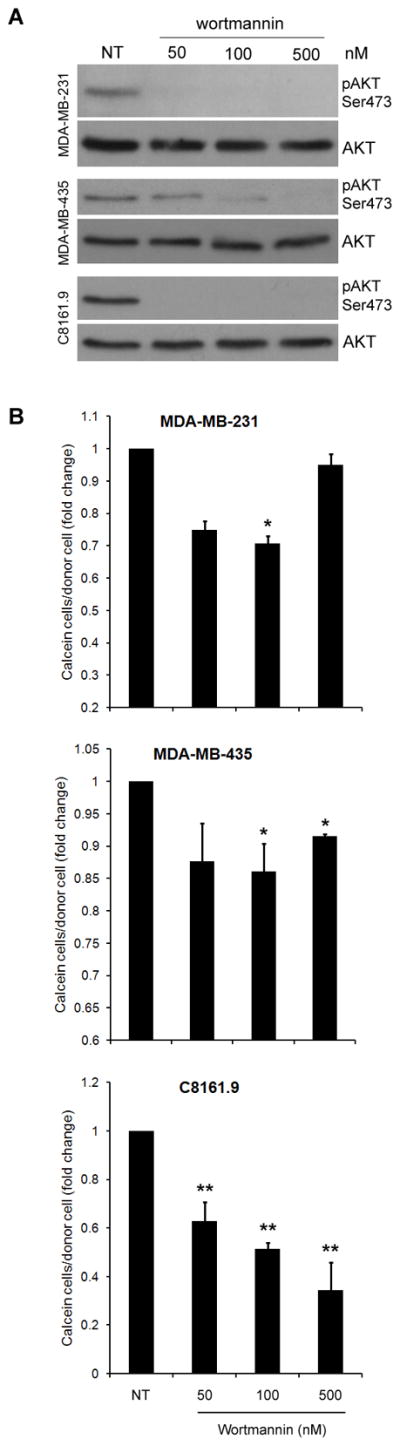
Inhibition of AKT is not responsible for increases in GJIC. (A) Nanomolar concentrations of Wortmannin were used to inhibit AKT phosphorylation at Ser 473. (B) Quantified results of calcein dye transfer assays showing no increases in GJIC using Wortmannin based phospho-AKT inhibition, while rather slight decreases were observed. [* = p<0.05, ** = p<0.01]
Figure 3.

Direct inhibition of AKT does not increase GJIC. (A) The AKT inhibitor, AKTinbVII, was used to decrease phospho-AKT levels directly (10 μM), without a corresponding increase in GJIC (B). Similar to AKT inhibition with Wortmannin, slight decreases in GJIC were observed.
LY303511 Increases GJIC Similar to LY294002
Since PI3K/AKT signaling was ruled out as the mechanism for the LY294002 mediated increase in GJIC, we examined if the stated effect was related to its chemical structure. LY303511 is structurally identical to LY294002 except for a substitution of -O for -NH in the morpholine ring (Supplementary Figure 2), and does not potently inhibit PI3K. Treatment of cells with LY303511 caused an increase in calcein spread similar to levels of LY294002 (Figure 4A). The ability of LY303511 to increase GJIC did not occur concomitant with inhibition of phosphorylation of AKT as measured by immunoblotting (Figure 4B).
Figure 4.

Both LY303511 and LY294002 increase GJIC. LY303511 treatment (10 μM) increases calcein transfer in MDA-MB-231, MDA-MB-435 and C8161.9 cells (A), despite not potently inhibiting PI3K (B). [* = p<0.05, ** = p<0.01]
Cx43 is Relocalized After Treatment With LY294002 and LY303511
Since connexins are the functional units of connexon channels, we examined whether LY294002 and LY303511 altered regulation of Cx43, a connexin frequently dysregulated in breast and other cancers (39–42). Immunoblot analysis of Cx43 levels in C8161.9 and MDA-MB-231 showed a consistent decrease in Cx43 levels after treatment with LY294002, however this effect was not observed with LY303511 (Figure 5A,B). This result may be due to other non-specific effects of LY294002 that are not shared by LY303511, or the simultaneous inhibition of PI3K.. However, significant changes in trafficking of Cx43 from the cytosol to the plasma membrane were observed following treatment with the LY compounds (Figure 5C). These results show that both LY294002 and LY303511 are capable of causing a dramatic subcellular relocalization of Cx43, and that this effect is likely responsible for the increases in GJIC.
Figure 5.

Cx43 localizes to the plasma membrane after LY294002 or LY303511 treatment. (A) Representative immunoblot analysis from three independent experiments of Cx43 in C8161.9 and MDA-MB-231 6 hr post treatment with 10μM LY294002 and LY303511. (B) Quantification of Cx43 immunoblot analyses (n=3) [** = p<0.01]. (C) Exposure of C8161.9 or MDA-MB-231 cells to LY294002 (10 μM) or LY303511 (10 μM) for 6 hr resulted in substantially more plasma membrane localization of Cx43 than untreated (NT) cells. Arrows highlight Cx43 plaques between cells.
PKA Activity is Upregulated in Response to LY294002 and LY303511
PKA activity leads to phosphorylation of specific residues of Cx43 promoting its transport from the Golgi apparatus to the plasma membrane and increasing GJIC. (43–47). Since LY294002 and LY303511 do not dramatically alter protein levels of Cx43, but rather change Cx43 subcellular localization, we set out to determine if an increase in PKA activity was responsible for the enhancement in GJIC. PKA phosphorylates the transcription factor CREB at Ser 133, which was used as an indicator of PKA activity. Using an Alexa Fluor 488 conjugated antibody that binds CREB when phosphorylated at Ser 133, a significant increase in fluorescence within the nucleus following a 2 hr treatment with LY294002 or LY303511 was observed (Figure 6A), indicative of PKA activation. To determine how quickly PKA is activated, nuclear extracts were isolated following treatment with LY294002 at varying time points. Increased phosphorylation of CREB was evident as early as 15 min post-treatment (Figure 6B). In support of PKA playing a role in increased GJIC in these cells, MDA-MB-231 were treated with increasing concentrations of 8-BR-cAMP, a cell permeable analog of cAMP that upregulates PKA activity. 8-BR-cAMP increased GJIC in a dose dependent manner in these cells (Figure 6C). To determine if PKA activity was responsible for the LY294002 mediated enhancement in GJIC, cells were co-treated with LY294002 (10 μM) and the PKA inhibitor H89 (10 μM). Addition of H89 caused a significant reduction in GJIC induced by LY294002 (Figure 6D,E), indicating that upregulation of PKA by LY294002 is at least, in part, responsible for the increase in GJIC.
Figure 6.

PKA activity is increased by LY294002 and LY303511 and required for enhanced GJIC. (A) Immunofluorescence analysis of CREB phosphorylated at Ser 133 in MDA-MB-231 revealed an increase in phosphorylation after 2 hr treatment with LY294002 (10 μM) and LY303511 (10 μM). (B) Analysis of phosphorylation of CREB at Ser 133 in nuclear extracts from MDA-MB-231 treated with LY294002 (10 μM) showed phosphorylation of CREB within 15 min. Treatment with the PKA agonist 8-BR-cAMP (0.1 mM, 0.5 mM, 1.0 mM) increased GJIC in a dose dependent manner in MDA-MB-231 (C), while inhibition of PKA activity by H89 (10 μM) when used in co-treatments with LY294002 (10 μM) inhibited GJIC compared to LY294002 alone (D, calcein) (E, quantified). [*=p<0.05]. Pretreatment of cells with the adenylate cyclase inhibitors 2′5′-dideoxyadenosine (F) or SQ 22,536 (G) (10 μM,75 μM,100 μM) did not reduce the ability of LY294002 (10 μM) to increase GJIC.
Discussion
GJIC is a complex process capable of inhibiting or promoting the migratory and invasive qualities of cancer cells. Importantly, GJIC is defined in a context-dependent manner, since tumor cells can form homotypic (tumor cell-tumor cell) or heterotypic (tumor cell-host cell) interactions using multiple permutations of the >20 mammalian connexins (48). Further complexity exists since connexin proteins are now known to mediate multiple cellular effects independent of their involvement in gap junctions. Since a number of studies show that restoration of gap junctional proteins and/or GJIC can reverse tumorigenicity or metastatic potential (18, 19, 21, 23–25), understanding how GJIC is dysregulated in cancer cells and delineating such (dys)regulatory signaling pathways will allow insight into defining the role(s) of GJIC in tumor progression and metastasis.
Since the metastasis suppressor BRMS1 inhibits metastasis in numerous models, and has previously been reported to inhibit AKT phosphorylation at Ser 473 while increasing GJIC, we initially set out to determine whether a link between these two effects existed. We utilized two PI3K inhibitors, LY294002 and Wortmannin, to recapitulate the effect of BRMS1 on AKT phosphorylation in cells that do not express detectable endogenous BRMS1, followed by measuring GJIC.
Treatment of cells with the relatively more ‘selective’ PI3K inhibitor LY294002 (49) significantly increased GJIC in multiple cancer cell lines. No clear trends were observed in metastatic potential and baseline GJIC between the cell lines used. For example, MDA-MB-231 and C8161.9 are both highly metastatic in murine models, however MDA-MB-231 exhibited low baseline GJIC while C8161.9 displayed moderate levels (Fig. 1A) which was higher in comparison to some of the non-metastatic cell lines (i.e. MDA-MB-468). However, regardless of baseline communication rates, all cell lines experienced a significant increase in GJIC when treated with LY294002. Unexpectedly, treatment of the same cell lines with Wortmannin did not increase GJIC, raising doubts regarding the connection between PI3K and GJIC. This was further substantiated when cells treated with a direct AKT inhibitor failed to induce GJIC as well. Interestingly, inhibition of AKT with these treatments caused decreases in GJIC, signifying that AKT appears to be partially involved in regulation of GJIC, however the magnitude of these changes did not match the level of GJIC increase observed with LY294002. Reasoning that the effect of LY294002 could be due to signaling through the inositol trisphosphate arm, cells were treated with phospholipase C inhibitors without increasing GJIC (Data not shown). Taken together, these data required exploration of potential PI3K-independent effects of LY294002. To facilitate dissection of the possible non-specific effects, a chemically related molecule, LY303511 (50), was used. We found that LY303511 enhanced GJIC similarly to LY294002.
We examined a number of published dose-dependent, non-specific effects reported for these compounds including inhibition of casein kinase 2 (51) (Supplementary Figure 3) and increased intracellular calcium levels (52) (Supplementary Figure 4A,B), however none of the effects tested corresponded with GJIC enhancement.
Although there is a correlation between connexin levels and GJIC in many cancer cells, functional connexons are assembled by, among other post-translational modifications, phosphorylation through protein kinases (53, 54). Since we observed relatively slight changes in Cx43 protein levels after treatment with LY294002 and LY303511, but highly substantial changes in Cx43 localization to the plasma membrane, we hypothesized that Cx43 was being modified post-translationally and that these effects were likely to be inducing the effect on GJIC. Among the most well-defined phosphorylating molecules resulting in connexin trafficking to the plasma membrane is PKA (43–47). While Davies et al. previously reported that LY294002 did not directly affect PKA activity in cell-free kinase assays, it is important to emphasize that they could not preclude indirect effects of LY294002 on PKA activation (as would occur in whole cells). We consistently found a robust increase in CREB phosphorylation at Ser 133 by both immunoblot and immunofluorescence analysis, confirming that PKA was indeed activated by both LY294002 and LY303511. These results agree with our data involving relocalization of Cx43 to the plasma membrane and a corresponding increase in GJIC which could be expected upon activation of PKA.
Further strengthening our hypothesis that PKA activation by LY294002/LY303511 enhanced homotypic GJIC are observations that cells treated with 8-BR-cAMP (an agonist of PKA) or H89 (a PKA inhibitor) increased or decreased GJIC, respectively. Since Davies et al. showed that LY294002 did not directly affect PKA activity, we set out to determine if LY294002/LY303511 induced activation of adenylate cyclase, which would lead to increased cAMP levels and PKA activation. Pretreatment of cells with the adenylate cyclase inhibitors 2′5′-dideoxyadenosine and SQ 22,536 did not reduce the ability of LY294002 to induce GJIC, in contrast to direct PKA inhibition with H89. These data suggest that LY294002/LY303511 are acting downstream of adenylate cyclase, most likely through other indirect cellular interactions that have yet to be determined.
Since H89 significantly reduced LY294002/303511 mediated increase in GJIC, it appears that activation of PKA is at least in part, responsible for the changes in GJIC. Collectively, our results highlight the fact that cancer cells may reduce GJIC not by causing a downregulation of connexin expression, but rather by altering the regulatory pathways related to connexin function and/or localization. This can readily be appreciated since we induced an increase in GJIC in seven cancer cell lines without exogenous expression of a connexin gene. Additionally, with literature building on membrane independent roles of connexin proteins, it is possible that cancer cells may not just cause a relocalization of connexins away from the plasma membrane, but utilize these proteins for other membrane-independent tasks related to cancer cell function. Although this report is limited to observations with Cx43, these results warrant further investigation of other connexins and highlight an important principle to consider for future studies in this area.
Although not a central tenet for the studies recorded here, our data highlight the caution necessary when interpreting results using any pharmaceutical reagent (in this case LY294002), no matter how selective that agent is expected to be. More importantly, the findings also have important therapeutic implications for adjuvant cancer therapies. Since GJIC restoration was possible by exogenous drug treatment, it may be possible to accomplish the same in vivo. Cell permeable compounds such as LY294002 and LY303511 that can induce GJIC in cancer cells may be further developed for treatments aimed at increasing the penetrance of chemotherapeutic agents throughout a tumor via an increase in gap junction activity. Doing so would be much easier to accomplish with a drug, than by attempting to re-express or over-express connexins in tumor cells. However, whether PKA acts as a true convergence point for dysregulation in cancer cells remains to be determined.
Supplementary Material
Acknowledgments
This work was supported primarily by the U.S. Army Medical Research and Materiel Command grants W81-XWH-07-1-0399 (to D.R.W.) and W81-XWH-08-1-0779 (to T.M.B) with additional support by U.S. Public Health Service Grants CA87728 and CA134981 (to D.R.W.) and a grant from the National Foundation for Cancer Research - Center for Metastasis Research (to D.R.W.). We thank Drs. Janet Price (University of Texas M.D. Anderson Cancer Center) for providing the MDA-MB-231 and -435 cell lines and Frank Meyskens for initially providing the C8161 cell line. This manuscript is submitted in partial fulfillment of the requirements for the Ph.D. degree in the Molecular and Cellular Pathology Graduate Program at UAB (T.M.B.).
Abbreviations
- BRMS1
breast cancer metastasis suppressor 1
- CREB
cAMP response element binding
- Cx43
connexin 43
- GJIC
gap junctional intercellular communication
- PI3K
phosphoinositide 3-kinase
- PKA
protein kinase A
- IP3
inositol tris phosphate
- ATP
adenosine triphosphate
Footnotes
Disclosure of Potential Conflicts of Interest
The authors declare no potential conflicts of interest.
References
- 1.Talmadge JE, Fidler IJ. AACR Centennial Series: The biology of cancer metastasis: Historical Perspective. Cancer Res. 2010;70:5649–69. doi: 10.1158/0008-5472.CAN-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bodenstine TM, Welch DR. Metastasis suppressors and the tumor microenvironment. Cancer Microenviron. 2008;1:1–11. doi: 10.1007/s12307-008-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369:1742–57. doi: 10.1016/S0140-6736(07)60781-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samant RS, Debies MT, Hurst DR, et al. Suppression of murine mammary carcinoma metastasis by the murine ortholog of breast cancer metastasis suppressor 1 (Brms1) Cancer Lett. 2006;235:260–5. doi: 10.1016/j.canlet.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 5.Samant RS, Debies MT, Shevde LA, Verderame MF, Welch DR. Identification and characterization of murine ortholog (Brms1) of breast cancer metastasis suppressor 1 (BRMS1) Int J Cancer. 2002;97:15–20. doi: 10.1002/ijc.1569. [DOI] [PubMed] [Google Scholar]
- 6.Samant RS, Seraj MJ, Saunders MM, et al. Analysis of mechanisms underlying BRMS1 suppression of metastasis. Clin Exp Metastasis. 2001;18:683–93. doi: 10.1023/a:1013124725690. [DOI] [PubMed] [Google Scholar]
- 7.Seraj MJ, Samant RS, Verderame MF, Welch DR. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res. 2000;60:2764–9. [PubMed] [Google Scholar]
- 8.Shevde LA, Samant RS, Goldberg SF, et al. Suppression of human melanoma metastasis by the metastasis suppressor gene, BRMS1. Exp Cell Res. 2002;273:229–39. doi: 10.1006/excr.2001.5452. [DOI] [PubMed] [Google Scholar]
- 9.Zhang S, Lin QD, Di W. Suppression of human ovarian carcinoma metastasis by the metastasis-suppressor gene, BRMS1. Int J Gynecol Cancer. 2006;16:522–31. doi: 10.1111/j.1525-1438.2006.00547.x. [DOI] [PubMed] [Google Scholar]
- 10.Smith PW, Liu Y, Siefert SA, et al. Breast cancer metastasis suppressor 1 (BRMS1) suppresses metastasis and correlates with improved patient survival in non-small cell lung cancer. Cancer Lett. 2009;276:196–203. doi: 10.1016/j.canlet.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silveira AC, Hurst DR, Vaidya KS, Ayer DE, Welch DR. Over-expression of the BRMS1 family member SUDS3 does not suppress metastasis of human cancer cells. Cancer Lett. 2009;276:32–7. doi: 10.1016/j.canlet.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurst DR, Xie Y, Vaidya KS, et al. Alterations of BRMS1-ARID4A interaction modify gene expression but still suppress metastasis in human breast cancer cells. J Biol Chem. 2008;283:7438–44. doi: 10.1074/jbc.M709446200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meehan WJ, Samant RS, Hopper JE, et al. Breast cancer metastasis suppressor 1 (BRMS1) forms complexes with retinoblastoma-binding protein 1 (RBP1) and the mSin3 histone deacetylase complex and represses transcription. J Biol Chem. 2004;279:1562–9. doi: 10.1074/jbc.M307969200. [DOI] [PubMed] [Google Scholar]
- 14.Saunders MM, Seraj MJ, Li ZY, et al. Breast cancer metastatic potential correlates with a breakdown in homospecific and heterospecific gap junctional intercellular communication. Cancer Res. 2001;61:1765–7. [PubMed] [Google Scholar]
- 15.Mese G, Richard G, White TW. Gap junctions: Basic structure and function. J Invest Dermatol. 2007;127:2516–24. doi: 10.1038/sj.jid.5700770. [DOI] [PubMed] [Google Scholar]
- 16.Nicolson GL, Dulski KM, Trosko JE. Loss of intercellular junction communication correlates with metastatic potential in mammary adenocarcinoma cells. Proc Natl Acad Sci. 1988;85:473–6. doi: 10.1073/pnas.85.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carystinos GD, Bier A, Batist G. The role of connexin-mediated cell-cell communication in breast cancer metastasis. J Mamm Gland Biol Neopl. 2001;6:431–40. doi: 10.1023/a:1014787014851. [DOI] [PubMed] [Google Scholar]
- 18.Mehta PP, Bertram JS, Loewenstein WR. Growth-inhibition of transformed-cells correlates with their junctional communication with normal-cells. Cell. 1986;44:187–96. doi: 10.1016/0092-8674(86)90497-6. [DOI] [PubMed] [Google Scholar]
- 19.Mesnil M, Crespin S, Avanzo JL, Zaidan-Dagli ML. Defective gap junctional intercellular communication in the carcinogenic process. Biochim Biophys Acta Biomemb. 2005;1719:125–45. doi: 10.1016/j.bbamem.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 20.Nicolson GL, Gallick GE, Dulski KM, et al. Lack of correlation between intercellular junctional communication, p21rasEJ expression, and spontaneous metastatic properties of rat mammary cells after transfection with c-H-rasEJ or neo genes. Oncogene. 1990;5:747–53. [PubMed] [Google Scholar]
- 21.Cesen-Cummings K, Fernstrom MJ, Malkinson AM, Ruch RJ. Frequent reduction of gap junctional intercellular communication and connexin43 expression in human and mouse lung carcinoma cells. Carcinogenesis. 1998;19:61–7. doi: 10.1093/carcin/19.1.61. [DOI] [PubMed] [Google Scholar]
- 22.Zhu D, Cheng C-F, Pauli BU. Mediation of lung metastasis of murine melanomas by a lung-specific endothelial cell adhesion molecule. Proc Natl Acad Sci. 1991;88:9568–72. doi: 10.1073/pnas.88.21.9568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin W, Zempel G, Hulser D, Willecke K. Growth inhibition of oncogene-transformed rat fibroblasts by cocultured normal cells: relevance of metabolic cooperation mediated by gap junctions. Cancer Res. 1991;51:5348–54. [PubMed] [Google Scholar]
- 24.Li Z, Zhou Z, Welch DR, Donahue HJ. Expressing connexin 43 in breast cancer cells reduces their metastasis to lungs. Clin Exp Metastasis. 2008;25:893–901. doi: 10.1007/s10585-008-9208-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yano T, Fujimoto E, Hagiwara H, et al. Connexin 32 as an anti-invasive and anti-metastatic gene in renal cell carcinoma. Biol Pharm Bull. 2006;29:1991–4. doi: 10.1248/bpb.29.1991. [DOI] [PubMed] [Google Scholar]
- 26.Naus CC, Laird DW. Implications and challenges of connexin connections to cancer. Nature Rev Cancer. 2010;10:435–41. doi: 10.1038/nrc2841. [DOI] [PubMed] [Google Scholar]
- 27.Czyz J. The stage-specific function of gap junctions during tumourigenesis. Cellular & Molecular Biology Letters. 2008;13:92–102. doi: 10.2478/s11658-007-0039-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cronier L, Crespin S, Strale PO, Defamie N, Mesnil M. Gap junctions and cancer: New functions for an old story. Antioxidants & Redox Signaling. 2009;11:323–38. doi: 10.1089/ars.2008.2153. [DOI] [PubMed] [Google Scholar]
- 29.Sirnes S, Kjenseth A, Leithe E, Rivedal E. Interplay between PKC and the MAP kinase pathway in Connexin43 phosphorylation and inhibition of gap junction intercellular communication. Biochem Biophys Res Comm. 2009;382:41–5. doi: 10.1016/j.bbrc.2009.02.141. [DOI] [PubMed] [Google Scholar]
- 30.Klotz LO, Patak P, Ale-Agha N, et al. 2-Methyl-1,4-naphthoquinone, vitamin K-3, decreases gap-junctional intercellular communication via activation of the epidermal growth factor receptor/extracellular signal-regulated kinase cascade. Cancer Res. 2002;62:4922–8. [PubMed] [Google Scholar]
- 31.DeWald DB, Torabinejad J, Samant RS, et al. Metastasis suppression by breast cancer metastasis suppressor 1 involves reduction of phosphoinositide signaling in MDA-MB-435 breast carcinoma cells. Cancer Res. 2005;65:713–7. [PubMed] [Google Scholar]
- 32.Schubert AL, Schubert W, Spray DC, Lisanti MP. Connexin family members target to lipid raft domains and interact with caveolin-1. Biochem. 2002;41:5754–64. doi: 10.1021/bi0121656. [DOI] [PubMed] [Google Scholar]
- 33.van Zeijl L, Ponsioen B, Giepmans BNG, et al. Regulation of connexin43 gap junctional communication by phosphatidylinositol 4,5-bisphosphate. J Cell Biol. 2007;177:881–91. doi: 10.1083/jcb.200610144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ross DT, Scherf U, Eisen MB, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24:227–35. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Q, Fan H, Shen J, Hoffman RM, Xing HR. Human breast cancer cell lines co-express neuronal, epithelial, and melanocytic differentiation markers in vitro and in vivo. Plos One. 2010;5:e9712. doi: 10.1371/journal.pone.0009712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chambers AF. MDA-MB-435 and M14 cell lines: Identical but not M14 melanoma? Cancer Res. 2009;69:5292–3. doi: 10.1158/0008-5472.CAN-09-1528. [DOI] [PubMed] [Google Scholar]
- 37.Montel V, Suzuki M, Galloy C, Mose ES, Tarin D. Expression of melanocyte-related genes in human breast cancer and its implications. Differentiation. 2009;78:283–91. doi: 10.1016/j.diff.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 38.DeRosier LC, Buchsbaum DJ, Oliver PG, et al. Combination Treatment with TRA-8 Anti–Death Receptor 5 Antibody and CPT-11 Induces Tumor Regression in an Orthotopic Model of Pancreatic Cancer. Clin Cancer Res. 2007;13:5535s–43s. doi: 10.1158/1078-0432.CCR-07-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crow DS, Kurata WE, Lau AF. Phosphorylation of connexin 43 in cells containing mutant Src oncogenes. Oncogene. 1992;7:999–1003. [PubMed] [Google Scholar]
- 40.Bani-Yaghoub M, Bechberger JF, Naus CCG. Reduction of Connexin43 expression and dye-coupling during neuronal differentiation of human NTera2/clone D1 cells. J Neurosci Res. 1997;49:19–31. doi: 10.1002/(sici)1097-4547(19970701)49:1<19::aid-jnr3>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 41.Sato K, Gratas C, Lampe J, et al. Reduced expression of the P-2 form of the gap junction protein connexin 43 in malignant meningiomas. J Neuropathol Exp Neurol. 1997;56:835–9. [PubMed] [Google Scholar]
- 42.Husoy T, Knutsen HK, Cruciani W, et al. Connexin 43 is overexpressed in Apc(Min/+)-mice adenomas and colocalises with COX-2 in myofibroblasts. Int J Cancer. 2005;116:351–8. doi: 10.1002/ijc.21025. [DOI] [PubMed] [Google Scholar]
- 43.Godwin AJ, Green LM, Walsh MP, et al. In-situ regulation of cell-cell communication by the camp-dependent protein-kinase and protein-kinase-C. Mol Cell Biochem. 1993;128:293–307. doi: 10.1007/BF01076779. [DOI] [PubMed] [Google Scholar]
- 44.TenBroek EM, Lampe PD, Solan JL, Reynhout JK, Johnson RG. Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J Cell Biol. 2001;155:1307–18. doi: 10.1083/jcb.200102017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atkinson MM, Lampe PD, Lin HH, et al. cAMP modifies the cellular-distribution of connexin 43 and induces A persistent increase in the junctional permeability of mouse mammary tumor cells. J Cell Sci. 1995;108:3079–90. doi: 10.1242/jcs.108.9.3079. [DOI] [PubMed] [Google Scholar]
- 46.Burghardt RC, Barhoumi R, Sewall TC, Bowen JA. Cyclic AMP induces rapid increases in gap junction permeability and changes in the cellular distribution of Connexin 43. J Membr Biol. 1995;148:243–53. doi: 10.1007/BF00235042. [DOI] [PubMed] [Google Scholar]
- 47.Paulson AF, Lampe PD, Meyer RA, et al. Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. J Cell Sci. 2000;113:3037–49. doi: 10.1242/jcs.113.17.3037. [DOI] [PubMed] [Google Scholar]
- 48.Laird DW. The gap junction proteome and its relationship to disease. Trends Cell Biol. 2010;20:92–101. doi: 10.1016/j.tcb.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 49.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylnositol 3-Kinase, 2-(4-Morpholinyl)-8-Phenyl-4H-1-Benzopyran-4-One (LY294002) J Biol Chem. 1994;269:5241–8. [PubMed] [Google Scholar]
- 50.Ding JB, Vlahos CJ, Liu RC, Brown RF, Badwey JA. Antagonists of phosphatidylinositol 3-kinase block activation of several novel protein-kinases in neutrophils. J Biol Chem. 1995;270:11684–91. doi: 10.1074/jbc.270.19.11684. [DOI] [PubMed] [Google Scholar]
- 51.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochemical Journal. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ethier MF, Madison JM. LY294002, but not wortmannin, increases intracellular calcium and inhibits calcium transients in bovine and human airway smooth muscle cells. Cell Calcium. 2002;32:31–8. doi: 10.1016/s0143-4160(02)00111-2. [DOI] [PubMed] [Google Scholar]
- 53.Solan JL, Lampe PD. Connexin 43 phosphorylation: structural changes and biological effects. Biochemical Journal. 2009;419:261–72. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Intl J Biochem Cell Biol. 2004;36:1171–86. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.