

Abstract
Botulinum neurotoxins (BoNT) are the etiological agents responsible for botulism, a disease characterized by peripheral neuromuscular blockade and a characteristic flaccid paralysis of humans. BoNT/A is the most toxic protein known to man and has been classified by the Centers of Disease Control (CDC) as one of the six highest-risk threat agents for bioterrorism. Of particular concern is the apparent lack of clinical interventions that can reverse cellular intoxication. Efforts to uncover molecules that can act within an intoxicated cell so as to provide symptomatic relief to BoNT/A are paramount. Aminopyridines have shown clinical efficacy for multiple sclerosis treatment as well as BoNT/A intoxication; yet, aminopyridines for BoNT/A treatment has been abandoned because of blood brain barrier (BBB) penetration producing undesired neurotoxic side effects. Two aminopyridines, (5 and 11), exhibited inhibitory activity toward Shaker-IR voltage-gated potassium (KV1.x) channels with potencies similar to that of the previous “gold-standard”, 3,4-diaminopyridine (3,4-DAP), including reversal of symptoms from BoNT-induced paralysis in phrenic nerve-hemidiaphragm preparations. Importantly, pharmacokinetic experiments revealed a lack of BBB penetration of 5, which is a significant advancement toward resolving the neurotoxicity issues associated with prolonged 3,4-DAP treatments. Finally, 5 was found to be as effective as 3,4-DAP in rescuing BoNT-poisoned mice in the mouse lethality assay, signifying an optimized balance between the undesired permeability across the BBB, and the required permeability across lipid cellular membranes. The results demonstrate that 5 is the most promising small molecule K+ channel inhibitor discovered to date for the treatment of BoNT/A intoxication.
Keywords: Botulinum Neurotoxin, aminopyridine, K+ channel inhibitors
INTRODUCTION
Botulinum neurotoxins (BoNTs A-G) are a family of seven immunologically distinct proteins derived from strains of Clostridium botulinum, a gram-positive, sporulating anaerobic bacillus.(1) Botulinum neurotoxins are responsible for botulism, a disease historically associated with food poisoning. Furthermore, the BoNTs are also some of the most deadly toxins known to man, approximately 10 million times that of cyanide,(2) and are characterized by peripheral neuromuscular blockade and a characteristic flaccid paralysis.
The BoNTs potent toxicity coupled with their highly specific mechanism of action render these neurotoxins both useful to medicine and a public threat. Indeed, the BoNTs are classified by the US Centers for Disease Control (CDC) as bioterrorism agents due to a relative ease of production, their extreme lethal potency (1 ng/kg),(3) and the duration of their paralytic activity. With the current concerns of biological warfare around the world, a vastly enhanced defense against potential bioterrorist weapons is of high priority.(4, 5) Currently, there are no approved pharmacological treatments for BoNT intoxication.(6) We also note that due to the protracted paralysis and necessity for intubation and mechanical respiration, the numbers of medical care units capable of proving supportive care for recovery in the event of a bioterrorism incident would be limited.
Botulinum toxins induce muscle paralysis through inhibition of acetylcholine release at neuromuscular junctions by proteolytic cleavage of SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins.(7, 8) This effect can be reduced or reversed by enhancing phasic Ca2+ influx by blocking voltage-dependent K+ channels in nerve terminals, allowing acetylcholine release within the synaptic cleft. To our knowledge, only a handful of compounds, including TEA (tetraethylammonium) 1,(9–11) 4-aminopyridine (4-AP) 2, (12, 13) and 3,4 diaminopyridine (3,4-DAP) 3,(13) (Figure 1) have been shown to act as reversible K+ channel blockers with an ability to allow synaptic response recovery from serotype A BoNT poisoning, generating a muscle action potential and inducing muscle contraction in vitro and in animal models. It is important to note that prior literature reports include conflicting and often puzzling accounts of clinical treatment of patients suffering of severe serotype A and B BoNT intoxications with 3,4-DAP. Intriguingly 3,4-DAP was reportedly effective in treatment of BoNT/B poisoning,(14) but, what is even more puzzling, failed to provide symptomatic relief to BoNT/A-intoxicated patients.(15) One possible explanation for the apparent inconsistency is the low tolerance of the patients to high doses of 3,4-DAP, which are necessary for effective restoration of muscle function.(16)
Figure 1.
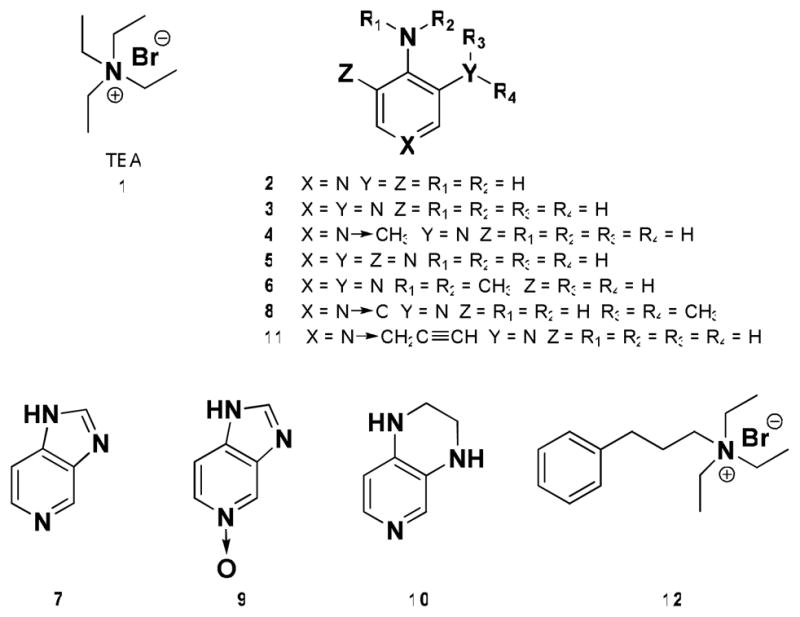
Structures of the leading small molecule K+ channel inhibitors 1–3, and the experimental analogues 4–12.
Interestingly, these aminopyridines have also been considered as therapeutics for symptomatic treatment of such neurological disorders as myasthenia gravis,(17) multiple sclerosis,(18) spinal cord injuries,(19) and Alzheimer’s disease,(20) providing an additional incentive for further exploration of the aminopyridine template. Yet, while 4-AP and 3,4-DAP have produced impressive results as potential therapeutics they also have presented certain toxicity issues. Seizures have been reported previously with these aminopyridines that arise from their penetration across the blood-brain barrier (BBB). Discovery of molecules that could obviate BBB crossing while still bringing synaptic relief in the form of a rapid, albeit temporary, reversal of paralysis could provide inroads to an alternative form of care for BoNT intoxication.
Among aminopyridines previously tested for K+ channel antagonism and subsequent reversal of BoNT/A-induced paralysis, 3,4-DAP was found to be highly effective while possessing the least toxicity.(13) The observed central nervous system (CNS) toxicity of 3,4-DAP has been associated with its ability to cross the BBB,(21–23) and lipid membranes in general, while in its neutral form, which in turn is requisite for its efficacy in blocking K+ channels. An attempt to reduce lipid membrane penetrability by generating a cationic quarternary analogue of 3,4-DAP produced 3,4-diamino-1-methylpyridinium 4 (Figure 1), unfortunately, 4 was determined to be inactive in rat phrenic nerve-hemidiaphragm preparations. This latter result led a leading botulinum research group in the mid 90’s to state that “it does not appear possible to produce structural modifications of 3,4-DAP that would completely eliminate its CNS toxicity”.(13) However, ample crystallographic and modeling literature data has been amassed to challenge this assertion,(24–26) which we saw as an opportunity to discover new molecules, potentially with an improved therapeutic index.
In the context of K+ channel modulation, a substantial body of experimental(27, 28) and theoretical(24–26) evidence indicates that aminopyridine pharmacophore requirements include a positive charge, usually a result of protonation, and at least one amino group suitable for hydrogen bonding. Observations of pH dependence of aminopyridine activity(28) as well as modeling reports,(24–26) indicate that aminopyridines cross cellular membrane in their neutral form, and then are re-ionized and block K+ channels from the intracellular side of the pore. Docking simulations suggest that protonated aminopyridines form several hydrogen bonds with the amide group oxygen atoms of the four Thr107 and Ala111 residues located on separate chains of the tetrameric assembly comprised of four K+ channel α-subunits, while orienting the molecular plane of the ligand perpendicular to the axis of the pore, thus effectively blocking it.(29) Thus, manipulations of the 3,4-DAP structure, affecting its pKa and the predisposition for hydrogen bonding seemed viable as a route towards aminopyridine analogues with improved BoNT/A-induced paralysis-alleviating activity, while using the same technique to simultaneously reduce the transient brain penetration, which is responsible for the neurotoxicity of 3,4-DAP.
RESULTS AND DISCUSSION
Conceptual design of aminopyridine analogues
3,4,5-Triaminopyridine 5, preparation was contemplated to test the effects of augmented hydrogen bonding as well as the potential influence of enhancement of basicity of the pyridine moiety. The pKa values of 4-aminopyridine, 3,4-diaminopyridine and 3,4,5-triaminopyridine were estimated to be 9.2, 8.9 and 8.8, respectively (see Supporting Information for details), which is in a reasonable agreement with the experimental pKa value of 4-aminopyridine reported at 9.114.(ref) While the differences among the pKa values of these compounds are relatively small, they may influence not only the interaction with Kv channels, but also the rate of intracellular penetration, thus affecting their therapeutic values. We also surmised that the reduced lipophilicity would result in decreased brain penetration, and diminished neurotoxicity, compared to 3,4-DAP. N-Alkylation of the 4-amino group (3-amino-4-(dimethylamino)pyridine 6) was anticipated to result in deconjugation of that group from the pyridine moiety due to steric effects, allowing investigations into the significance of electronic effects of 4-aminogroup as well as to probe the ramifications of the N-alkylation on hydrogen bonding interactions with the K+ channel. In contrast, rigidified 5-azabenzimidazole 7 was projected to force full conjugation of both amino groups with the pyridine moiety, potentially affecting the hydrogen bonding pattern during the K+ channel binding, and to amplify π-cation and π-π interactions, which are responsible for the binding of aminopyridines to the receptor site in the K+ channel, as suggested by literature reports.(25) N-oxide analogues 8 and 9 would be prepared, as they were readily accessible, are highly polar molecules, have excellent water solubility and the N-O moiety of pyridine N-oxides possesses unique chemical/biological functionality, which can act effectively as a push electron donor and as a pull electron acceptor group. The pyrido[4,3-b]piperazine analogue 10 was of interest as it possesses a somewhat less rigid scaffold than that of azabenzimidazole 7, while maintaining the conjugation of the amino groups with the pyridine moiety. The 3,4-diamino-1-(prop-2-ynyl)pyridinium analogue 11, was to be prepared and tested to assess the previously held view that pyridinium salts cannot effectively function as K+ channel blockers.(13) Importantly, this compound, should the prior held tenet be proven incorrect, could serve as a second molecular scaffold for aminopyridinium structures using the Huisgen 1,3-dipolar reaction or as it has been popularized and re-termed by Sharpless as the “click” reaction; which is an ideal method to install diversity within a library, especially one biologically based in nature.(30) Finally, modification of TEA 1 via introduction of an aromatic moiety, would permit examination of potentially crucial π-π interactions found within the receptor combining site; such interactions could be explored through triethyl-3-phenylpropylammonium 12.
Chemistry
Analogues 5–12 (Figure 1) were synthesized using modified procedures from the corresponding literature reports (Supporting Information, S3-S5). Thus, nitration of 4-aminopyridine and N,N-dimethyl-4-aminopyridine followed by catalytic hydrogenation of the nitro-intermediates yielded 3,4,5-triaminopyridine 5, (31) and 3-amino-4-(dimethylamino)pyridine 6, (32), respectively. 5-Azabenzimidazole 7 was used as commercially available, and the corresponding N-oxide 9 was prepared by ReV-mediated hydrogen peroxide oxidation(33) of 7. Compound 8 was synthesized by amination of 3-bromo-4-nitropyridine-1-oxide(34) with dimethylamine,(35, 36) and the subsequent catalytic hydrogenation of the product. Stepwise reductive amination of glyoxal with 3,4-diaminopyridine 3 produced pyrido[4,3-b]piperazine 10.(37) The 3,4-diamino-1-(prop-2-ynyl)pyridinium analogue 11 and triethyl-3-phenylpropylammonium compound 12 were obtained by N-alkylations of 3,4-diaminopyridine 3 with propargyl bromide, and of triethylamine with (3-bromopropyl)benzene, respectively.
Mouse Phrenic Nerve-Hemidiaphragm Assay
With a series of molecules in hand their efficacy was initially queried in the reversal of BoNT/A-induced paralysis using mouse phrenic nerve-hemidiaphragm preparations. Since BoNT markedly alters evoked transmitter release at neuromuscular junctions, the hemidiaphragm assay is an ideal technique to examine agents that can modulate nerve activity.(38) Under this semblance we considered two complementary routes to examine our molecules; the first involved measurement of reduced in vivo BoNT lethality, while the second would measure a compound’s effect in delaying the onset and slowing the progression paralysis, which is a well-characterized model that has been used to benchmark 4-AP and 3,4-DAP in the past.(39) At 330 μM, the compounds 5, 6 and 10 exhibited potencies, slightly lower than that of 3,4-DAP, with the differences found to be statistically significant at 0.05 level (2-tailed tests), while at the lower concentrations the differences became negligible. As evident from Table 1, 3,4,5-triaminopyridine 5 was found to be quite potent in comparison to the benchmark compound 3,4-DAP, which can be rationalized by augmented hydrogen bonding between ligand and the K+ channel receptor site, as well as essentially the same pKa with respect to 3,4-DAP. N-methylation of the 4-aminogroup (analogue 6) resulted in a slight decline in efficacy, compared to 3,4-DAP, possibly due to steric interference from the N-methyl groups, as well as a diminished pKa of the pyridine moiety, which was estimated to be 8.2. Azabenzimidazole 7 and the N-oxide compounds 8 and 9 were found to be unable to reverse BoNT/A-induced muscle paralysis, likely due to steric hindrance of the amino groups thus weakening the requisite hydrogen bonding with the K+ channel receptor site, as well as the possibly low tolerance of the receptor for the modification of the pyridine moiety via N-oxidation. The pyrido[4,3-b]piperazine analogue 10 was determined to possess a modest potency in this assay, indicating a limited benefit from N-alkylation of the 3- and 4-aminogroups of the 3,4-DAP template in the search for potent K+ channel blockers. The 3,4-diamino-1-(prop-2-ynyl)pyridinium analogue 11, on the other hand, demonstrated a high efficacy in paralysis reversal, statistically indistinguishable from that of 3,4-DAP, in sharp contrast with the previously reported inactive N-methylpyridinium, 4. It seems plausible that the analogue 11 benefits from the increased hydrophobicity of the N-propynyl group, compared to the N-methyl analogue 4, which facilitates the transport of this compound through cellular membranes, while the increased steric bulk of the N-alkyl group having little or no effect on this analogue’s potency. Finally, the apparent neurotoxicity of the compound 12 prevented determination of its ability to reverse BoNT/A-induced muscle paralysis.
Table 1.
Paralysis times at 10 pM BoNT/A (minutes).
| 0 μM | 30 μM | 100 μM | 330 μM | |
|---|---|---|---|---|
| Control | 102±6 | - | - | - |
| 3 (3,4-DAP) | - | 110±6 | 303±14 | 408±23 |
| 5 | - | 108±6 | 230±19 | 304±15 |
| 6 | - | 107±7 | 223±17 | 285±12 |
| 10 | - | 104±9 | 200±21 | 243±19 |
| 11 | - | 106±4 | 207±10 | 332±6 |
Paralysis is defined as a 90% reduction in twitch amplitude. The data represents the mean ± S.E. (n = 10).
From the phrenic nerve-hemidiaphragm results there are several key points, the most salient being that there is a window of opportunity to find new aminopyridines that can be of therapeutic interest. Of further importance is that both substitution on the aminopyridine ring and pyridinium salts are viable options. The latter discovery is especially exciting as previously it was disclosed (vide supra) that such structures were not thought to be feasible for treating BoNT intoxication.(13) Finally, we note that pyridines 5 and 11 at first glance appear not as potent as 3,4-DAP (Table 1); however, if we compare apparent toxicity (Table 2) for all three compounds at 3.3 × 10−4 M, where the toxin’s time to paralysis becomes in essence equally attenuated, these compounds become equivalent to each other with respect to therapeutic value.
Table 2.
Apparent toxicity at 10 pM BoNT/A (per cent).
| 0 μM | 30 μM | 100 μM | 330 μM | |
|---|---|---|---|---|
| Control | 100 | - | - | - |
| 3 (3,4-DAP) | - | 99 | 9 | 6 |
| 5 | - | 99 | 10 | 9 |
| 11 | - | 99 | 20 | 8 |
Apparent toxicity = percent of toxicity (as measured by paralysis time) of tissues exposed to 10 pM BoNT/A, e.g., 10% toxicity is equivalent to the toxicity of a 1 pM BoNT/A dose.
Electrophysiological Studies
The biological effects of the leading aminopyridines 5 and 11 as well as the 3,4-DAP 3 control were investigated in X. laevis oocytes heterologously expressing Shaker-IR KV channel variant(40, 41) using the two-electrode voltage clamp (TEVC) technique.(42) The IC50 values for the compounds 3, 5 and 11 were determined to be 22.2, 697 and 26.7 μM, respectively (Figure 3). These findings unambiguously show that the aminopyridines 5 and 11 are inhibitors of KV1.x channels, with the potencies comparable to that of 3,4-DAP. This result also correlates well with the results from the BoNT-induced paralysis reversal in phrenic nerve-hemidiaphragm preparations, where 3,4-DAP 3 exhibited the highest potency, while compounds 5 and 11 compounds had similar, albeit slightly lower efficacy. The lower potency of 5 compared to that of 3,4-DAP is likely to be a reflection of the different sensitivity of rat neuronal channels and Drosophila Shaker channels, and, perhaps, in part due to complexity of the expression pattern of Kv channels in the nervous system.
Figure 3.

Dose-response curves for inhibition of Shaker channels by 3,4-DAP, 5, and 11. The data represents the mean ± S.E. (n = 5).
Pharmacokinetics and Mouse Brain Penetration After i.p. Injection
Since the neurotoxic side effects of 3,4-DAP had been attributed to its transient brain penetration, it was imperative to retain the inhibitory potency of 3,4-DAP towards K+ channels, but also to limit its brain exposure. The earlier literature reports noted that 3,4-DAP exhibited somewhat less brain penetration than 4-aminopyridine, probably due to reduced lipophilicity, as well as the increased propensity towards hydrogen bonding.(21, 22, 43) It therefore seemed plausible that introduction of additional hydrogen bond donor or acceptor groups into 3,4-DAP (3,4,5-triaminopyridine 5), or its N-alkylation (yielding the cationic compound 11) would result in the desired reduction in brain exposure. The pharmacokinetics results for the tested compounds 3, 5 and 11 showed rapid absorption into the plasma from the i.p. site of injection with the Tmax occurring at 0.50 h (first time point collected) for all three compounds (Figure 4). The plasma halflives for 5 and 3 (3,4-DAP) of 0.31 h and 0.34 h, respectively, are quite short and would have produced little accumulation in plasma, while the plasma half-life of 11, determined to be at 1.2 h, was long enough to produce some plasma accumulation under the repeat dose regimen necessary for an effective reversal of BoNT-induced paralysis. Compound 11 produced the highest plasma exposure with a Cmax of 402 ng/mL and an AUC(0-infinity) of 620 h-ng/mL, with a low brain exposure that was 13% of the plasma exposure. At the same time, triaminopyridine 5 gave both the lowest plasma exposure and no detectable brain exposure at 3.0 mg/kg i.p., although the possibility of a significant brain exposure at a higher dose cannot be eliminated at this point. The control compound 3 (3,4-DAP) with a brain to plasma ratio of 0.38 showed significant brain penetration, which seems to correlate with the literature reports.(21, 22)
Figure 4.

Concentration (ng/mL) of substrate in mouse plasma and brain tissue after i.p. administration of 3,4-DAP and analogues 5 and 11 at 3.0 mg/kg (non-serial sampling). The data represents the mean ± S.E. (n = 3).
Mouse Lethality Assay
To further examine the therapeutic relevance of these compounds a mouse lethality assay was carried out. The mouse toxin neutralization uses death as an endpoint, and is the only animal model approved for the study of antagonists with botulinum neurotoxins. The importance of this experiment is that the results obtained will show definitively whether a molecule can provide symptomatic relief from BoNT/A intoxication. Initially, both leading compounds 5 and 11 were examined for toxicity issues by administering 200 μg of each compound and observing the animals over a 48 hr period; no distress was observed at this dose level. The rescue experiments were conducted at 10 LD50’s of BoNT/A (n = 10) for both the control (BoNT/A alone) and animals that were to be treated with 5, 11, or 3,4-DAP. An arbitrary dose of 75 μg of each aminopyridine compound was used, as this dose of 3,4-DAP was shown to relieve the effects of BoNT/A poisoning. At 120 minutes first signs of BoNT distress was observed and 5, 11 or 3,4-DAP was administered i.p. Subsequently, every 75 minutes each of these compounds were administered for a total of 5 injections; the results are shown in Figure 2. Remarkably, 3,4,5-triaminopyridine 5 was found to be as effective as 3,4-DAP, whereas 3,4-diamino-1-(prop-2-ynyl)pyridinium 11 was not. At this juncture the reasons for the observed low efficacy of 11 remains unclear, however, we surmise that metabolic clearance might be involved and thus the ability to functionalize this scaffold using “click chemistry” will afford us ample opportunity to improve on this result.
Figure 2.

Rescue of BoNT/A intoxicated mice. The data represents the mean ± S.E. (n = 10). The differences between all group pairs are statistically significant (P ≤ 0.001), with the exception of the 3,4-DAP and 5 groups with respect to one another.
SUMMARY AND OUTLOOK
Intense research efforts have been ongoing to discover small molecules that can effectively treat BoNT/A intoxication. Virtually all efforts have centered upon inhibiting enzymatic activity of the neurotoxin, unfortunately little success has been achieved.(6, 8) Our current research has taken an alternative tactic, the use of K+ channel blockers. The aminopyridines 4-AP and 3,4-DAP have both been shown to be K+ channel blockers and have provided symptomatic treatment of MS patients.(44) However, both 4-AP and 3,4-DAP cross the blood-brain barrier readily, hence, their potential usefulness has been questioned.(45) Capitalizing on crystallographic and modeling data, we were able to identify two new leads for a small molecule K+ channel inhibitor-based therapeutic approach for the rescue of BoNT/A intoxicated cells/animals, thus marking the first advancement in this area in almost 20 years. Triaminopyridine 5 proved to be effective in the reversal of BoNT/A-induced muscle paralysis in both phrenic nerve-hemidiaphragm preparations and in a mouse toxin neutralization assay; significantly, it showed potency rivaling that of the long-standing benchmark compound 3,4-DAP. Most gratifyingly, this compound’s blood-brain-barrier entry was restricted, which suggests that this triaminopyridine is likely to exhibit significantly reduced in vivo neurotoxicity, compared to 3,4-DAP. Lastly, of significant note was pyridinium salt 11, while inactive in the mouse lethality study, its potent efficacy in paralysis reversal as seen in the phrenic nerve-hemidiaphragm assay, coupled with its ability to inhibit K+ channels appears to contradict previous studies suggesting that pyridinium salts cannot be utilized as effective K+ channel blockers. Further advancement of these new leads will pave the way for the development of new small molecule K+ channel inhibitors for the treatment of BoNT/A poisoning, as well as other neurological disorders.
EXPERIMENTAL SECTION
Materials and Methods
ACS grade organic solvents were purchased from VWR Scientific, and other reagents were obtained from Sigma-Aldrich and used as commercially available. The TLC chromatograms were visualized by UV light, and by dipping in potassium permanganate solution followed by heating (hot plate). Final purifications were accomplished by preparative RP-HPLC on a C18-bonded silica column (Vydac 218TP152022, 250×22 mm, 15–20 μm, 300 Å) using a Shimadzu SCL-10A HPLC system. The compounds were eluted with a linear gradient of 1%–10% (v/v) of 0.09% (v/v) TFA/acetonitrile in 0.1% v/v aqueous TFA solution over 40 min with 10 mL/min flow rate. The purity of the compounds 1–12 was checked by analytical reverse-phase HPLC using a Vydac C18 218TP104 column (Western Analytical Products) monitored at 230 and 254 nm, and was determined to be >95% for all compounds reported herein.
Synthetic procedures for compounds 5–12
See the Supporting Information.
Mouse phrenic nerve-hemidiaphragm assay
Phrenic nerve-hemidiaphragms from mice were incubated (4°C, 45 min) with serotype A botulinum neurotoxin, which, along with the serotype C, is sensitive to aminopyridine treatment. Then the phrenic nerve-hemidiaphragms were washed and transferred to tissue baths without added toxin. For control tissues, the phrenic nerve was stimulated, and the elapsed time for onset of paralysis was monitored. Experimental tissues were treated identically, except that the bathing medium contained 3,4-DAP or the analogue 5–12. The paralysis time for control tissues (n = 10) was 54 min; that for experimental tissues (n = 10) was 102 min, Table 1. A narrow concentration range, (0.3 × 10−4 to 3.3 × 10−4 M) was used to produce an enhancement in the muscle twitch response due to nerve stimulation (Table 1). This is a known concentration range reported previously for experiments on reversal of BoNT/A-induced paralysis with 3,4-DAP, while at higher concentrations, there was substantial and sustained depression of muscle responses with 3,4-DAP.
Electrophysiological Studies
Xenopus laevis oocytes were isolated and injected with 20 nl of RNase-free water containing 0.5 ng cRNA. Whole-cell currents were measured 1–3 days after injection by the two-electrode voltage clamp technique, using a GeneClamp 500B amplifier (Axon Instruments). Data were sampled at 2 kHz and filtered at 0.5 kHz with Clampex 9.0 software (Axon Instruments). The pipette contained 3 M KCl and the bath solution contained (in mM): 4 KCl, 96 NaCl, 1 MgCl2, 0.3 CaCl2, 5 HEPES, pH 7.4, with NaOH. Oocyte membrane potential was held at −80 mV and currents were measured at 0 mV. Currents were measured before and after 30–40 min-long incubation in the desired dilution of the tested compound in bath solution. Data points were mean ± SEM for 5–10 cells (Figure 3). The solid line was a fit of the data to the equation: I = 1/(1/([C]/K0.5)h) where I is the measured current, [C] was the compound concentration, K0.5 was the compound concentration required to achieve 50% inhibition, and h was the Hill coefficient. K0.5 values were 26.7 ± 4.0, 697 ± 80 and 22.2 ± 4.3 μM for 3,4-DAP, 5 and 11, respectively. The Hill coefficient was 1.04 ± 0.18, 1.09 ± 0.13 and 1.28 ± 0.34 for 3,4-DAP, 5 and 11, respectively.
Pharmacokinetics and Mouse Brain Penetration
1. Compound Administration
Pharmacokinetic studies were performed in accordance with the Guide for the Care and Use of Laboratory Animals, at a facility whose program is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International. Male CD-1 mice (Charles River) of 25 to 35 g in weight were divided into six groups of three animals each for measurements at six time points including pre-dose, 0.5, 1.0, 2.0, 4.0 and 8.0 h. This was done for each of the three compounds tested for a total of 54 mice. Each animal was dosed by intra-peritoneal injection at 5.0 mL/kg with either the saline vehicle (pre-dose) or compound dissolved in vehicle at a dose of 3.0 mg/kg. Animal groups were sacrificed using carbon dioxide at the described time points followed by removal of blood by cardiac puncture and brain by dissection. Blood was collected in tubes containing heparin and the plasma separated by centrifugation before being frozen on dry ice. Brains were frozen on dry ice as soon as removed. Pharmacokinetic parameters were calculated using WinNonlin version 5.2 (Pharsight Corporation) from the bioanalytical data.(46)
2. Sample Preparation
The mouse plasma and brain samples were stored at −20 °C until analysis. Mouse plasma samples (25.0 μL) were transferred into three micro-centrifuge tubes (1.7 mL) and mixed with 50 μL of Internal Standard solution (200 ng/mL of 11 in acetonitrile for 3 and 5; 200 ng/mL of 5 in acetonitrile for 11). The tubes were vortex mixed for 1 min and centrifuged at 10,000 g for 4 min. A 50 μL sample of the supernatant was transferred into an autosampler vial and mixed with 50 or 150 μL (for 3) of water.
Each of the mouse brain samples was first homogenized with 3.0 ml of 25% acetonitrile in water. Mouse brain homogenate samples (50.0 μL) were transferred into microcentrifuge tubes (1.7 mL) and mixed with 100 μL of Internal Standard solution (200 ng/mL of 11 in acetonitrile for 5 and 3; 200 ng/mL of 5 in acetonitrile for 11).The tubes were vortex mixed for 1 min and centrifuged at 10,000 g for 5 min. An aliquot of the supernatant (50.0 μL) was transferred into an autosampler vial and mixed with 50 μL (for 11) and 150 μL (for 3) water or 100 μL of 50% acetonitrile (for 5 brain homogenate) for LC/MS/MS analysis.
3. Chromatography
The LC/MS/MS analyses were performed on an Agilent 1100 Binary Pump HPLC system equipped with a Sciex API 3000 mass-spectrometer (compound 3), or a Shimadzu LC-20AD system with a Sciex API 5000 mass-spectrometer (compounds 5 and 11), using a Polaris C-18, 3 μm, 50 mm × 3.0 mm HPLC column. 0.1% Formic acid and 45% methanol-45% acetonitrile in water were used as the solvents A and B, respectively, with the following gradient: (0 min) 30% B, (0.1 min) 100% B, (2.9 min) 100% B, (3.0 min) 30% B, (6.0 min) 30% B; flow rate of 200 μL/min.
4. Preparation of Quality Control and Calibration Samples
Quality Control Samples and Calibration Samples were prepared by spiking blank mouse plasma and brain homogenate (from control animal of this study) with working solutions prepared from a single weighing. The standard samples were prepared in the following dilutions: 0.500, 1.00, 2.500, 5.00, 10.0, 25.0, 50.0, 100, 250, 500 ng/mL, and the QC samples used 0.500, 5.00, 50.0, 500 ng/mL concentrations for the plasma samples, and 2.50, 25.0, 250 ng/mL concentrations for the brain tissue samples. The calibration curves were weighed by 1/χ2 using a linear regression without being forced through the origin.
5. Sensitivity and Linearity of the Method
The lower limits of quantitation (LLOQ) for 3, 5 and 11 were determined to be 1.000 ng/mL, 0.500 ng/mL, 0.500 ng/mL, respectively, in plasma; 1.000 ng/mL, 1.000 ng/mL, 0.500 ng/mL, respectively, in brain homogenate; and 6.000 ng/mL, 6.000 ng/mL, and 3.000 ng/mL, respectively, in brain tissue. The calibration curves were linear in the above prepared concentration ranges.
BoNT/A mouse lethality assay
The C. botulinum type A neurotoxin was prepared and purified as previously described.(47) Female CD-1 out bred mice (17–23 g; Harlan Sprague Dawley, Madison, WI) animals that were included in the toxin challenge group were injected with 0.5 ml of a solution of neurotoxin containing 10 i.p. LD50/mL. At the first signs of BoNT distress (120 min) the K+ channel blockers were administered i.p. and subsequently every 75 minutes for a total of 5 injections. Control animals did not receive toxin challenge. Animals were observed for signs of botulism, and the time of death in minutes was recorded.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge W. Metz of Sanofi-Aventis for his role in bridging support between the research groups and A. Moreno of TSRI for her expert assistance in HPLC analysis of the reported compounds. Support of this project was partially provided by the National Institute of Allergy and Infectious Diseases AI080671.
Footnotes
SUPPORTING INFORMATION AVAILABLE Synthetic and computational procedures, HPLC traces and 1H/13C NMR spectra of compounds 4–12; additional details for the biological evaluation of compounds 5 and 11. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Johnson EA, Bradshaw M. Clostridium botulinum and its neurotoxins: a metabolic and cellular perspective. Toxicon. 2001;39:1703–1722. doi: 10.1016/s0041-0101(01)00157-x. [DOI] [PubMed] [Google Scholar]
- 2.Singh BR. Intimate details of the most poisonous poison. Nat Struct Biol. 2000;7:617–619. doi: 10.1038/77900. [DOI] [PubMed] [Google Scholar]
- 3.Schantz EJ, Johnson EA. Properties and use of botulinum toxin and other microbial neurotoxins in medicine. Microbiol Mol Biol Rev. 1992;56:80–99. doi: 10.1128/mr.56.1.80-99.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnon SS, Schechter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Hauer J, Layton M, Lillibridge S, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Swerdlow DL, Tonat K. Botulinum Toxin as a Biological Weapon: Medical and Public Health Management. JAMA. 2001;285:1059–1070. doi: 10.1001/jama.285.8.1059. [DOI] [PubMed] [Google Scholar]
- 5.Osborne SL, Latham CF, Wen PJ, Cavaignac S, Fanning J, Foran PG, Meunier FA. The janus faces of botulinum neurotoxin: Sensational medicine and deadly biological weapon. J Neurosci Res. 2007;85:1149–1158. doi: 10.1002/jnr.21171. [DOI] [PubMed] [Google Scholar]
- 6.Willis B, Eubanks Lisa M, Dickerson Tobin J, Janda Kim D. The Strange Case of the Botulinum Neurotoxin: Using Chemistry and Biology to Modulate the Most Deadly Poison. Angew Chem Int Ed. 2008;47:8360–8379. doi: 10.1002/anie.200705531. [DOI] [PubMed] [Google Scholar]
- 7.Simpson LL. Identification of the Major Steps in Botulinum Toxin Action. Annu Rev Pharmacol Toxicol. 2004;44:167–193. doi: 10.1146/annurev.pharmtox.44.101802.121554. [DOI] [PubMed] [Google Scholar]
- 8.Dickerson TJ, Janda KD. The Use of Small Molecules to Investigate Molecular Mechanisms and Therapeutic Targets for Treatment of Botulinum Neurotoxin A Intoxication. ACS Chem Biol. 2006;1:359–369. doi: 10.1021/cb600179d. [DOI] [PubMed] [Google Scholar]
- 9.Lundh H, Cull-Candy SG, Leander S, Thesleff S. Restoration of transmitter release in botulinum-poisoned skeletal muscle. Brain Res. 1976;110:194–198. doi: 10.1016/0006-8993(76)90222-5. [DOI] [PubMed] [Google Scholar]
- 10.Lundh H, Leander S, Thesleff S. Antagonism of the paralysis produced by botulinum toxin in the rat: The effects of tetraethylammonium, guanidine and 4-aminopyridine. J Neurol Sci. 1977;32:29–43. doi: 10.1016/0022-510x(77)90037-5. [DOI] [PubMed] [Google Scholar]
- 11.Metezeau P, Desban M. Botulinum toxin type A: kinetics of calcium dependent paralysis of the neuromuscular junction and antagonism by drugs and animal toxins. Toxicon. 1982;20:649–654. doi: 10.1016/0041-0101(82)90058-7. [DOI] [PubMed] [Google Scholar]
- 12.Molgo J, Lemeignan M, Lechat P, Peradejordi F. Increase in evoked transmitter release from motor nerve terminals by some amino N-heterocyclic compounds. I. Comparative experimental activities and extracellular pH-dependence. Eur J Med Chem. 1985;20:149–153. [Google Scholar]
- 13.Adler M, Scovill J, Parker G, Lebeda FJ, Piotrowski J, Deshpande SS. Antagonism of botulinum toxin-induced muscle weakness by 3,4-diaminopyridine in rat phrenic nerve-hemidiaphragm preparations. Toxicon. 1995;33:527–537. doi: 10.1016/0041-0101(94)00183-9. [DOI] [PubMed] [Google Scholar]
- 14.Doc M, Ali AB, Karras A, Misset B, Garrouste-Orgeas M, Deletie E, Goldstein F, Carlet J. Treatment of severe botulism with 3,4-diaminopyridine. Presse Med. 2002;31:601–602. [PubMed] [Google Scholar]
- 15.Davis LE, Johnson JK, Bicknell JM, Levy H, McEvoy KM. Human type A botulism and treatment with 3,4-diaminopyridine. Electromyogr Clin Neurophysiol. 1992;32:379–383. [PubMed] [Google Scholar]
- 16.Adler M, Sheridan RE, Manley HA, Manley A, Apland J, Deshpande SS, Romano J. Development of Treatments for Intoxication by Botulinum Neurotoxin. In: Flora SJS, Romano JA, Baskin SI, Sekhar K, editors. Pharmacological Perspectives of Toxic Chemicals and Their Antidotes. Narosa Publishing House; New Delhi, India: 2004. pp. 359–381. [Google Scholar]
- 17.Carlsson C, Rosen I, Nilsson E. Can 4-aminopyridine be used to reverse anaesthesia and muscle relaxation? Acta Anaesthesiol Scand. 1993;27:87–90. doi: 10.1111/j.1399-6576.1983.tb01911.x. [DOI] [PubMed] [Google Scholar]
- 18.Schwid SR, Petrie MD, McDermott MP, Tierney DS, Maso DH, Goodman AD. Quantitative assessment of sustained-release 4-aminopyridine for symptomatic treatment of multiple sclerosis. Neurology. 1997;48:817–821. doi: 10.1212/wnl.48.4.817. [DOI] [PubMed] [Google Scholar]
- 19.McEvoy KM, Windebank AJ, Daube JR, Low PA. 3,4-Diaminopyridine in the treatment of Lambert-Eaton myasthenic syndrome. N Engl J Med. 1989;321:1567–1571. doi: 10.1056/NEJM198912073212303. [DOI] [PubMed] [Google Scholar]
- 20.Davidson M, Zemishlany Z, Mohs RC, Horvath TB, Powchik P, Blass JP, Davis KL. 4-Aminopyridine in the treatment of Alzheimer’s disease. Biol Psychiatry. 1988;23:485–490. doi: 10.1016/0006-3223(88)90020-0. [DOI] [PubMed] [Google Scholar]
- 21.Uchiyama T, Lemeignan M, Lechat P. Equipotency of Anti-Curare Activity of 4-Aminopyridine and 3, 4-Diaminopyridine in Anesthetized Rats. Japan J Pharmacol. 1985;38:329–333. doi: 10.1254/jjp.38.329. [DOI] [PubMed] [Google Scholar]
- 22.Plewa M, Martin T, Menegazzi J, Seaberg D, Wolfson A. Hemodynamic effects of 3,4-diaminopyridine in a swine model of verapamil toxicity. Ann Emergency Med. 1994;23:499–507. doi: 10.1016/s0196-0644(94)70069-9. [DOI] [PubMed] [Google Scholar]
- 23.Damsma G, Biessels PTM, Westerink BHC, De Vries JB, Horn AS. Differential effects of 4-aminopyridine and 2,4-diaminopyridine on the in vivo release of acetylcholine and dopamine in freely moving rats measured by intrastriatal dialysis. Eur J Pharmacol. 1988;145:15–20. doi: 10.1016/0014-2999(88)90343-3. [DOI] [PubMed] [Google Scholar]
- 24.Nino A, Munoz-Caro C. Theoretical analysis of the molecular determinants responsible for the K+ channel blocking by aminopyridines. Biophys Chem. 2001;91:49–60. doi: 10.1016/s0301-4622(01)00151-x. [DOI] [PubMed] [Google Scholar]
- 25.Munoz-Caro C, Nino A. The nature of the receptor site for the reversible K+ channel blocking by aminopyridines. Biophys Chem. 2002;96:1–14. doi: 10.1016/s0301-4622(02)00002-9. [DOI] [PubMed] [Google Scholar]
- 26.Nino A, Munoz-Caro C, Carbo-Dorca R, Girones X. Rational modelling of the voltage-dependent K+ channel inactivation by aminopyridines. Biophys Chem. 2003;104:417–427. doi: 10.1016/s0301-4622(03)00030-9. [DOI] [PubMed] [Google Scholar]
- 27.Lippiat JD, Standen NB, Davies NW. A residue in the intracellular vestibule of the pore is critical for gating and permeation in Ca2+-activated K+ (BKCa) channels. J Physiol. 2000;529:131–138. doi: 10.1111/j.1469-7793.2000.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choquet D, Korn H. Mechanism of 4-aminopyridine action on voltage-gated potassium channels in lymphocytes. J Gen Physiol. 1992;99:217–240. doi: 10.1085/jgp.99.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caballero NA, Meléndez FJ, Niño A, Muñoz-Caro C. Molecular docking study of the binding of aminopyridines within the K+ channel. J Mol Model. 2007;13:579–586. doi: 10.1007/s00894-007-0184-9. [DOI] [PubMed] [Google Scholar]
- 30.Kolb HC, Sharpless KB. The growing impact of click chemistry on drug discovery. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 31.Graboyes H, Day AR. Metabolite Analogs. VIII. Syntheses of Some Imidazopyridines and Pyridotriazoles. J Am Chem Soc. 1957;79:6421–6426. [Google Scholar]
- 32.Burton AG, Frampton RD, Johnson CD, Katritzky AR. The kinetics and mechanism of the electrophilic substitution of heteroaromatic compounds. Part XXVIII. The preparation and kinetic nitration of 2-, 3-, and 4-dimethylaminopyridines and their 1-oxides in sulphuric acid. J Chem Soc, Perkin Trans. 1972;2:1940–1949. [Google Scholar]
- 33.Jiao Y, Yu H. Methyltrioxorhenium (MeReO3) Catalyzed Selective Oxidation of Purine and Related Compounds into Their N-Oxides. Synlett 2001. 2001:0073–0074. [Google Scholar]
- 34.Clark-Lewis JW, Singh RP. 459. Preparation of 3,4-diamino-, 3-amino-4-methylamino-, and 4-amino-3-methylamino-pyridine. J Chem Soc. 1962:2379–2382. [Google Scholar]
- 35.Essery JM, Schofield K. 961. Some derivatives of 4-amino- and 4-nitro-pyridine. J Chem Soc. 1960:4953–4959. [Google Scholar]
- 36.Vohra MM, Pradhan SN, Jain PC, Chatterjee SK, Anand N. Synthesis and Structure-Activity Relationships of Some Aminopyridines, Imidazopyridines, and Triazolopyridines. J Med Chem. 1965;8:296–304. doi: 10.1021/jm00327a006. [DOI] [PubMed] [Google Scholar]
- 37.Singh S, Das G, Singh OV, Han H. Development of More Potent 4-Dimethylaminopyridine Analogues. Org Lett. 2007;9:401–404. doi: 10.1021/ol062712l. [DOI] [PubMed] [Google Scholar]
- 38.Simpson LL. Studies on the binding of botulinum toxin type A to the rat phrenic nerve-hemidiaphragm preparation. Neuropharmacology. 1974;13:683–691. doi: 10.1016/0028-3908(74)90014-8. [DOI] [PubMed] [Google Scholar]
- 39.Simpson L. Kinetic studies on the interaction between botulinum toxin type A and the cholinergic neuromuscular junction. J Pharmacol Exp Ther. 1980;212:16–21. [PubMed] [Google Scholar]
- 40.Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- 41.Claydon TW, Vaid M, Rezazadeh S, Kehl SJ, Fedida D. 4-Aminopyridine Prevents the Conformational Changes Associated with P/C-Type Inactivation in Shaker Channels. J Pharmacol Exp Ther. 2007;320:162–172. doi: 10.1124/jpet.106.110411. [DOI] [PubMed] [Google Scholar]
- 42.Kozminsky-Atias A, Somech E, Zilberberg N. Isolation of the first toxin from the scorpion Buthus occitanus israelis showing preference for Shaker potassium channels. FEBS Letters. 2007;581:2478–2484. doi: 10.1016/j.febslet.2007.04.065. [DOI] [PubMed] [Google Scholar]
- 43.Baud FJ, Megarbane B, Deye N. 4-Aminopyridine Derivatives for Calcium Channel Blocker Poisonings. Clin Toxicol. 2008;46:356. [Google Scholar]
- 44.Bever CT, Leslie J, Camenga DL, Panitch HS, Johnson KP. Preliminary trial of 3,4-diaminopyridine in patients with multiple sclerosis. Annals Neurol. 1990;27:421–427. doi: 10.1002/ana.410270411. [DOI] [PubMed] [Google Scholar]
- 45.Judge SIV, Bever CT. Potassium channel blockers in multiple sclerosis: Neuronal Kv channels and effects of symptomatic treatment. Pharmacol & Therapeutics. 2006;111:224–259. doi: 10.1016/j.pharmthera.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 46.Gabrielsson J, Weiner D. Pharmacokinetic and Pharmacodynamic Data Analysis: Concepts and Applications. 4. Swedish Pharmaceutical Press; Stockholm, Sweden: 2001. [Google Scholar]
- 47.Eubanks LM, Hixon MS, Jin W, Hong S, Clancy CM, Tepp WH, Baldwin MR, Malizio CJ, Goodnough MC, Barbieri JT, Johnson EA, Boger DL, Dickerson TJ, Janda KD. An in vitro and in vivo disconnect uncovered through high-throughput identification of botulinum neurotoxin A antagonists. Proc Natl Acad Sci USA. 2007;104:2602–2607. doi: 10.1073/pnas.0611213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.