

Abstract
Herpesviruses enter cells by membrane fusion either at the plasma membrane or in endosomes, depending on the cell type. Glycoprotein B (gB) is a conserved component of the multiprotein herpesvirus fusion machinery and functions as a fusion protein, with two internal fusion loops, FL1 and FL2. We determined the crystal structures of the ectodomains of two FL1 mutants of herpes simplex virus type 1 (HSV-1) gB to clarify whether their fusion-null phenotypes were due to global or local effects of the mutations on the structure of the gB ectodomain. Each mutant has a single point mutation of a hydrophobic residue in FL1 that eliminates the hydrophobic side chain. We found that neither mutation affected the conformation of FL1, although one mutation slightly altered the conformation of FL2, and we conclude that the fusion-null phenotype is due to the absence of a hydrophobic side chain at the mutated position. Because the ectodomains of the wild-type and the mutant forms of gB crystallized at both low and neutral pH, we were able to determine the effect of pH on gB conformation at the atomic level. For viruses that enter cells by endocytosis, the low pH of the endosome effects major conformational changes in their fusion proteins, thereby promoting fusion of the viral envelope with the endosomal membrane. We show here that upon exposure of gB to low pH, FL2 undergoes a major relocation, probably driven by protonation of a key histidine residue. Relocation of FL2, as well as additional small conformational changes in the gB ectodomain, helps explain previously noted changes in its antigenic and biochemical properties. However, no global pH-dependent changes in gB structure were detected in either the wild-type or the mutant forms of gB. Thus, low pH causes local conformational changes in gB that are very different from the large-scale fusogenic conformational changes in other viral fusion proteins. We propose that these conformational changes, albeit modest, play an important functional role during endocytic entry of HSV.
Herpes simplex virus type 1 (HSV-1) is the prototype of the diverse herpesvirus family that includes many notable human pathogens (35). In addition to the icosahedral capsid and the tegument that surround its double-stranded DNA genome, herpesviruses have an envelope—an outer lipid bilayer—bearing a number of surface glycoproteins. During infection, HSV-1 must fuse its envelope with a cellular membrane in order to deliver the capsid into a target host cell, thereby initiating infection. Among its viral glycoproteins, only glycoprotein B (gB), gD, gH, and gL are required for fusion (40). gD, a receptor-binding protein (7, 38), is found only in alphaherpesviruses. However, gB, gH, and gL are present in all herpesviruses and constitute their core fusion machinery (17). Of these three proteins, gB is the most highly conserved and is a class III viral fusion protein (3, 18), presumably directly involved in bringing the viral and host cell membranes together to enable their fusion. gH/gL does not resemble a viral fusion protein and, instead, probably regulates the fusogenic activity of gB (8).
Two fusion loops in gB, FL1 and FL2, are critical for its function. Mutations within the fusion loops, e.g., W174R or Y179S in FL1, abolish both the fusion and the virus complementation activities of gB (2, 15) and cause severe defects in nuclear egress (42). Moreover, the respective mutant ectodomains, unlike the wild-type (wt) ectodomain, are unable to associate with model membranes, e.g., liposomes (14). Given the dramatic effect of these single amino acid substitutions on gB function, we wanted to determine whether the mutations affected the conformation of the fusion loops, or perhaps, even the overall structure of gB, which could help explain the loss of function in these mutants.
We previously determined the crystal structure of the trypsin-cleaved ectodomain of HSV-1 gB, gB730 (18). Trypsin treatment was necessary to obtain well-diffracting crystals, but it removed ∼80 residues from the N terminus of the ectodomain and cleaved two internal loops, causing disorder in adjacent regions. Thus, we also wanted to locate regions of the gB ectodomain missing from the available structure.
To this end, we crystallized the intact, uncleaved ectodomains of wild-type gB730 and two gB mutants, W174R and Y179S, and determined their respective structures. Overall, the structures are similar to the previously determined structure of trypsin-cleaved gB730, although several previously unresolved regions can now be seen. Neither mutation significantly affects the conformation of the gB ectodomain, be it globally or locally within the fusion loops, and we conclude that the presence of bulky hydrophobic residues at positions 174 and 179 is necessary for proper function of the fusion loops.
Interestingly, the structures were obtained at more than one pH, which allowed us to investigate the effect of pH on gB structure. We found that low pH did not cause changes in the overall conformation of the gB ectodomain either in the crystals or in solution, as shown using electron microscopy and size exclusion chromatography. However, low pH significantly changed the conformation of FL2. Based on our results, we propose that upon exposure of gB to low pH, a key histidine residue in FL2 is protonated and expelled from an uncharged groove, thereby forcing the entire FL2 to relocate. We hypothesize that this conformational change plays a functional role during endosomal entry of HSV.
MATERIALS AND METHODS
Protein expression and purification.
Generation of the gB730 construct for recombinant baculovirus expression in insect cells was described previously (6, 18). This construct ends at residue 730, just prior to the hydrophobic membrane-proximal region. For improved expression in insect cells, the endogenous signal sequence was replaced with honeybee mellitin signal sequence, which is cleaved during protein maturation. The resulting protein contains residues 30 to 730 of the gB ectodomain, plus N-terminal residues DP due to cloning strategy (37). Protein was purified from supernatants of insect cells infected with recombinant baculovirus by using immunoaffinity and size exclusion chromatography, as described previously (6, 18). Construction of fusion loop mutants was described previously (14), and mutants W174R and Y179S were purified in the same way as wt gB730. For crystallization and biochemical experiments, gB samples in 10 mM Tris-HCl (pH 7.6), 100 mM NaCl, and 1 mM EDTA were concentrated to 3.5 to 4.5 mg/ml using Millipore Ultra-4 (molecular mass cutoff, 50 kDa).
Protein crystallization and data collection.
Crystals of wt or mutant gB730 were grown by vapor diffusion at room temperature in hanging drops with 2 μl of protein sample and 2 μl of reservoir solution. Crystals of the W174R mutant were grown using 7% PEG 4000, 0.5 M NaCl, and 0.1 M sodium citrate (pH 5.5) (W174R-acidic). Crystals of the Y179S mutant were grown using either 10% PEG 4000, 0.5 M NaCl, and 0.1 M sodium citrate (pH 5.5) (Y179S-acidic) or 10% PEG 4000, 0.2 M NaCl, and 0.1 M Tris-HCl (pH 8.5) (Y179S-basic). Crystals of wt gB730 were grown using 15% PEG 4000, 0.3 M NaCl, and 0.1 M sodium citrate (pH 5.5) (wt-gB-acidic). Hexagonal rods typically appeared after several days and grew to their final size over 2 to 3 weeks.
For data collection, crystals were transferred stepwise to the solution identical to the well solution plus 15 or 17% mesoerythritol, 0.1 M NaCl, and 1 mM EDTA and plunged into liquid N2. The data for the mutant crystals were collected at 100 K at X25 beamline at the National Synchrotron Light Source. The data for the wt-gB-acidic crystals were collected at 100 K at F1 beamline at the Cornell High Energy Synchrotron Source. All datasets were processed using HKL2000 (31) and indexed in space group P3 (Table 1) .
TABLE 1.
Data collection and refinement statisticsa
| Parameter | W174R-acidic | Y179S-basic | Y179S-acidic | wt-gB-acidic |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P3 | P3 | P3 | P3 |
| Cell dimensions (Å, °) | 117.09, 117.09, 321.38, 90, 90, 120 | 117.99, 117.99, 321.54, 90, 90, 120 | 117.76, 117.76, 318.56, 90, 90, 120 | 117.80, 117.80, 318.32, 90, 90, 120 |
| Wavelength (Å) | 0.9795 | 1.0809 | 1.0809 | 0.9186 |
| Resolution (Å) | 48.35-2.26 (2.30-2.26) | 48.69-2.76 (2.81-2.76) | 48.56-2.88 (2.98-2.88) | 50.37-2.80 (2.90-2.80) |
| Total reflections | 512,855 | 538,533 | 516,278 | 399,393 |
| Unique reflections | 227,148 (8,362) | 118,749 (5,373) | 111,383 (10,786) | 112,550 (8,016) |
| Completeness (%) | 98 (72) | 92 (83) | 100 (97) | 95 (68) |
| I/σI | 7.57 (1.06) | 12.94 (1.38) | 10.27 (1.04) | 8.98 (0.98) |
| Redundancy | 2.3 (1.7) | 4.5 (3.8) | 4.6 (3.5) | 3.5 (2.7) |
| Rmerge | 0.12 (0.732) | 0.115 (0.907) | 0.132 (1.000) | 0.125 (0.995) |
| Refinement | ||||
| Resolution range (Å) | 47.3-2.26 | 48.7-2.76 | 48.6-2.88 | 41.535-3.00 |
| Rwork/Rfree | 0.1724/0.2270 | 0.2015/0.2425 | 0.2381/0.2829 | 0.1972/0.2492 |
| No. of atoms | ||||
| Protein | 19,578 | 19,454 | 19,205 | 19,116 |
| Ligand/ion | 88 | 295 | 192 | 112 |
| Solvent | 1,803 | 209 | 84 | 46 |
| B-factors | ||||
| Protein | 37.34 | 59.21 | 86.19 | 64.63 |
| Ligand/ion | 55.05 | 95.79 | 98.15 | 88.81 |
| Solvent | 34.43 | 28.11 | 39.53 | 24.80 |
| RMSD | ||||
| Bond length (Å) | 0.007 | 0.003 | 0.002 | 0.003 |
| Bond angle (°) | 1.004 | 0.601 | 0.493 | 0.658 |
| Ramachandran plot (%) | ||||
| Most favored region | 93.62 | 93.57 | 92.92 | 92.62 |
| Additionally allowed region | 6.17 | 6.18 | 6.91 | 7.30 |
| Disallowed region | 0.21 | 0.25 | 0.17 | 0.08 |
Values in parentheses are for the highest-resolution shell.
Structure phasing and refinement.
Initially, we focused on the W174R-acidic data set because it had the highest resolution. Molecular replacement using Phaser (25) and the previously determined structure of trypsin-cleaved gB730 (PDB 2GUM) (18), which we refer to as wt-gB-neutral, located four protomers of gB, henceforth named chain A, chain B, chain C, and chain D. Each protomer belongs to one of four crystallographic trimers, generated by a 3-fold crystallographic symmetry operation (see Fig. S1 in the supplemental material). The MR solution was used as a starting model for refinement in phenix.refine (1) using data from 47.3 to 2.26 Å resolution.
Prior to refinement, 5% of the data were set aside for cross-validation. The model was refined in phenix.refine (1) with iterative rounds of rebuilding in Coot (12). Refinement included rigid-body refinement, gradient minimization, torsion-angle simulated annealing, and individual atomic displacement parameter refinement. Analysis using phenix.xtriage (1) detected merohedral twinning, and the twin law (h, -h-k, -l) was applied throughout refinement.
Refinement resulted in difference maps of excellent quality that clearly showed the locations of residues missing from the previously determined structure of wt-gB-neutral (PDB 2GUM). The density in the vicinity of FL2, residues 257 to 264, was weak, but extra density was visible nearby. To clarify the location of the polypeptide chain in this area, residues 174 to 180 and residues 255 to 265 were omitted, and the model was subjected to a single round of simulated annealing refinement. The omit maps clearly showed that in the W174R-acidic structure, FL1, residues 174 to 180, adopted the same overall conformation as in the wt-gB-neutral structure, whereas FL2, residues 257 to 264, had a different conformation. The omit density was clear enough to build the new conformation for residues 257 to 264 (see Fig. S2A in the supplemental material).
Prior to refinement of the Y179S-acidic, Y179S-basic, and wt-gB-acidic structures, test set flags were transferred from the W174R-acidic data set to these datasets. The molecular replacement solution for W174R-acidic was used as a starting model for refinement of the other three structures. Models were refined using phenix.refine (1) with iterative rounds of rebuilding in Coot (12) essentially in the same way as was done for W174R-acidic structure except that group atomic displacement parameter refinement was used due to lower resolution of Y179S-acidic, Y179S-basic, and wt-gB-acidic data. The omit density was used to check and, when necessary, rebuild the conformation of residues 257 to 264 (see Fig. S2B to D in the supplemental material). Molprobity (10) was used to assess the stereochemical quality of all models. Final statistics are listed in Table 1. The models are missing some N-terminal and C-terminal residues as well as residues in a single linker region (see Table S1 in the supplemental material). Some models contain carbohydrate moieties (N-acetylglucosamine) modifying residues N141, N398, N430, or N674, and mesoerythritol, a cryoprotectant.
All structure figures were created by using Pymol (http://www.pymol.org/). The pKa of H263 was calculated by using PROPKA 2.0 (4, 23).
EM.
Protein was incubated on electron microscopy (EM) grids and stained with a 0.75% solution of uranyl formate (pH ∼4.5). EM images were taken at room temperature by using a Tecnai G2 Spirit BioTWIN equipped with an AMT 2k charge-coupled device camera microscope at the Harvard Medical School EM core facility.
Size exclusion chromatography.
Size exclusion chromatography was performed on a Biologic Duoflow chromatography system (Bio-Rad) equipped with a Superdex S200 column (GE Healthcare) equilibrated with either 0.1 M Tris-HCl (pH 7.6) or 0.1 M sodium citrate (pH 3.0), plus 0.1 M NaCl. The Superdex S200 column was calibrated using thyroglobulin (670 kDa), ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), and ovalbumin (44 kDa); blue dextran was used to determine the void volume (GE Healthcare).
Analysis of pH effects on gB by SDS-PAGE and Western blots.
Concentrated protein samples were diluted into 0.1 M buffer at the indicated pH—CAPS (N-cyclohexyl-3-aminopropanesulfonic acid; pH 10.5), bicine (pH 9.0), Tris (pH 8.0 or 8.5), HEPES (pH 7.0), MES (morpholineethanesulfonic acid; pH 6.0), citrate (pH 5.5, 5.0, or 3.0), or acetate (pH 4.0)—and incubated at 25°C for 30 min. Where indicated, acidified samples were neutralized by adding a precalculated volume of 1 N NaOH. For SDS-PAGE analysis of the pH-treated wt gB730, all samples were prepared under mildly denaturing conditions (nonreducing, unboiled samples containing 0.1% SDS) to preserve the trimeric species of gB730 (9). For Coomassie blue staining, 4 μg of protein was used per lane. For Western blot analysis with monoclonal antibody (MAb) DL16, 1.2 μg of protein was used per lane. Samples were then resolved by 4 to 15% Tris-SDS-PAGE (Bio-Rad) and either stained with Coomassie blue or transferred to a nitrocellulose membrane. For Western blotting, membranes were blocked with 5% skimmed milk in Tris-buffered saline with 0.05% Tween and then probed with a trimer-specific MAb DL16 at a 1:1,000 dilution. This was followed by incubation with anti-mouse IgG-horseradish peroxidase conjugate at a 1:10,000 dilution. The blots were developed using a Pierce Western Pico chemiluminescence kit.
Accession numbers.
Atomic coordinates and structure factors have been deposited to the RCSB Protein Data Bank under accession numbers 3NWA (W174R-acidic), 3NWF (wt-gB-acidic), 3NWD (Y179S-acidic), and 3NW8 (Y179S-basic).
RESULTS
Structure determination.
wt and mutant gB730 proteins crystallized in PEG 4000 in the presence of 0 to 0.5 M NaCl at a pH ranging from 5.5 to 8.5. However, conditions that yielded diffraction-quality crystals varied depending on the mutation. Whereas wt gB730 and both mutants, W174R and Y179S, yielded diffraction-quality crystals at pH 5.5, only the Y179S mutant yielded diffraction-quality crystals at pH 8.5. We refer to these crystals and the four corresponding structures as wt-gB-acidic, W174R-acidic, Y179S-acidic, and Y179S-basic. All crystals belong to space group P3. W174R-acidic crystals diffracted to the highest resolution, 2.26 Å, and thus were analyzed first (see the supplemental material). The W174R-acidic structure was solved by using molecular replacement (25) and chain A of the trypsin-cleaved gB730 structure (PDB 2GUM), determined previously (18), as a search model. The W174R-acidic structure was used as a starting model for refinement of the other three structures. All refined structures have excellent geometry (10) (Table 1).
Overall structures.
The original structure of trypsin-cleaved wt gB730 (PDB 2GUM) (18) was obtained at neutral pH and will be referred to as wt-gB-neutral. Due to trypsin cleavage used in sample preparation, residues 28 to 102 were absent from the crystallized protein sample, while residues 103 to 108, 331 to 336, 462 to 490, and 726 to 730 were disordered in the structure. In contrast, all four structures reported here were obtained from the uncleaved gB ectodomain (residues 28 to 730), and thus we were able to locate some previously unresolved residues, i.e., residues 103 to 108, 331 to 336, and 462 to 477 (Fig. 1A; see also Table S1 in the supplemental material). N-terminal residues 103 to 108 pack into a groove in domain IV such that residue 103 ends up at the top of the molecule (Fig. 1A). Loop 331 to 336 is now completely ordered and packs against a side of domain I, to which it belongs. Residues 462 to 477 form a helix αX in domain II. This domain is structurally homologous to pleckstrin homology domains (18), all of which contain a similarly located helix (22). Knowing the location of residues 462 to 477 allowed us to visualize the epitope of the neutralizing MAb H1781 within an important functional region (5, 32) (Fig. 1B). Thus, the structures of the uncleaved wt and mutant gB730 proteins, especially, the high-resolution W174R-acidic structure, provide a more complete view of the gB ectodomain.
FIG. 1.

(A) W174R-acidic structure (chain A). A single protomer is colored by domain with domain I, pale blue; domain II, pale green; domain III, yellow; domain IV, orange; and domain V, red. Domains are labeled. New regions, unresolved in the previously determined wt-gB-neutral structure (PDB 2GUM), are shown in purple. Fusion loops FL1, residues 175 to 180, and FL2, residues 258 to 264, are shown in cyan and green, respectively. (B) The region of gB containing the now-visible epitope of the neutralizing MAb H1781, residues 454 to 473 (5), is shown in green. The view in panel B is shown in the same orientation as in panel A. (C) Superposition of the four structures determined here—Y179S-basic (chain C), W174R-acidic (chain B), Y179S-acidic (chain A), and wt-gB-acidic (chain A)—onto the previously determined wt-gB-neutral structure (chain A). The same chains are shown in all subsequent structure figures. Single protomers are shown for clarity. All superpositions were performed with Coot (12) using the entire wt-gB-neutral structure as reference molecule. For any pairwise superposition, the root mean square deviation (RMSD) over all Cα atoms is less than 1.25 Å (see Table S2 in the supplemental material). Relative to the colored protomer in panel A, the protomers in panel C are rotated 40° to the viewer's left around the vertical axis.
Comparison of each of the four new structures with the wt-gB-neutral structure revealed no major differences in their overall architecture (Fig. 1C). Given that the five available gB structures differ with respect to the presence of mutations, proteolytic cleavage, or pH used in crystallization, we conclude that none of these differences cause any global conformational changes in the structure of the gB ectodomain.
FL2 undergoes a pH-dependent conformational change.
Despite their overall similarity, the five gB structures analyzed here differ dramatically in the conformation of FL2, residues 257 to 264 (Fig. 2; see also Fig. S3A in the supplemental material). In contrast, FL1, residues 174 to 180, which contains mutations Y179S and W174R, maintains similar conformations in all structures (Fig. 2; see also Fig. S3B in the supplemental material). Specifically, FL2 has two main conformations: the inward and the outward. FL2 points inward, toward the 3-fold symmetry axis of the gB trimer, in the structure of wt-gB crystallized at neutral pH (wt-gB-neutral) and in the structure of Y179S obtained at basic pH (Y179S-basic) (Fig. 2). Thus, we refer to it as the neutral/inward conformation. In contrast, in all structures obtained under acidic conditions, FL2 points outward, assuming the acidic/outward conformation (Fig. 2). The neutral/inward and the acidic/outward conformations of FL2 are very different. Relative to the neutral/inward conformation, the positions of the individual Cα atoms in the acidic/outward conformation shift by up to 10.6 Å, with significant differences in side chain orientations (see Fig. S3A in the supplemental material). For example, the Cα atom of residue H263 shifts by as much as 8.9 Å between the wt-gB-neutral and wt-gB-acidic structures. The acidic/outward conformation is similar in the wt-gB-acidic and the Y179S-acidic structures but is slightly different in the W174R-acidic structure, as described in more detail below. Due to the higher resolution of the wt-gB-neutral and the Y179S-acidic structures, all detailed comparisons of the neutral/inward and the acidic/outward conformations were carried out with them.
FIG. 2.

Low-pH-dependent conformational change in fusion loop FL2. (A) Overlay of the fusion loop regions, in side view. For clarity, only single protomers are shown. The inward and the outward conformations of the FL2 are labeled. The color scheme is the same as in Fig. 1C. The curved arrow indicates low-pH-induced movement of FL2. W174 and Y179 on FL1 are indicated by red asterisks. Residues 261 to 262 are missing in the wt-gB-acidic structure, and the flanking residues 260 and 263 are connected with a black dotted line. (B) Surface view of the fusion loop region. The wt-gB-neutral structure, with residues 258 to 264 and the nitrogen of Y265 omitted, is shown as a white surface. FL2 in each of the five structures is shown as a Cα trace with H263 shown as sticks. All superpositions were performed on residues 185 to 250 using Coot (12) and wt-gB-neutral trimer (chain A) as a reference molecule.
Histidines are well known as pH-dependent switch residues in viral fusion proteins (13, 19, 20, 34). The typical pKa of the histidine side chain in proteins is 6.5 (39), making them sensitive to pH changes. Residue H263 is likely a “switch” residue that is key to pH-dependent conformational changes in FL2. In the neutral/inward conformation of FL2, the side chain of H263 rests in an uncharged groove, buttressed by van der Waals interactions with the side chains of W174 and Y265 (Fig. 3A and B). In this conformation, its estimated pKa is 3.5 (4, 23), which makes protonation unfavorable. Thus, we propose that the neutral/inward conformation of FL2 is stabilized by the van der Waals interactions involving the uncharged side chain of H263. Additional stabilization is provided by a hydrogen bond between the side chain of E260 in FL2 and the side chain of H177 in FL1 (Fig. 3A and B), and two main chain hydrogen bonds between FL1 and FL2, specifically, between residues F175 and A261 and residues V173 and R264.
FIG. 3.
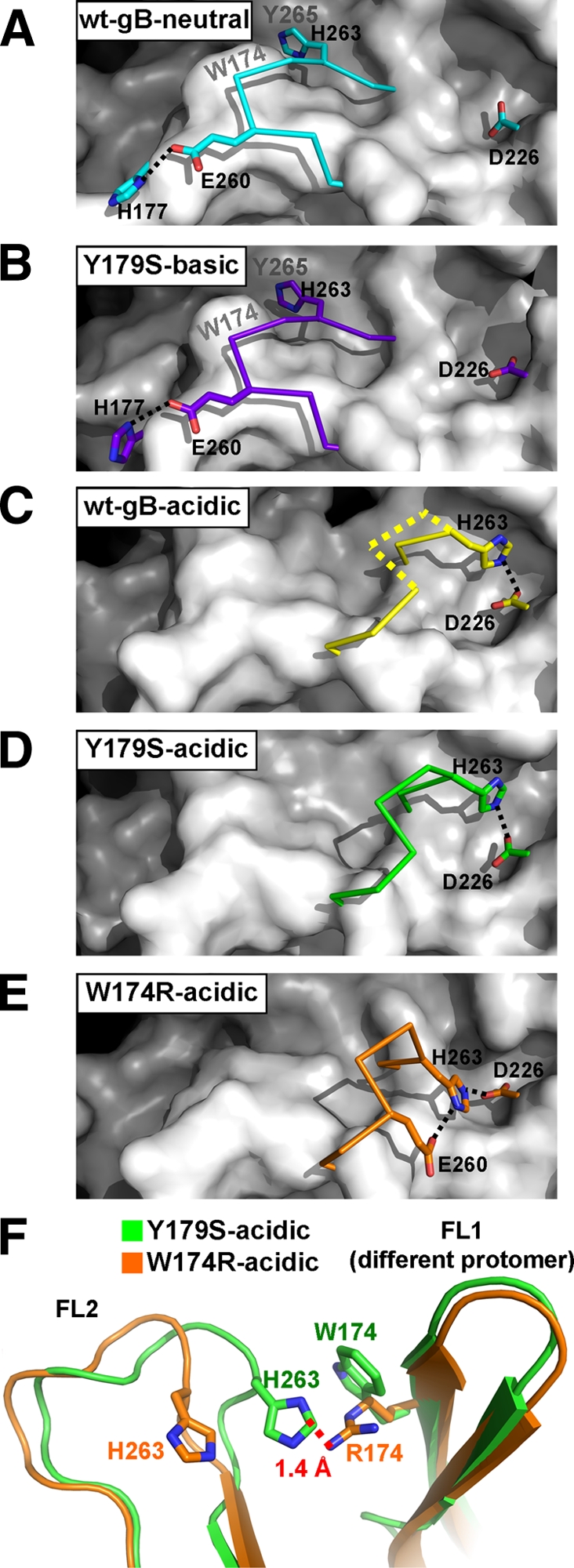
Interactions stabilizing the conformation of FL2. (A to E) FL2, residues 258 to 264, is shown as a Cα trace with important side chains shown as sticks. The color scheme is the same as in Fig. 1C and 2. wt-gB-acidic (C) is missing residues 261 to 262; their presumable Cα trace is shown as a dotted yellow line, based on their positions in Y179S-acidic. The gB trimer is shown as a white surface, with residues 258 to 264 and the nitrogen of Y265 omitted. wt-gB-neutral (A) and Y179S-basic (B) structures have FL2 in the inward conformation, whereas wt-gB-acidic (C), Y179S-acidic (D), and W174R-acidic (E) structures have FL2 in the outward conformation. Salt bridges are shown with black dotted lines. The locations of residues Y265 and W174, which form van der Waals interactions with the H263 side chain in the inward conformation of FL2, are labeled in panels A and B. All structures were superposed as described in Fig. 2, and the views in panels A to E are identical. (F) FL2 has distinct conformations in the Y179S-acidic and W174R-acidic structures. Side chains of H263, W174, and R174 are shown as sticks. A red dotted line shows the distance between the closest atoms of R174 from W174R-acidic and H263 from Y179S-acidic.
In contrast, in the acidic/outward conformation, the side chain of H263 rests in an acidic site, where it forms a salt bridge with the side chain of D226 (Fig. 3C to E). This electrostatic interaction is only possible if the H263 side chain is protonated. Since the estimated pKa of the H263 side chain in the acidic/outward conformation is ∼8.0, it is protonated at pH 5.5. In addition, the acidic/outward conformation of FL2 is stabilized by a main chain hydrogen bond between two FL2 residues, V259 and A261. Thus, the neutral/inward and the acidic/outward conformations are stabilized by different sets of interactions, and yet both involve H263. We hypothesize that the protonation of H263 under acidic conditions causes FL2 to relocate from its inward to its outward conformation.
Effect of mutations on the conformation of FL2.
The mutation Y179S, located in FL1, has little if any effect on the conformation of FL2. Specifically, the conformation of FL2 is nearly identical in the wt-gB-neutral and Y179S-basic structures (Fig. 2). Likewise, the conformation of FL2 is nearly identical in the wt-gB-acidic and Y179S-acidic structures (Fig. 2). Therefore, we attribute the difference in FL2 conformation between Y179S-acidic and Y179S-basic structures to the effect of pH.
In contrast, the W174R mutation does have a small effect on the conformation of FL2, beyond changes that can be accounted for by the effect of the pH. The outward conformation of FL2 in W174R-acidic structure is distinct from the acidic/outward conformation found in both the Y179S-acidic and the wt-gB-acidic structures (Fig. 2 and 3). In the W174R-acidic structure, in addition to a salt bridge between the side chains of H263 and D226, the outward conformation of FL2 is also stabilized by a second salt bridge between the side chains of H263 and E260 (Fig. 3E). This distinct conformation of the FL2 in the W174R-acidic structure appears to be caused by the W174R mutation. The arginine side chain at position 174 in the mutant is larger than the side chain of tryptophan in the wt protein (or in the Y179S mutant) and, therefore, R174 in one protomer likely repels the protonated side chain of H263 in a neighboring protomer, pushing H263, along with the rest of FL2, farther outward (Fig. 3F).
Neither the Y179S nor the W174R mutation significantly affects the conformations of FL1 and FL2, and even the small change in the conformation of FL2 in the W174R mutant cannot explain its nonfunctional phenotype. Thus, we conclude that bulky hydrophobic residues at positions 174 and 179 are necessary for proper function of gB fusion loops, and their removal causes the nonfunctional phenotype.
Two histidines in gB have altered conformations under low-pH conditions.
To determine whether pH subtly altered additional regions of gB, we examined all histidine residues. Apart from FL2 residue H263, only H308 was affected by pH. Under neutral or basic conditions, the side chain of H308 points slightly up and out (Fig. 4A), but it flips downward, toward the fusion loops, under acidic conditions (Fig. 4B). It was recently reported that exposure of gB to low pH altered the binding of a monoclonal antibody, H126 (11). The epitope for this antibody includes residue Y303, which is in the immediate vicinity of H308 (21). We propose that low pH alters the epitope of H126 by changing the conformation of the H308 side chain, thereby diminishing the binding of this antibody to gB.
FIG. 4.

Low-pH conformational changes affect MAb epitopes. Side-by-side surface views of domains I and V of the wt-gB-neutral (A) and W714R-acidic structures (B) (bottom panels) and an enlarged view of the H126 epitope in these structures (top panels). Regions are colored by epitope, and FL2 is colored orange. The side chain of H308 is shown as yellow sticks. The nearby Y303 side chain is shown in purple. Y303N is the MAb resistance mutant (mar) for the H126 neutralizing antibody. Views in panel A and B are shown in the same orientation, obtained by superposing the entire W174R-acidic trimer onto the wt-gB-neutral trimer.
pH does not affect the overall conformation of gB.
We used several approaches to determine whether the crystal structures of gB at either neutral or low pH accurately reflect its conformation in solution. First, we inspected gB730 at pH 7.6 or 3.0 using negative-stain electron microscopy. gB730 has a characteristic rod-like shape that can be easily discerned in EM images (36). Moreover, the two ends are readily distinguishable, namely, the “crown,” which corresponds to domain IV, and the “base,” which contains the fusion loops (36) (Fig. 1A). We observed no differences in the overall shape of gB730 under different pH conditions (Fig. 5A). Under acidic conditions, the length of the gB730 particles, 16.0 ± 0.8 nm (n = 6), was the same as under neutral conditions, 16.1 ± 0.8 nm (n = 7). This number correlated well with the length of the gB ectodomain calculated from its atomic coordinates, 16.7 nm (18). Therefore, the overall conformation of gB730 remains the same under acidic and neutral conditions. In addition, in size exclusion chromatography, gB730 had similar elution profiles at either pH 7.6 or 3.0, further confirming that pH had no effect on the overall size or conformation of the protein in solution (Fig. 5B).
FIG. 5.

Effect of low pH on the overall structure of the gB ectodomain. (A) Electron micrographs of wt gB730 at pH 7.6 (top) and pH 3.0 (bottom). The distinct “crown” and “base” ends are labeled with red arrows in two representative particles, as well as on the surface representation of wt-gB-neutral structure. (B) An overlay of the size exclusion chromatograms for wt gB730 at pH 7.6 (blue) or 3.0 (red) shows that the protein elutes in the same volume at either pH. (C and D) Effect of pH on the electrophoretic mobility of gB730. Purified protein was incubated at the indicated pH and analyzed by SDS-PAGE (0.1% SDS). Samples labeled with “N” were neutralized to pH 7.5 prior to loading. Identical protein amounts were loaded into each lane. ***, Trimer; *, monomer. (C) Coomassie blue-stained gel. (D) Western blot using trimer-specific MAb DL16.
We next tested whether pH had an effect on the electrophoretic mobility of gB730. We incubated gB730 at different pH and tested its oligomeric state using mildly denaturing SDS-PAGE (9) and either Coomassie blue stain (Fig. 5C) or Western blotting with a trimer-specific MAb DL16 (Fig. 5D). We found that as the pH decreased, the band corresponding to the gB730 trimer disappeared, and a band corresponding to the monomer appeared (Fig. 5C). This effect was reversible because after neutralization of low-pH-treated gB730, the trimeric band reappeared. A similar result was also observed with both the Y179S and the W174R mutant proteins (data not shown). This apparent dissociation could only be detected by SDS-PAGE but not in solution. We hypothesize that when gB is incubated at low pH, the interprotomer contacts within the gB trimers are weakened. These weakened trimers remain intact in the absence of detergent, be it in solution or on an EM grid, but dissociate in the presence of detergent (Fig. 5C and D). The fact that the low-pH effect is reversible, as revealed by SDS-PAGE, further suggests that the trimer does not dissociate in solution under acidic conditions.
DISCUSSION
Effect of mutations on the gB conformation.
Initially, we wanted to determine whether the fusion-null phenotype of mutants Y179S and W174R was due to the effect of these mutations on the structure of the gB ectodomain. We determined two structures for the Y179S mutant and one structure for the W174R mutant and compared them to the structures of wt gB. Surprisingly, we found that these mutations, both in FL1, did not change either the overall conformation of gB or the conformation of FL1. Further, the Y179S mutation had no effect on the conformation of FL2 at either acidic or basic pH. The W174R mutation had a small effect on FL2 under acidic pH, however, which probably resulted from steric and electrostatic repulsion caused by arginine in position 174. On the basis of our structural analysis, we conclude that the fusion-null phenotypes of the W174R and Y179S mutants, along with their inability to associate with liposomes, are not due to large changes in conformation of either fusion loop. Rather, it is more likely that the presence of bulky aromatic residues at positions 174 and 179 is necessary for the proper function of FL1, most likely membrane insertion, by analogy with fusion loops of other viral fusion proteins (27, 33). Other bulky hydrophobic residues in these positions would be predicted to function, and indeed, the W174Y mutant is able to mediate cell-cell fusion, albeit at a 2-fold lower level (15).
pH-dependent conformational changes in gB.
The wt and mutant ectodomains crystallized at acidic, as well as at neutral (or basic) pH, which allowed us to investigate the effect of pH on the structure of the gB ectodomain. We found that pH causes a significant conformational change within the second fusion loop, FL2. This loop adopts two different conformations depending on pH, the inward conformation under neutral or basic pH, or the outward conformation under acidic pH (Fig. 2). We propose that the relocation of FL2 from its inward to its outward conformation is mediated by the protonation and expulsion of residue H263 (Fig. 3; see also Fig. S4 in the supplemental material).
How does FL2 change its conformation upon exposure to low pH? We envision the following sequence of events. At pH 7.5, H263 is uncharged but becomes protonated upon exposure to low pH (Fig. 3A). The estimated pKa of the H263 side chain in the neutral/inward conformation is 3.5, so at pH 5.5, only 1% of the H263 side chains would be protonated. However, the presence of a protonated histidine side chain within an uncharged groove flanked by aromatic residues is unfavorable. So, the side chain of H263 would be forced to move out of the groove into the solvent (see Fig. S4A in the supplemental material), pulling the entire FL2 with it. This, in turn, would displace E260 and break its hydrogen bond with H177 (see Fig. S4B in the supplemental material). Once the H263 side chain is exposed to the solvent, its pKa would rise to a more typical value for histidines, probably ∼6.5. This rise in pKa would ensure that H263 stays protonated at pH 5.5. The mobility of an unanchored, displaced FL2 would allow the now positively charged H263 side chain to move into the acidic docking site, which contains the D226 side chain (see Fig. S4C and E in the supplemental material). This acidic site is exposed and unoccupied under neutral conditions, making it available for binding (Fig. 3A; see also Fig. S4D in the supplemental material). Once H263 forms the salt bridge with D226, FL2 will be locked in place in the acidic/outward conformation (Fig. 3D; see also Fig. S4E in the supplemental material). Thus, in summary, we propose that H263 acts as a pH “switch” residue in gB.
Effect of pH on the biochemical properties of gB.
The ectodomain of gB does not undergo any global pH-dependent changes either in crystals or in solution, as seen by EM and size exclusion chromatography. Nevertheless, low-pH-treated gB has a faster electrophoretic mobility in SDS-PAGE, migrating as a monomer as opposed to a trimer at neutral pH. These results are in agreement with previous results obtained for soluble HSV-2 gB (s-gB, a form lacking only the transmembrane region) (11), but our explanation for these observations is different. Based on the pH-induced changes in electrophoretic mobility, Dollery et al. concluded that s-gB dissociates at low pH. However, in our hands, this dissociation can only be detected when gB is subjected to PAGE in the presence of SDS and does not occur in solution or in the crystal structures. We suggest that at low pH, the interprotomer contacts within the gB trimer are weakened. Trimers remain intact when the protein is under detergent-free conditions, in solution or on an EM grid or in crystals, but dissociate when the protein is incubated with detergent, as seen for both s-gB (11) and gB730 (Fig. 5). Given the extensive trimerization interface in the gB ectodomain, complete dissociation and reassociation of the trimer is unlikely. Moreover, the fact that the low pH effect seen by SDS-PAGE is reversible yields further evidence that the trimer never completely dissociates in solution.
Second, it was suggested that gB undergoes significant conformational changes at low pH based on diminished reactivity of several neutralizing MAbs (11). Our results provide a likely explanation for this effect. Epitopes of three of these antibodies are located in the vicinity of the fusion loops, within domain I (SS55) or the C terminus of domain V (SS106 and SS144) (5). The low-pH-induced conformational change in FL2 likely affects the epitopes of these antibodies (Fig. 4), reducing antibody affinity. Although the epitope of another neutralizing antibody, H126, which includes residue Y303 (21), is located farther away from the fusion loops, it, too, could be affected by the change in FL2 conformation. However, a better explanation for the reduced reactivity of H126 is the conformational change in the H308 side chain (Fig. 4). H308 is located near Y303, and this change, albeit small, could alter the H126 epitope enough to reduce H126 binding under acidic conditions, especially considering that a single point mutation Y303N is sufficient to convey resistance to neutralization of virus by H126 (21). Thus, relatively small pH-induced conformational changes in gB account well for the decrease in antibody reactivity at low pH.
Finally, it was observed that at low pH, s-gB partitioned into the detergent phase of Triton X114, suggesting an increase in the overall hydrophobicity of s-gB (11). However, none of the three low-pH gB structures show any noticeable alteration in surface hydrophobicity (see Fig. S5 in the supplemental material). We suggest that low pH weakens interprotomer contacts within gB trimers, causing them to dissociate in the presence of detergent. This dissociation would expose previously buried hydrophobic interfaces, resulting in detergent partitioning. As a nonionic detergent, Triton X-114 is a milder denaturant than ionic detergents such as SDS. However, with a critical micelle concentration (CMC) of 0.011% in water, 2% Triton X114 would probably cause dissociation of a number of trimers, already partly destabilized by low pH. Indeed, approximately one-third of the gB was detected in the detergent phase (11). Moreover, previous data suggest that gB trimers dissociate under conditions that are far from being denaturing. For example, when gB is in the presence of 0.2% SDS on SDS-PAGE, some trimers dissociate, but many conformational epitopes remain intact (5). Thus, just like the increased electrophoretic mobility, the apparent increase in hydrophobicity is probably due to detergent-induced trimer dissociation rather than to any overall conformational change to gB.
Role of the pH-dependent conformational changes.
The role of pH in herpesvirus entry is of considerable interest given that herpesviruses enter a number of cells by endocytosis (26, 28, 29), in addition to entering some cells at the plasma membrane (41). Endosomal entry of HSV requires four entry glycoproteins—gB, gD, and the gH/gL heterodimer—as well as the low pH of endosomes (30). The observed conformational changes in gB occur at pH 5.5, which corresponds to the pH of late endosomes (24) and thus may be relevant for endosomal entry.
The effect of pH on viral fusion proteins has been well documented in different viruses that enter cells by endocytosis. In these viruses, the low pH of the endosomes triggers fusogenic changes within a viral fusion protein. Notable examples include influenza virus HA and flavivirus glycoprotein E, both of which undergo large conformational changes induced by low pH (16). These conformational changes are typically irreversible. Interestingly, reversible pH-dependent conformational changes characterize the two other class III fusion proteins: glycoprotein G from vesicular stomatitis virus and the baculovirus gp64 (3). In all of these cases, the initial prefusion conformation is destabilized at low pH and converts into the more stable postfusion conformation. These large conformational changes in the viral fusion proteins are thought to be the driving force behind membrane fusion (16).
In contrast, the pH-dependent conformational changes observed in HSV-1 gB are small and clearly different from the fusogenic conformational changes that characterize other viral fusion proteins. It is tempting to speculate that the conformational change in FL2 is needed during endosomal entry. Recently, it was shown that soluble gH/gL associates with liposomes only in the presence of gB730 and low pH (T. M. Cairns, J. C. Whitbeck, M. Samanta, H. Lou, E. E. Heldwein, R. J. Eisenberg, and G. H. Cohen, unpublished data). Having FL2 in the acidic/outward conformation may be necessary for this interaction, which could help explain why HSV requires low pH during endosomal entry. Future studies are necessary to test this hypothesis. For example, endosomal entry can be prevented by using lysosomotropic agents (29), and it would be interesting to test whether such agents can also prevent the gB-gH/gL interaction during endosomal entry. Further, our analysis suggests that protonation of H263 mediates the pH-dependent conformational change in FL2. Although an H263A mutation inhibits fusion by only 50% (14), suggesting that H263 is not critical for cell-cell fusion, whether the H263A mutation affects endosomal entry has not yet been tested.
All of the crystal structures analyzed here most likely represent the postfusion conformation of gB. What is the possible function of the pH-dependent conformational changes in FL2 in the postfusion structure? The exposure of gB to acidic conditions in endosomes probably occurs prior to fusion, so acidic pH is more likely to affect gB in its prefusion form. Although the structure of the prefusion form is currently unknown, FL2 could, in principle, undergo similar changes in both the prefusion and the postfusion forms. In fact, a mild acid pretreatment (pH 5.5), can irreversibly impair infection by 50%, independent of the entry route (29). So, impaired entry following acid pretreatment of virions could be due to a conformational change in FL2 even in the prefusion gB structure. If the conformational change in FL2 under acidic conditions indeed promotes the interaction between gB and gH/gL, acid pretreatment could prematurely induce this interaction. In that case, in the absence of gD/receptor interaction, gH/gL could then, perhaps, act as a “fusion trigger” by binding gB, prematurely activating it, and converting it irreversibly to its postfusion conformation. In this manner, acidic pretreatment of virions could prevent viral entry regardless of the entry route. Nevertheless, we cannot exclude the formal possibility that low pH may cause other, perhaps, larger conformational changes.
pH-dependent conformational changes in gB, be they limited to FL2 or involving larger regions, cannot be required in all viral entry routes. Certainly, they could not be triggered by low pH during entry at the plasma membrane or during cell-cell spread. However, if such conformational changes accompany the transition of gB from its prefusion form to its postfusion conformation, then a yet-unknown viral or host accessory protein would be necessary to trigger the same conformational changes during plasma membrane entry.
Although the precise role of pH during endocytic entry of herpesviruses has not yet been fully characterized, understanding it is critical for revealing the complex mechanism of herpesvirus cell entry. Our work serves as an important step toward this goal by providing the structural basis for low-pH induced conformational changes in gB, the herpesvirus fusion protein.
Supplementary Material
Acknowledgments
We thank A. Héroux of the National Synchrotron Light Source for collecting X-ray diffraction data on crystals of mutant gB proteins and the beamline scientists at CHESS beamline F1 for help with data collection on wt-gB-acidic crystals. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under contract DE-AC02-98CH10886. We also thank M. Eriksson at the HMS EM Core Facility for help with sample preparation and imaging and E. Vogan for helpful discussions during structure determination.
This study was funded by the NIH grant 1DP20D001996 and by the Pew Scholars Program in Biomedical Sciences (E.E.H.) and by NIH grants AI18289 (G.H.C.), AI056045 (R.J.E.), and AI076231 (R.J.E.).
H.L. expressed and purified wt and mutant gB730 proteins, S.D.S. and E.E.H. carried out all steps of structure determination and analysis, E.E.H. conducted biochemical experiments, and S.D.S., G.H.C., R.J.E., and E.E.H. wrote the manuscript.
Footnotes
Published ahead of print on 13 October 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Adams, P. D., R. W. Grosse-Kunstleve, L. W. Hung, T. R. Ioerger, A. J. McCoy, N. W. Moriarty, R. J. Read, J. C. Sacchettini, N. K. Sauter, and T. C. Terwilliger. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58:1948-1954. [DOI] [PubMed] [Google Scholar]
- 2.Atanasiu, D., J. C. Whitbeck, M. P. de Leon, H. Lou, B. P. Hannah, G. H. Cohen, and R. J. Eisenberg. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J. Virol. 84:3825-3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Backovic, M., and T. S. Jardetzky. 2009. Class III viral membrane fusion proteins. Curr. Opin. Struct. Biol. 19:189-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bas, D. C., D. M. Rogers, and J. H. Jensen. 2008. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins 73:765-783. [DOI] [PubMed] [Google Scholar]
- 5.Bender, F. C., M. Samanta, E. E. Heldwein, M. P. de Leon, E. Bilman, H. Lou, J. C. Whitbeck, R. J. Eisenberg, and G. H. Cohen. 2007. Antigenic and mutational analyses of herpes simplex virus glycoprotein B reveal four functional regions. J. Virol. 81:3827-3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bender, F. C., J. C. Whitbeck, M. Ponce de Leon, H. Lou, R. J. Eisenberg, and G. H. Cohen. 2003. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 77:9542-9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carfi, A., S. H. Willis, J. C. Whitbeck, C. Krummenacher, G. H. Cohen, R. J. Eisenberg, and D. C. Wiley. 2001. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell 8:169-179. [DOI] [PubMed] [Google Scholar]
- 8.Chowdary, T. K., T. M. Cairns, D. Atanasiu, G. H. Cohen, R. J. Eisenberg, and E. E. Heldwein. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 17:882-888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen, G. H., V. J. Isola, J. Kuhns, P. W. Berman, and R. J. Eisenberg. 1986. Localization of discontinuous epitopes of herpes simplex virus glycoprotein D: use of a nondenaturing (“native” gel) system of polyacrylamide gel electrophoresis coupled with Western blotting. J. Virol. 60:157-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis, I. W., A. Leaver-Fay, V. B. Chen, J. N. Block, G. J. Kapral, X. Wang, L. W. Murray, W. B. Arendall III, J. Snoeyink, J. S. Richardson, and D. C. Richardson. 2007. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35:W375-W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dollery, S. J., M. G. Delboy, and A. V. Nicola. 2010. Low pH-induced conformational change in herpes simplex virus glycoprotein B. J. Virol. 84:3759-3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126-2132. [DOI] [PubMed] [Google Scholar]
- 13.Fritz, R., K. Stiasny, and F. X. Heinz. 2008. Identification of specific histidines as pH sensors in flavivirus membrane fusion. J. Cell Biol. 183:353-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hannah, B. P., T. M. Cairns, F. C. Bender, J. C. Whitbeck, H. Lou, R. J. Eisenberg, and G. H. Cohen. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 83:6825-6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hannah, B. P., E. E. Heldwein, F. C. Bender, G. H. Cohen, and R. J. Eisenberg. 2007. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 81:4858-4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrison, S. C. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heldwein, E. E., and C. Krummenacher. 2008. Entry of herpesviruses into mammalian cells. Cell Mol. Life Sci. 65:1653-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heldwein, E. E., H. Lou, F. C. Bender, G. H. Cohen, R. J. Eisenberg, and S. C. Harrison. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217-220. [DOI] [PubMed] [Google Scholar]
- 19.Kadlec, J., S. Loureiro, N. G. Abrescia, D. I. Stuart, and I. M. Jones. 2008. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 15:1024-1030. [DOI] [PubMed] [Google Scholar]
- 20.Kampmann, T., D. S. Mueller, A. E. Mark, P. R. Young, and B. Kobe. 2006. The role of histidine residues in low-pH-mediated viral membrane fusion. Structure 14:1481-1487. [DOI] [PubMed] [Google Scholar]
- 21.Kousoulas, K. G., B. Huo, and L. Pereira. 1988. Antibody-resistant mutations in cross-reactive and type-specific epitopes of herpes simplex virus 1 glycoprotein B map in separate domains. Virology 166:423-431. [DOI] [PubMed] [Google Scholar]
- 22.Lemmon, M. A., and K. M. Ferguson. 2000. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem. J. 350(Pt. 1):1-18. [PMC free article] [PubMed] [Google Scholar]
- 23.Li, H., A. D. Robertson, and J. H. Jensen. 2005. Very fast empirical prediction and rationalization of protein pKa values. Proteins 61:704-721. [DOI] [PubMed] [Google Scholar]
- 24.Maxfield, F. R., and T. E. McGraw. 2004. Endocytic recycling. Nat. Rev. Mol. Cell. Biol. 5:121-132. [DOI] [PubMed] [Google Scholar]
- 25.McCoy, A. J., R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni, and R. J. Read. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milne, R. S., A. V. Nicola, J. C. Whitbeck, R. J. Eisenberg, and G. H. Cohen. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 79:6655-6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Modis, Y., S. Ogata, D. Clements, and S. C. Harrison. 2004. Structure of the dengue virus envelope protein after membrane fusion. Nature 427:313-319. [DOI] [PubMed] [Google Scholar]
- 28.Nicola, A. V., J. Hou, E. O. Major, and S. E. Straus. 2005. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 79:7609-7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicola, A. V., A. M. McEvoy, and S. E. Straus. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77:5324-5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicola, A. V., and S. E. Straus. 2004. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J. Virol. 78:7508-7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307-326. [DOI] [PubMed] [Google Scholar]
- 32.Qadri, I., C. Gimeno, D. Navarro, and L. Pereira. 1991. Mutations in conformation-dependent domains of herpes simplex virus 1 glycoprotein B affect the antigenic properties, dimerization, and transport of the molecule. Virology 180:135-152. [DOI] [PubMed] [Google Scholar]
- 33.Roche, S., S. Bressanelli, F. A. Rey, and Y. Gaudin. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187-191. [DOI] [PubMed] [Google Scholar]
- 34.Roche, S., F. A. Rey, Y. Gaudin, and S. Bressanelli. 2007. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 315:843-848. [DOI] [PubMed] [Google Scholar]
- 35.Roizman, B., and P. E. Pellett. 2001. The family Herpesviridae: a brief introduction, p. 2381-2398. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott/The Williams & Wilkins Co., Philadelphia, PA.
- 36.Silverman, J. L., S. Sharma, T. M. Cairns, and E. E. Heldwein. 2010. Fusion-deficient insertion mutants of herpes simplex virus type 1 glycoprotein B adopt the trimeric postfusion conformation. J. Virol. 84:2001-2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sisk, W. P., J. D. Bradley, R. J. Leipold, A. M. Stoltzfus, M. Ponce de Leon, M. Hilf, C. Peng, G. H. Cohen, and R. J. Eisenberg. 1994. High-level expression and purification of secreted forms of herpes simplex virus type 1 glycoprotein gD synthesized by baculovirus-infected insect cells. J. Virol. 68:766-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spear, P. G., R. J. Eisenberg, and G. H. Cohen. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275:1-8. [DOI] [PubMed] [Google Scholar]
- 39.Stryer, L. 1988. Biochemistry, 3rd ed. W. H. Freeman & Company, New York, NY.
- 40.Turner, A., B. Bruun, T. Minson, and H. Browne. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 72:873-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wittels, M., and P. G. Spear. 1991. Penetration of cells by herpes simplex virus does not require a low pH-dependent endocytic pathway. Virus Res. 18:271-290. [DOI] [PubMed] [Google Scholar]
- 42.Wright, C. C., T. W. Wisner, B. P. Hannah, R. J. Eisenberg, G. H. Cohen, and D. C. Johnson. 2009. Fusion between perinuclear virions and the outer nuclear membrane requires the fusogenic activity of herpes simplex virus gB. J. Virol. 83:11847-11856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.