

Abstract
BACKGROUND AND PURPOSE
Vasculopathies represent the main cause of morbidity and mortality in diabetes. Vascular malfunctioning in diabetes is associated with abnormal vasoconstriction and Ca2+ handling by smooth muscle cells (SMC). Phosphatidylinositol 3-kinases (PI3K) are key mediators of insulin action and have been shown to modulate the function of voltage-dependent L-type Ca2+ channels (CaV1.2). In the present work, we investigated the involvement of PI3K signalling in regulating Ca2+ current through CaV1.2 (ICa,L) and vascular dysfunction in a mouse model of type I diabetes.
EXPERIMENTAL APPROACH
Changes in isometric tension were recorded on myograph. Ca2+ currents in freshly dissociated mice aortic SMCs were measured using the whole-cell patch-clamp technique. Antisense techniques were used to knock-down the PI3Kδ isoform.
KEY RESULTS
Contractile responses to phenylephrine and KCl were strongly enhanced in diabetic aorta independent of a functional endothelium. The magnitude of phenylephrine-induced ICa,L was also greatly augmented. PI3Kδ expression, but not PI3Kα, PI3Kβ, PI3Kγ, was increased in diabetic aortas and treatment of vessels with a selective PI3Kδ inhibitor normalized ICa,L and contractile response of diabetic vessels. Moreover, knock-down of PI3Kδin vivo decreased PI3Kδ expression and normalized ICa,L and contractile response of diabetic vessels ex vivo.
CONCLUSIONS AND IMPLICATIONS
Phosphatidylinositol 3-kinase δ was essential to the increased vascular contractile response in our model of type I diabetes. PI3Kδ signalling was up-regulated and most likely accounted for the increased ICa,L, leading to increased vascular contractility. Blockade of PI3Kδ may represent a novel therapeutic approach to treat vascular dysfunction in diabetic patients.
LINKED ARTICLE
This article is commented on by Sturek, pp. 1455–1457 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2010.00997.x
Keywords: diabetes, vascular dysfunction, PI3Kδ, L-type Ca2+ channels, Ca2+ currents
Introduction
Abnormal vasoconstriction plays an important role in the pathophysiology of diabetes (Piercy and Taylor, 1998; Mayhan et al., 1999; Lagaud et al., 2001; Okon et al., 2003) and greatly contributes to increased morbidity and mortality in patients with this disease. The contraction of vascular smooth muscle cells (VSMC) is tightly coupled to increases in the intracellular calcium concentration ([Ca2+]i). In smooth muscle, voltage-dependent L-type Ca2+ channels (CaV1.2; nomenclature follows Alexander et al., 2009) represent the major pathway for Ca2+ entry and play a crucial role in excitation-contraction coupling (Jackson, 2000) and their activity is modulated by signal transduction mechanisms in VSMC (McDonald et al., 1994). Phosphatidylinositol 3-kinases (PI3K) are a family of ubiquitously expressed enzymes, which possess both lipid and protein kinase activities (Anderson et al., 1999). Class I PI3Ks are enzymes that selectively phosphorylate the 3′-OH position of the inositol in phosphatidylinositol 4,5-bisphosphate (PIP2). Class I PI3Ks have been subdivided further according to their structure and mode of activation by cell surface receptors. The Ia subgroup consists of p110α, p110β and p110δ catalytic subunits associated with a p85 adapter subunit to form a heterodimeric complex (Cantrell, 2000). All three isoforms of this class are activated by the binding of specific phospho-tyrosyl motifs to the two SH2 domains of the regulatory subunits. Class Ib PI3K is composed of the p110γ catalytic subunit associated with a p101 regulatory protein. PI3Kγ is stimulated by Gβγ heterodimers released after G protein-coupled receptor (GPCR) activation. The convenient classification of class I PI3K isoforms according to their mechanism of activation has been challenged by evidence that PI3Kβ can also be activated by GPCRs (Maier et al., 1999; Murga et al., 2000). PI3Ks have been implicated in the modulation of vascular smooth muscle contractility (Kawanabea et al., 2004; Kim et al., 2006; Morello et al., 2009). However, there is still limited information on the role of individual PI3Ks in the control of vascular functions. It is known that class Ia and class Ib PI3Ks modulate CaV1.2 channnels and increase Ca2+ current (Macrez et al., 2001; Le Blanc et al., 2004). Angiotensin II activates Ca2+ entry by stimulating L-type Ca2+ channels through Gβγ-sensitive PI3Kγ in portal vein myocytes (Quignard et al., 2001). Interestingly, PI3Kδ has been reported as being involved in the increased basal tonus in aorta and mesenteric arteries from hypertensive rats (Northcott et al., 2002; 2004; 2005;).
Because abnormal vasoconstriction is one of the many vascular complications of diabetes we hypothesized that PI3Kδ could be involved in the vascular dysfunction present in diabetes. In this study, we bring new insights on the role and signalling pathways involving PI3Kδ in vascular function and disease. Here we provide evidence for the first time that: (i) PI3Kδ participates in the control of vascular contractile response elicited by α1-adrenoceptor stimulation; (ii) PI3Kδ levels are increased in a model of type I diabetes; and (iii) stimulation of a GPCR increases Ca2+ current through CaV1.2 channels (ICa,L) in a PI3Kδ-dependent manner, and accounts for the increased vascular contractility in type I diabetes.
Methods
Animals and induction of diabetes
All animal care and experimental protocols complied with guidelines for the humane use of laboratory animals and were approved by the animal ethics committee of the Federal University of Minas Gerais (protocol # 26/2007). We used male C57BL/6J mice. Diabetes was induced in 3-week-old mice by a single intraperitoneal injection of 120 mg·kg−1 streptozotocin, freshly dissolved in 10 mmol·L−1 citrate buffer, pH 4.5–5. Control mice were treated identically with vehicle alone. Mice that did not become diabetic within 3–4 days were excluded from the study. Experiments were started when mice had been hyperglycaemic for 9 weeks. The diabetic state was defined as a plasma glucose concentration exceeding 19.0 mmol·L−1 the day of the experiment. Average body weight was 20.94 ± 1.0 g and 26.97 ± 0.5 g for diabetic and control animals, respectively; and daily food intake was 5.7 ± 0.5 and 4.5 ± 0.08 (g per mouse), for diabetic and control animals respectively.
Organ chamber experiments
Rings from the thoracic aorta were obtained and set up as previously described (Rabelo et al., 2003). Vessels that responded to 10 µmol·L−1 acetylcholine (ACh), with greater relaxation than 80% were considered as containing a functional endothelium. When necessary, the endothelium was removed by intraluminal perfusion with 0.5% CHAPS for 20 s followed by repeated washings. Phenylephrine or KCl was added in increasing cumulative concentrations. After 60 min washing, the vessels were pretreated with different drugs for 30 min and a second cumulative concentration–response curve for phenylephrine was constructed and compared with the first one. Mechanical activity was recorded isometrically, as previously described (Lemos et al., 2002).
Freshly dissociated smooth muscle cells preparation
Smooth muscle cells (SMCs) from mice aorta were enzymically dissociated (Murakami et al., 2003). Thereafter, the tissue was mechanically dispersed; three to five drops were plated onto glass coverslips containing PSS and the SMCs left for at least 20 min to adhere before the measurements of Ca2+ currents.
Whole-cell patch-clamp recording
CaV1.2 currents were isolated by eliminating K+ currents by inclusion of Cs+ and tetra-ethyl ammonium (TEA) in the patch pipette and Cs+ in the bath solution. Patch pipettes were filled with (in mmol·L−1): 130 CsCl, 10 TEA-Cl, 10 EGTA, 4 MgCl2, 4 ATPMg and 10 HEPES, pH 7.2 with CsOH. The external solution contained (in mmol·L−1): 130 CsCl, 20 BaCl2, 0.5 MgCl2, 10 HEPES and 5 glucose, pH 7.4 with CsOH, and osmolarity of 270–300 mOsmol·L−1. Currents through CaV1.2 channels were evaluated using Ba2+ as charge carrier and further stimulated with Bay-K8644 (1 µmol·L−1) or phenylephrine (1 µmol·L−1) to increase signal to noise ratio. The currents were generated by 100 ms steps to 10 mV from a holding potential of −80 mV every 15 s. All inhibitors were applied acutely during current recordings for 2.5 min, the time necessary to reach steady state conditions. Data were collected after the whole-cell configuration was obtained and current amplitude had stabilized.
Western blot analysis
Western blot was performed as previously described (Capettini et al., 2008). We used the following antibodies: mouse monoclonal IgG1 anti-PI3K p110δ (1:1000), goat polyclonal anti-p110α (1:200), rabbit polyclonal anti-p110β (1:200), rabbit polyclonal anti-p110γ (1:200) or rabbit polyclonal anti-actin (1:3000) at room temperature. All antibodies were purchased from Santa Cruz Biotechnology, Santa Cruz, CA, USA.
In vivo antisense oligonucleotides
To silence the PI3Kδ isoform we used in vivo antisense oligodeoxynucleotides (AS-ODNs) (Capettini et al., 2008). The 21-base phosphorothioated AS-ODNs were constructed based on the PI3K p110δ mouse sequence (5′-TAG.GCA.CCT.GCA.Gat.GTA.CTG-3′; GenBank access number NM-008840.2 purchased from Eurogentech North America Inc. (Fremont, CA, USA). The phosphorothioated mismatch oligodeoxynucleotides (MM-ODNs) sequence with the composition 5′-CCT.TCG.TAC.CCT.TTT.TCC-3′, was used as control ODNs. Male C57BL/6J mice received 2 nmol·L−1 AS-ODNs or MM-ODNs in the penile vein 12 h before the experiments (Capettini et al., 2008).
Histological procedure and staining
Aorta rings from control and diabetic animals were fixed in formalin (10% w/v) in isotonic saline. Sections (5 µm) were stained and processed for light microscopic studies and morphometric analysis. Haematoxylin and eosin was used for determining the tissue area. In order to quantify the vessel wall thickness, four fields of each aorta were measured and images obtained with a panapochromatic objective ×40 in light microscopy (final magnification = ×100). The images were digitized through a JVC TK-1270/JGB microcamera and transferred to an analyser (Carl Zeiss- KS300 version 2, Kontron Electronics GmbH, Eiching bei Munchen, Germany).
Statistical analysis
Data were analysed by the use of GraphPad Prism© software (GraphPad Software Inc., USA) or SigmaPlot (SPSS Inc., USA) and are expressed as mean ± SEM. Two-way anova with Bonferroni multiple comparisons post-test was used to compare concentration–response curves obtained in aortic rings and in Western blot experiments. Student's t-test was used on other experiments. The Δ area under the curves (AUC) was calculated as the difference between the contractile responses in the presence or in the absence of different drugs in control or diabetic mouse aortic rings. The relative % Δ AUC is the percentage of the Δ AUC correlated to the total area (in the absence of the drug). All statistical analyses were considered significant when P < 0.05.
Materials
Acetylcholine, Bay-K8644, bosentan, calphostin C, ibuprofen, L-NAME, LY294002, phenylephrine, streptozotocin, TEA and wortmannin were supplied by Sigma Aldrich (St Louis, MO, USA); IC87114 was from ICOS corporation (Bothell, WA, USA) and indomethacin from Calbiochem (San Diego, CA, USA).
Results
Vasoconstrictor response is increased in aorta of diabetic mice
Relaxation induced by ACh was not different between groups (Figure 1A) suggesting no endothelial dysfunction. Conversely, contractile responses to phenylephrine in diabetic mice were dramatically higher in either endothelium-containing or denuded vessels (Figure 1B), making the participation of endothelium-derived contractile factors in this increased vasoconstriction unlikely. Consistent with these results, two cyclooxygenase inhibitors, indomethacin (10 µmol·L−1; Figure 1C) and Ibuprofen (10 µmol·L−1; data not shown), and the antagonist of endothelin receptors, bosentan (10 µmol·L−1; Figure 1D), did not restore contractile responses in diabetic vessels to control levels. In an attempt to evaluate whether contractile dysfunction could be linked to an alteration in sensitivity or increased expression of α1-adrenoceptors, we used KCl as vasoconstrictor. Our results show that contractile response to KCl was also equally enhanced in vessels from diabetic mice (Figure 1E). In the presence of L-NAME (300 µmol·L−1), a nitric oxide synthase inhibitor, contractile responses to KCl and phenylephrine (Figure 1E,F) remained bigger in diabetic arteries compared with controls. Together, the above results point to a probable dysfunction in the aortic SMCs.
Figure 1.
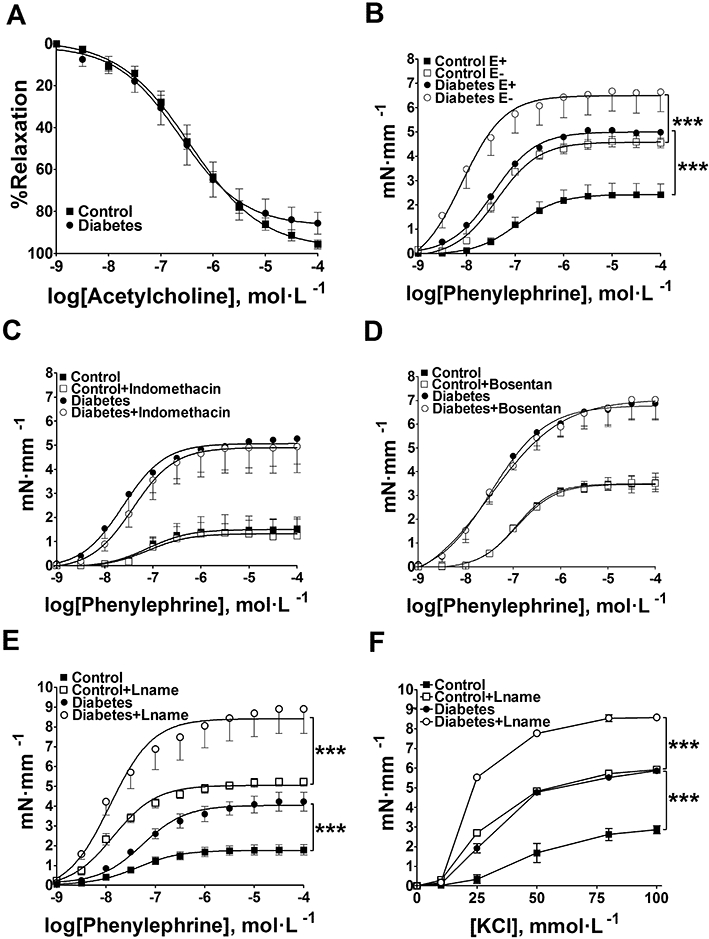
Concentration–response curves to acetylcholine (ACh), phenylephrine and KCl obtained in aortic rings from normoglycaemic (control) or diabetic mice. The data are represented as mean ± SEM. (A) ACh-induced relaxation in diabetic and control vessels with functional endothelium (n = 8). (B) Contractile responses to phenylephrine in endothelium-containing (E+) or denuded (E−) aortic rings (n = 5); ***P < 0.001. (C) Concentration–response curves to phenylephrine in endothelium-containing vessels with or without indomethacin (n = 5) or (D) bosentan (n = 4). Concentration–response curves to phenylephrine (E) and KCl (F) in endothelium-containing vessels L-NAME (n = 4). ***P < 0.001.
Morphological analysis
At this point it was interesting to analyse the possibility of morphological modifications in the structure of the vessel wall. Morphometrical analysis of sections showed decrease in the thickness and number of cells in the aorta of diabetic mice compared with the control group (Figure 2). Thus, these morphological alterations are not compatible with the greater vasoconstrictor response observed in aortas from diabetic mice.
Figure 2.
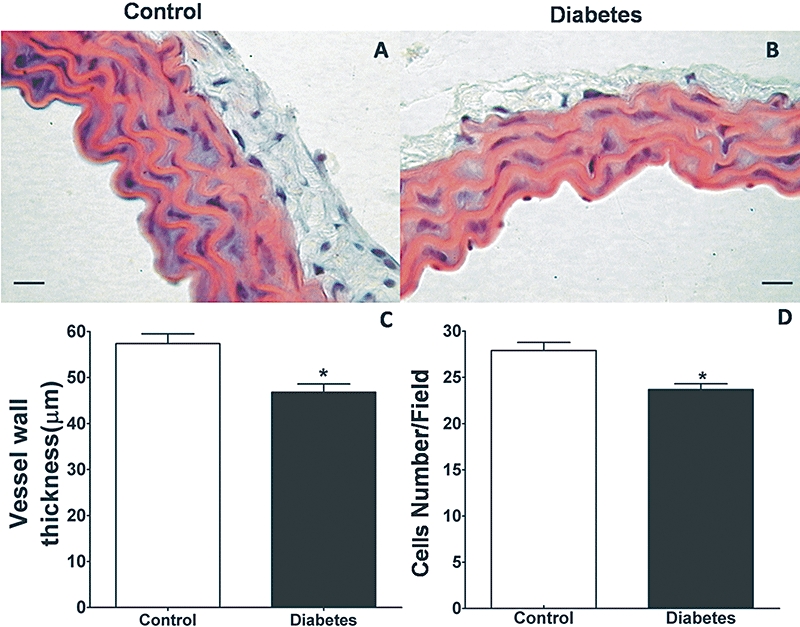
Morphometrical analyses in the structure of the vessel wall from normoglycaemic (control) or diabetic mice. Representative H&E-stained sections from (A) control and (B) diabetic mice aorta. (C) Average differences in vessel wall thickness (n = 7) and (D) cell numbers per field (n = 8) between control and diabetic mice. The data are represented as mean ± SEM; *P < 0.05. Scale bar: 10 µm.
Enhanced Ca2+ current through L-type voltage-dependent Ca2+ channels in diabetic smooth muscle cells
KCl-induced contraction has been shown to result in Ca2+ influx through CaV1.2 channels. To determine whether diabetes was associated with changes in L-type Ca2+ channels, we assessed whole-cell ICa,L measured in freshly dissociated aortic vascular myocytes. Consistent with the enhanced aortic contraction induced by KCl depolarization, diabetes resulted in higher density of Ca2+ channel currents (by 96%, at 10 mV, in the presence of Bay K8644). Figure 3A shows the current density–voltage relationship. The voltage at which 50% of channel population was activated (V50) was −14.24 ± 1.8 mV for diabetic and −11.20 ± 3.9 mV for control cells respectively (P = 0.47; n = 9). The slope factor was not different for diabetic (6.33 ± 0.91) and control (6.21 ± 1.59) cells (n = 9). Moreover, phenylephrine induced a large increase in current density (Figure 3A–C). The Ca2+ currents were abolished by nifedipine, confirming that Ca2+ entry was mainly through CaV1.2 channels (data not shown). No obvious differences in the kinetics of activation or inactivation were detected between control and diabetic SMCs. The cell capacitance measured in control and diabetic myocytes were 6.6 ± 0.3 pF (n = 37) and 6.0 ± 0.3 pF (n = 32), respectively, suggesting that there was no change in cell size induced by diabetes.
Figure 3.
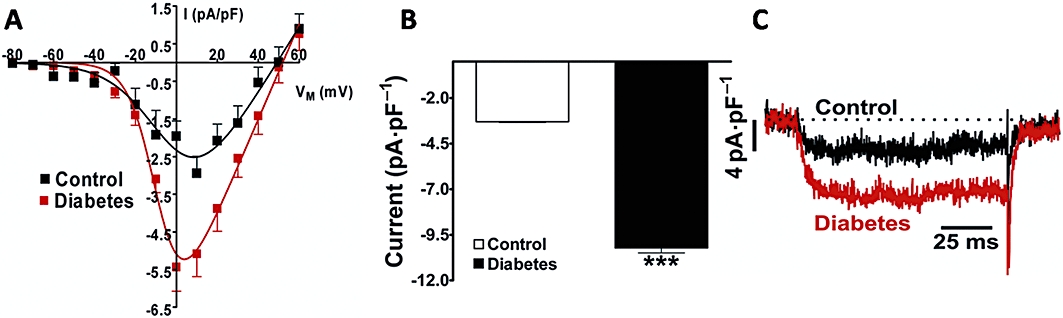
Vascular smooth muscle cells' L-type calcium current from normoglycaemic or diabetic mice. Panel (A) shows current density–voltage relationships in the presence of Bay-K8644 (control, n = 9 and diabetic, n = 9). (B) Average peak Ca2+ current density (ICa,L) ± SEM for control (n = 20) and diabetic (n = 10) mice. Here the vascular myocytes were stimulated with phenylephrine at 1 µM. (C) Representative original records of ICa,L in the presence of phenylephrine. ***P < 0.001.ICa,L, Ca2+ current through voltage-dependent L-type Ca2+ channel.
Mechanism involved in the augmentation of L-type Ca2+ current and contractile response
PI3K has been reported to modulate vascular contractility and CaV1.2 channels in SMCs (Le Blanc et al., 2004). LY294002 (20 µmol·L−1), an inhibitor that acts on all PI3K isoforms, prevented the phenylephrine-induced enhancement of ICa,L in diabetic aorta SMCs (Figure 4A). LY294002 (20 µmol·L−1) also decreased phenylephrine-induced contraction in diabetic animals. However, the decrease in contraction was greater in controls, as shown by the relative Δ % AUC (Figure 4B,C). We repeated the same experiment but using wortmannin (0.3 µmol·L−1), another inhibitor that acts on all PI3K isoforms, with similar results (Figure 5).
Figure 4.
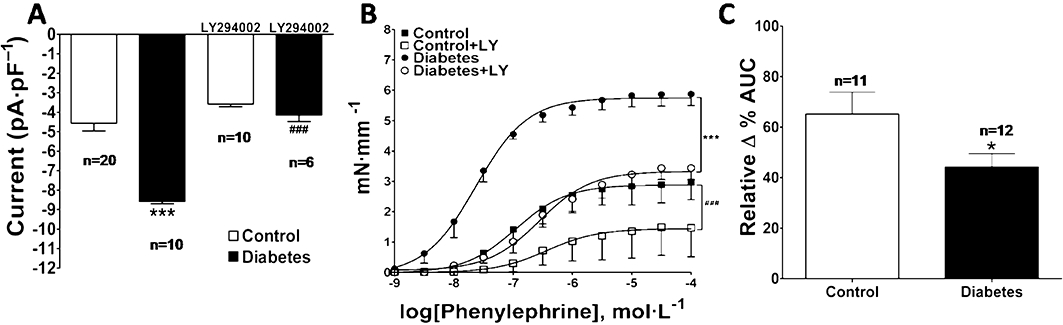
Effect of LY294002 on phenylephrine-induced ICa,L and contractile responses in normoglycaemic (control) and diabetic mice. The data are represented as mean ± SEM. Effect of LY294002 (20 µmol·L−1) on (A) ICa,L induced by phenylephrine (1 µmol·L−1) in aortic SMC; control versus diabetic ***P < 0.001, diabetic versus diabetic+LY294002 ###P < 0.001 and on (B) phenylephrine-induced contractile responses in aortic rings. (C) Inhibitory effect of LY294002 represented by the relative Δ % area under the curves shown in (B); *P < 0.05. ICa,L, Ca2+ current through voltage-dependent L-type Ca2+ channel.
Figure 5.
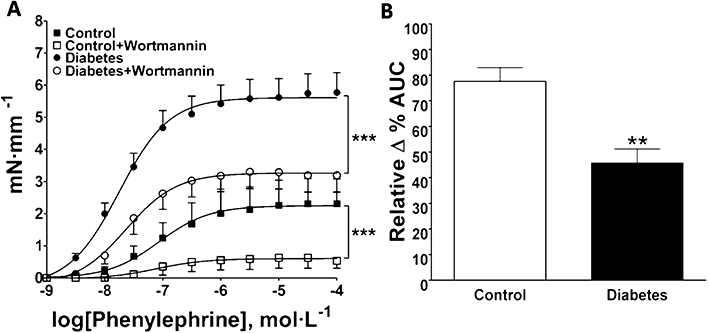
Effect of wortmannin on phenylephrine-induced contraction in aortic rings from normoglycaemic (control) and diabetic mice. The data are represented as mean ± SEM. (A) Concentration–response curves to phenylephrine with or without wortmannin (0.3 µmol·L−1; n = 6). (B) Inhibitory effect of wortmannin represented by the relative Δ % area under the curves shown in (A); **P < 0.01; ***P < 0.001.
Western blot analyses of aortic homogenates revealed the presence of PI3Kδ in control animals and increased expression of PI3Kδ was found in diabetic mice aorta (Figure 6A). PI3Kα was also expressed but in very low levels and no increase in expression was seen in diabetic animals (Figure 6B). Traces of PI3Kβ and PI3Kγ were also detected, and their expression was significantly decreased in diabetic aortas (Figure 6C,D).
Figure 6.
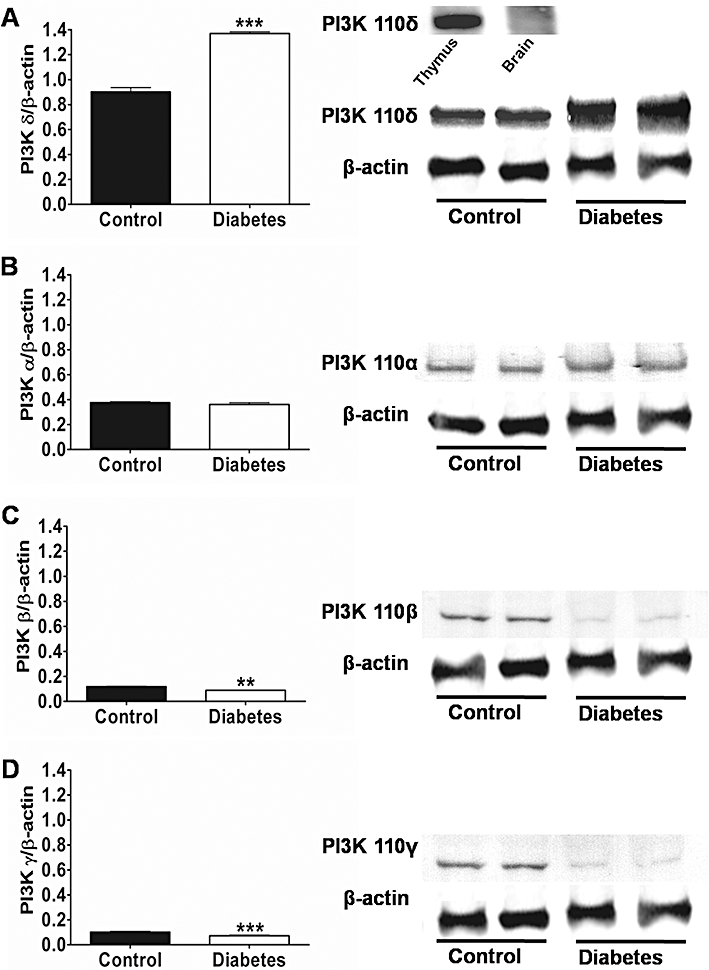
Western blot analysis of PI3Ks isoforms in aortas from normoglycaemic (control) and diabetic mice. On the left are shown the mean ± SEM values of three different experiments, and on the right representative gels. Results were normalized by β-actin content in samples. (A) PI3Kδ; (B) PI3Kα; (C) PI3Kβ and (D) PI3Kγ***P < 0.001 and **P < 0.01. PI3K, phosphatidylinositol 3-kinase.
To determine whether the PI3Kδ isoform is required for the unique response to phenylephrine in diabetic vascular myocytes, IC87114 (10 µmol·L−1) was used to selectively inhibit PI3Kδ (Sadhu et al., 2003a,b;); Interestingly, PI3Kδ inhibition did prevent phenylephrine-induced enhancement of ICa,L in diabetic vascular myocytes (Figure 7A) and normalized contractile response in the diabetic aorta (Figure 7B). These data suggest an important role of PI3Kδ in the pathophysiology of vascular disease in diabetes.
Figure 7.
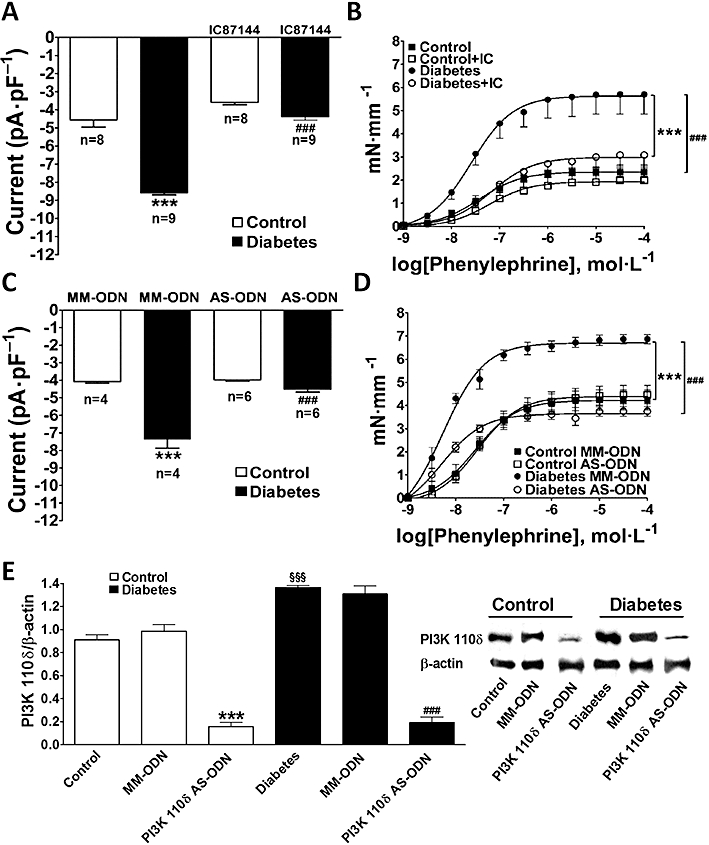
Effect of selective inhibition of the PI3Kδ isoform with IC87114 or antisense knock-down (AS-ODNs) on ICa,L and contractile responses from normoglycaemic (control) and diabetic mice. The data represents mean ± SEM. (A) Effect of IC87114 on ICa,L induced by phenylephrine in VSMC. Control versus diabetic ***P < 0.001, diabetic versus diabetic + IC87114 ###P < 0.001. (B) Effect of IC87114 on contractile responses to phenylephrine in endothelium-denuded aortic rings. Diabetes versus diabetes+IC87114 ***P < 0.001, control versus diabetes ###P < 0.001. (C) Effect of AS-ODN and its mismatch oligonucleotide (MM-ODN) on ICa,L induced by phenylephrine in VSMC. Control MM-ODN versus diabetic MM-ODN ***P < 0.001, diabetic MM-ODN versus diabetic AS-ODN ###P < 0.001. (D) Effect of AS-ODN and MM-ODN on contractile responses to phenylephrine in aortic rings. Control MM-ODN versus diabetic MM-ODN ***P < 0.001, diabetic MM-ODN versus diabetic AS-ODN ###P < 0.001. (E) Western blot analysis of the efficiency of AS-ODN to block the expression of the PI3kδ isoform in aortas from control and diabetic mice. Control versus diabetic §§§P < 0.001, control MM-ODN versus control AS-ODN ***P < 0.001 and diabetic MM-ODN versus diabetic AS-ODN ###P < 0.001. AS-ODNs, antisense oligodeoxynucleotides; ICa,L, Ca2+ current through voltage-dependent L-type Ca2+ channel; MM-ODNs, mismatch oligodeoxynucleotides; PI3K, phosphatidylinositol 3-kinase; VSMC, vascular smooth muscle cell.
To further support the hypothesis above, we specifically knocked down PI3Kδ expression by using AS-ODNs in vivo. The efficiency of the AS-ODNs to block expression of PI3Kδ isoform was evaluated by functional experiments in the organ bath and patch-clamp and by Western blot analysis. In diabetic vessels, there was decrease in ICa,L (Figure 7C) and contraction (Figure 7D) induced by α1-adrenoceptor stimulation after PI3Kδ knock-down to levels similar to those observed with pharmacological inhibition with IC87114 (Figure 7A,B). MM-ODNs were used as controls and had no significant effect (Figure 7C,D). The lowest level of PI3Kδ expression in the mouse aorta (∼86% reduction in control and 90% in diabetic vessels) was achieved 12 h after the AS-ODNs injection while MM-ODNs had no effect (Figure 7E). AS-ODNs knock-down of PI3Kδ did not affect expression of either PI3Kα, PI3Kβ and PI3Kγ (Figure 8). The increase in KCl-elicited vessel contraction (Figure 9A) and activation of voltage-dependent Ca2+ current (Figure 9B–D) in diabetic mice were also prevented by knocking down PI3Kδ. Taken together, our data provide compelling evidence that PI3Kδ signalling is up-regulated in diabetic aortic vessels and that its modulation clearly affects vascular function in our model of type 1 diabetes.
Figure 8.
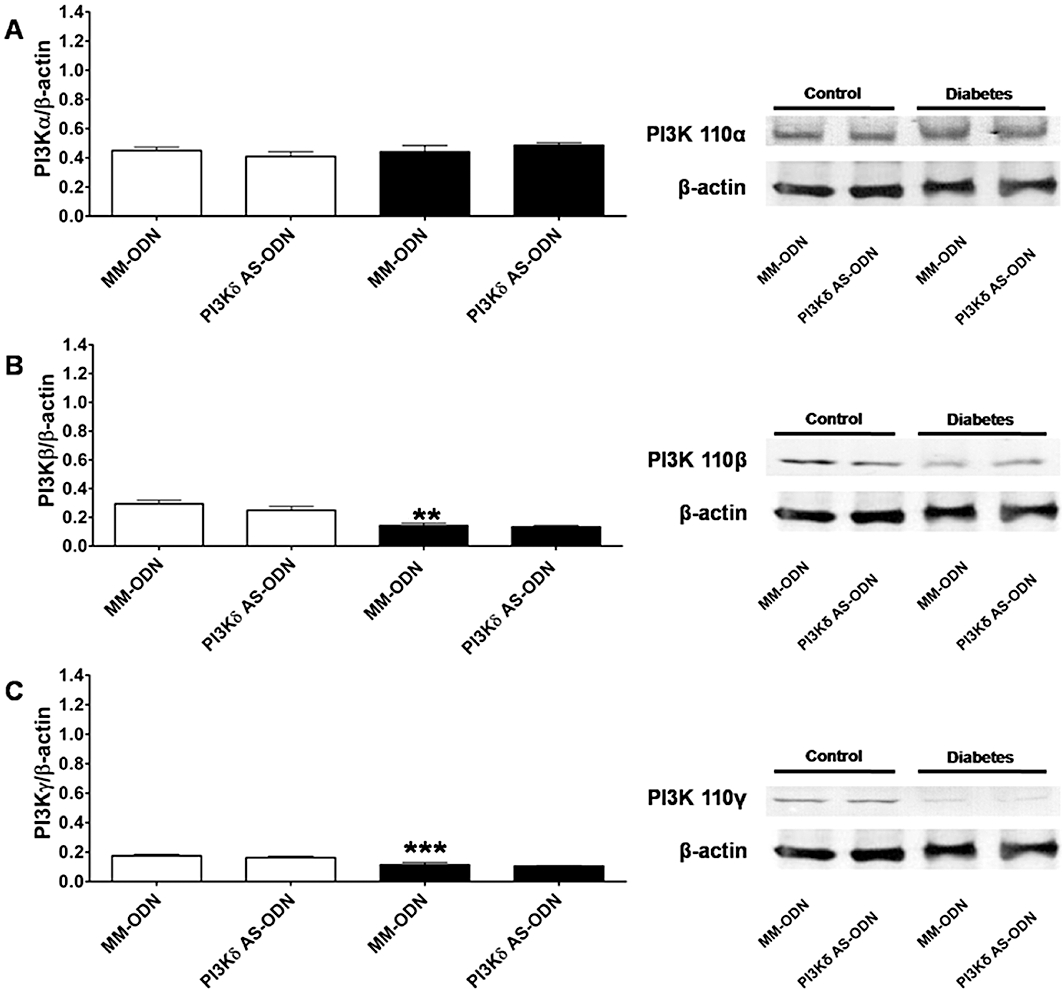
Western blot analysis of the effect of antisense knock-down (AS-ODNs) of PI3Kδ on the expression of PI3Kα (A), PI3Kβ (B) and PI3Kγ (C) isoforms in aortas from normoglycaemic (control) and diabetic mice. On the left are shown the mean ± SEM values of three different experiments, and on the right representative gels. Results were normalized by β-actin content in samples. Control MM-ODN versus diabetic MM-ODN ***P < 0.001, **P < 0.01. AS-ODNs, antisense oligodeoxynucleotides; MM-ODNs, mismatch oligodeoxynucleotides; PI3K, phosphatidylinositol 3-kinase.
Figure 9.
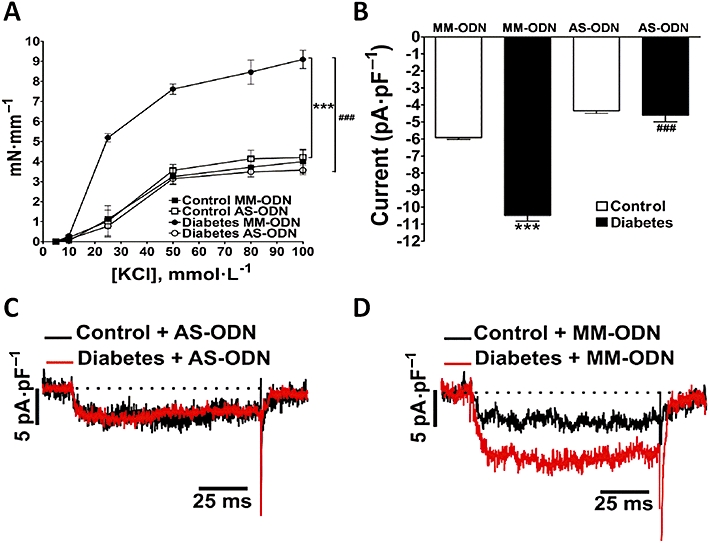
Effect of antisense oligonucleotides (AS-ODN) knock-down of PI3Kδ on KCl-induced contractions and on activation of ICa,L in the presence of Bay-K8644. PI3Kδ AS-ODN knock-down normalized (A) contractile responses to KCl in diabetic aortic rings (n = 7) and (B, C) ICa,L in smooth muscle cells in the presence of Bay-K8644 (1 µmol·L−1; n = 3). (B) Represents the average peak Ca2+ current density (n = 5), and (C, D) representative traces. Control mismatch oligonucleotide (MM-ODN) did not have any effect (A, B, D). In (A) diabetic MM-ODN versus control MM-ODN ***P < 0.001 and diabetic MM-ODN versus diabetic AS-ODN ###P < 0.001. In (B) control MM-ODN versus diabetic MM-ODN ***P < 0.001 and diabetic MM-ODN versus diabetic AS-ODN ###P < 0.001.ICa,L, Ca2+ current through voltage-dependent L-type Ca2+ channel; PI3K, phosphatidylinositol 3-kinase.
Protein kinase C (PKC) is well recognized to be involved on the control of SMCs contractility through modulation of the activity of CaV1.2 channels (Crozatier, 2006). Therefore, we also evaluated the involvement of PKC in the abnormal vasoconstriction of the diabetic mouse aorta. However, calphostin C (10 µmol·L−1), a selective inhibitor of PKC, did not modify the contractile response to phenylephrine either in control or in aorta of diabetic mice (Figure 10), while it inhibited contractions induced by phorbol ester (data not shown). These results suggest that the conventional and novel isoforms of PKC are not involved in the increased contractility of the aorta of diabetic mice.
Figure 10.
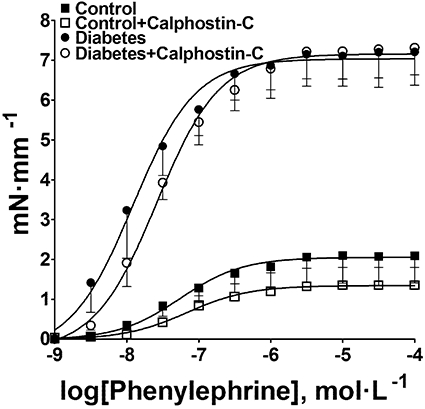
Effect of calphostin C (10 µmol·L−1) on contractile response to phenylephrine in aortas from control and diabetic mice. The data are means (±SEM) of five experiments.
Discussion
The major finding of this work is that the δ isoform of PI3K is specifically involved in the signal transduction pathway leading to increased Ca2+ currents through CaV1.2 in VSMCs and accounts for the increased vascular contractility in our mouse model of type I diabetes, elicited by stimulation of α1-adrenoceptors.
Macro- and microvascular disease conditions currently represent the principal causes of morbidity and mortality in patients with type I or type II diabetes mellitus (Nuzum and Pharm, 2009; Orasanu and Plutzky, 2009). Altered vascular contractility and impaired endothelium-dependent vasodilation have been demonstrated in various vascular beds in different animal models of diabetes and in humans with type I or type II diabetes (Kamata et al., 1988; Matsumoto et al., 2004). We show in this study that the responses of aortic rings from diabetic mice, to phenylephrine and KCl were about three times greater. Aortic pulse-wave velocity has been shown to be associated with cardiovascular risk (Blacher et al., 1999). Moreover, aortic stiffness has been shown as an independent predictor of all-cause and cardiovascular mortality (Laurent et al., 2001). Therefore, the comprehension of the mechanism(s) involved in increased contractility of aorta is crucial.
Increased arterial contractility in diabetes may be secondary to an abnormal response of smooth muscle (Kamata et al., 1988; Taylor et al., 1994) or endothelial dysfunction (Pieper et al., 1992; 1997;). In this work we have evaluated the role of the endothelium in many ways and show that: (i) vasodilation in response to ACh was not impaired in diabetic aorta; and (ii) removal of the endothelial layer, inhibition of prostaglandin synthesis with indomethacin or ibuprofen, inhibition of nitric oxide synthase with L-NAME or blockade of endothelin-1 receptors with bosentan did not normalize contraction of diabetic vessel to the level found in controls. Altogether these results suggest that the endothelium did not play a major role in mediating increased vascular contractility in our model of diabetes. A considerable body of evidence indicates a decreased endothelium-dependent relaxation in diabetes (Hink et al., 2001; Ding et al., 2005). However, our results agree with other reports that have shown unaltered or even augmented endothelium-dependent relaxation in diabetes (Brands and Fitzgerald, 1998; Pieper, 1999; Kobayashi et al., 2005). Moreover, some clinical data seem to be in agreement with these observations (Jaap and Tooke, 1995; Cipolla et al., 1996). Possible explanations for these apparent discrepancies may be related to time-dependent changes in endothelial function (Kobayashi and Kamata, 1999; Kobayashi et al., 2005), the vascular bed studied and model and severity of diabetes (De Vriese et al., 2000; Kobayashi et al., 2005).
An alternative explanation for the increased contractile response lies on an enhanced function of VSMC. The possibility that the morphological alterations in diabetic vascular tissue could be responsible for its increased vasoconstriction is unlikely, as the decrease in the vessel wall thickness and in the cell number observed here did not support this hypothesis.
A growing body of evidence indicates that the PI3K signalling pathway mediates agonist-induced contraction (Vecchione et al., 2005) and increase in calcium influx through CaV1.2 channels (Blair and Marshall, 1997; Viard et al., 1999; 2004; Macrez et al., 2001). Therefore, a series of experiments was undertaken to determine the role of PI3K and CaV1.2 channels on increased contractility in diabetes. Two inhibitors of PI3K, LY294002 and wortmannin produced a decrease in contraction in diabetic vessels and the selective inhibitor of the p110δ catalytic subunit of PI3K, IC87114, normalized contraction in aortic rings from diabetic mice suggesting an involvement of PI3Kδ in the increased contractility. These data were further corroborated by antisense specific knock-down of p110δ, which reduced the contractile responses in diabetic aortas following stimulation with phenylephrine, to the level of those observed in control vessels. Taken together, these data clearly show that the PI3Kδ isoform was implicated in the increased vasoconstriction found in diabetic aortic rings.
Knock-down of p110δ also normalized contraction to KCl in diabetic vessels. As KCl-induced contraction of aorta is mainly the result of Ca2+ influx through CaV1.2 channels we next evaluated the role of PI3K in regulating these channels. We used freshly dissociated mice aortic SMC to avoid adaptations of cells to culture conditions and modification in the pattern of PI3K isoforms expression, as reported in previous work (Macrez et al., 2001; Vecchione et al., 2005). Peak Ca2+ current density was higher in SMCs from diabetic animals. The increase in ICa,L was seen in cells stimulated with phenylephrine or Bay-K8644, as well. The Ca2+ signal is the primary determinant of the contraction of the vascular smooth muscle (Akata, 2007) and is in good agreement with the increased contractility found in diabetic vessels, in despite the small reduction (∼15%) of the number of cells.
A previous report showed that increased PI3Kδ activity leads to increased L-type Ca2+ channel activity in rat portal vein myocytes (Macrez et al., 2001). We fond here that LY294002, IC87114 and knock-down of p110δ normalized ICa,L in SMC from diabetic aorta to the level found in control SMC. Altogether, these results clearly indicated that PI3Kδ was responsible for the increased Ca2+ channel current and the consequently increased contractility in our model of diabetes.
The increased Ca2+ influx through CaV1.2 channels showed in this work are in agreement with data from the literature showing higher Ca2+ influx in cerebral artery myocytes during acute hyperglycaemia and in the dB/dB mouse (Navedo et al., 2010). However, our results are in disagreement with the reported decrease in Ca2+ influx observed in coronary smooth muscle from alloxan-induced diabetic Yucatan pigs (Witczak et al., 2006). The discrepancy between our results and theirs may well be due to the different species and vascular beds studied.
One mechanism by which PI3Kδ activity may be increased in diabetes is by enhancing protein expression. Thus, we used Western blot analyses to measure the expression of the catalytic subunits of the class I PI3K family and found that the main isoform present was p110δ with very small amounts of p110α, p110β and p110γ in control animals. However, an increased expression of one particular isoform of the catalytic subunit of PI3K, p110δ, in diabetic animals has been reported. Northcott et al. (2002; 2004; 2005;) showed p110δ expression to be increased in aortas from L-NNA, DOCA-salt and spontaneously hypertensive rats and to play a role in the increased spontaneous tone found in the aorta of these models of hypertension. The reasons for a decrease in expression of p110β and p110γ isoforms found in our study are not apparent and deserve further investigation. However, our data clearly indicate that these PI3K isoforms are not involved in the increased ICa,L and vasoconstriction found in our model of type I diabetes.
Notably, non-selective inhibition of PI3K with LY294002 and wortmannin was more effective in inhibiting contraction in control than in diabetic arteries, while PI3Kδ inhibition was only effective in diabetic animals. These results are consistent with the increased expression of PI3Kδ and fall in β and γ isoforms found in diabetic animals and suggest that different PI3K isoforms regulate contraction in normal and diabetic arteries. Another important point to consider is that LY294002 and wortmannin were much more effective in inhibiting contraction than CaV1.2 channels in control animals. These results suggest that an additional mechanism independent of CaV1.2 channels and of PI3Kδ would be contributing to the inhibition of the contraction. It is already known that activation of the Rho/Rho kinase pathway by PI3K leads to an increased level of myosin light chain phosphorylation and consequent vasoconstriction (Miao et al., 2002; Wang et al., 2006). In view of these considerations, it interesting to speculate that in control vessels, another isoform of PI3K could be contributing to the inhibition of the contraction via Rho activation.
It should be noted that the metabolic state of our animals may not completely reflect the pathogenesis of diabetic vascular complications in humans, as type 1 diabetic humans receiving insulin, have either normal or elevated body weight (Chaturvedi et al., 1995). However, it is also important to consider that often, symptoms of diabetes are not severe, or may be absent, and consequently hyperglycaemia of sufficient degree to cause pathological and functional changes may be present for a long time before the diagnosis is made (Alberti and Zimmet, 1998).
In summary, our data show that over-expression of the PI3Kδ isoform mediates the increased Ca2+ current through CaV1.2 channels that drives the increased contractility in diabetic vessels. Given that selective inhibition of the PI3Kδ isoform appears to be highly effective in blocking vasoconstriction in our model of type 1 diabetes, PI3Kδ could represent a novel target to expand the therapeutic strategy to treat vascular disease in diabetic patients.
Acknowledgments
This work was supported by FAPEMIG (Fundação de Apoio a Pesquisa do Estado de Minas Gerais)/PRONEX and CNPq/Brazil (Conselho Nacional de Desenvolvimento Científico e Tecnológico). JFP was supported by CAPES/Brazil (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior).
Glossary
Abbreviations
- [Ca2+]i
intracellular calcium concentration
- AS-ODNs
antisense oligodeoxynucleotides
- CaV1.2
voltage-dependent L-type Ca2+ channels
- GPCR
G protein-coupled receptor
- ICa,L
Ca2+ current through voltage-dependent L-type Ca2+ channel
- MM-ODNs
mismatch oligodeoxynucleotides
- PI3K
phosphatidylinositol 3-kinase
- PKC
protein kinase C
- SMCs
smooth muscle cells
- VSMCs
vascular smooth muscle cells
Conflict of interest
The authors state no conflict of interest.
References
- Akata T. Cellular and molecular mechanisms regulating vascular tone. Part 1: basic mechanisms controlling cytosolic Ca2+ concentration and the Ca2+-dependent regulation of vascular tone. J Anesth. 2007;21:220–231. doi: 10.1007/s00540-006-0487-5. [DOI] [PubMed] [Google Scholar]
- Alberti KGMM, Zimmet PZ. Definition diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus. Provisional report of a WHO Consultation. Diab Med. 1998;15:539–553. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158:S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RA, Boronenkov IV, Doughman SD, Kunz J, Loijens JC. Phosphatidylinositol phosphate kinases, a multifaceted family of signaling enzymes. J Biol Chem. 1999;274:9907–9910. doi: 10.1074/jbc.274.15.9907. [DOI] [PubMed] [Google Scholar]
- Blacher J, Asmar R, Djane S, London G, Safar M. Aortic pulse wave velocity as a marker of cardiovascular risk in hypertensive patients. Hypertension. 1999;33:1111–1117. doi: 10.1161/01.hyp.33.5.1111. [DOI] [PubMed] [Google Scholar]
- Blair LA, Marshall J. IGF-1 modulates N and L calcium channels in a PI3-kinase-dependent manner. Neuron. 1997;19:421–429. doi: 10.1016/s0896-6273(00)80950-2. [DOI] [PubMed] [Google Scholar]
- Brands MW, Fitzgerald SM. Acute endothelium-mediated vasodilation is not impaired at the onset of diabetes. Hypertension. 1998;32:541–547. doi: 10.1161/01.hyp.32.3.541. [DOI] [PubMed] [Google Scholar]
- Cantrell DA. Phosphoinositide 3-kinase signaling pathways. J Cell Sci. 2000;114:1439–1445. doi: 10.1242/jcs.114.8.1439. [DOI] [PubMed] [Google Scholar]
- Capettini LSA, Cortes SF, Gomes MA, Silva GAB, Pesquero JL, Lopes MJ, et al. Neuronal nitric oxide synthase-derived hydrogen peroxide is a major endothelium-dependent relaxing factor. Am J Physiol Heart Circ Physiol. 2008;295:H2503–H2511. doi: 10.1152/ajpheart.00731.2008. [DOI] [PubMed] [Google Scholar]
- Chaturvedi N, Stevens LK, Fuller JH. Mortality and morbidity associated with body weight in people with IDDM: the WHO multinational study of vascular disease in diabetes. Diabetes Care. 1995;18:761–765. doi: 10.2337/diacare.18.6.761. [DOI] [PubMed] [Google Scholar]
- Cipolla MJ, Harker CT, Porter JM. Endothelial function and adrenergic reactivity in human type-II diabetic resistance arteries. J Vasc Surg. 1996;23:940–949. doi: 10.1016/s0741-5214(96)70261-6. [DOI] [PubMed] [Google Scholar]
- Crozatier B. Central role of PKCs in vascular smooth muscle cell ion channel regulation Angiotensin II-induced aortic ring constriction is mediated by phosphatidylinositol 3-kinase/L-type calcium channel signaling pathway. J Mol Cell Cardiol. 2006;41:952–955. doi: 10.1016/j.yjmcc.2006.09.004. [DOI] [PubMed] [Google Scholar]
- De Vriese AS, Verbeuren TJ, Voorde JV, Lameire NH, Vanhoutte PM. Endothelial dysfunction in diabetes. Br J Pharmacol. 2000;130:963–974. doi: 10.1038/sj.bjp.0703393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Hashem M, Wiehler WB, Lau W, Martin J, Reid J, et al. Endothelial dysfunction in the streptozotocin-induced diabetic apoE-deficient mouse. Br J Pharmacol. 2005;146:1110–1118. doi: 10.1038/sj.bjp.0706417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res. 2001;88:14–22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- Jaap AJ, Tooke JE. Pathophysiology of microvascular disease in non-insulin-dependent diabetes. Clin Sci. 1995;89:3–12. doi: 10.1042/cs0890003. [DOI] [PubMed] [Google Scholar]
- Jackson WF. Ion channels and vascular tone. Hypertension. 2000;35:173–178. doi: 10.1161/01.hyp.35.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata K, Miyata N, Kasuya K. Mechanisms of increased responses of the aorta to alfa-adrenoceptor agonists in streptozotocin-induced diabetic rats. J Pharmacobiodyn. 1988;11:707–713. doi: 10.1248/bpb1978.11.707. [DOI] [PubMed] [Google Scholar]
- Kawanabea Y, Hashimotoa N, Masaki T. Effects of phosphoinositide 3-kinase on endothelin-1-induced activation of voltage-independent Ca2+ channels and vasoconstriction. Biochem Pharmacol. 2004;68:215–221. doi: 10.1016/j.bcp.2004.03.025. [DOI] [PubMed] [Google Scholar]
- Kim J, Lee CK, Park HJ, Kim HJ, So HH, Lee KS, et al. Epidermal growth factor induces vasoconstriction through the phosphatidylinositol 3-kinase-mediated mitogen-activated protein kinase pathway in hypertensive rats. J Pharmacol Sci. 2006;101:135–143. doi: 10.1254/jphs.fp0060021. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Kamata K. Relationship among cholesterol, superoxide anion and endothelium-dependent relaxation in diabetic rats. Eur J Pharmacol. 1999;367:213–222. doi: 10.1016/s0014-2999(98)00971-6. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Matsumoto T, Kamata K. The PI3-K/Akt pathway: roles related to alterations in vasomotor responses in diabetic models. J Smooth Muscle Res. 2005;41:283–302. doi: 10.1540/jsmr.41.283. [DOI] [PubMed] [Google Scholar]
- Lagaud GJL, Masih-Khan E, Kai S, Breemen CV, Dubé GP. Influence of type II diabetes on arterial tone and endothelial function in murine mesenteric resistance arteries. J Vasc Res. 2001;38:578–589. doi: 10.1159/000051094. [DOI] [PubMed] [Google Scholar]
- Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L, et al. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001;37:1236–1241. doi: 10.1161/01.hyp.37.5.1236. [DOI] [PubMed] [Google Scholar]
- Le Blanc C, Mironneau C, Barbot C, Henaff M, Bondeva T, Wetzker R, et al. Regulation of vascular L-type Ca2+ channels by phosphatidylinositol 3,4,5-trisphosphate. Circ Res. 2004;95:300–307. doi: 10.1161/01.RES.0000138017.76125.8b. [DOI] [PubMed] [Google Scholar]
- Lemos VS, Côrtes SF, Silva DM, Campagnole-Santos MJ, Santos RA. Angiotensin-(1-7) is involved in the endothelium-dependent modulation of phenylephrine-induced contraction in the aorta of mRen-2 transgenic rats. Br J Pharmacol. 2002;135:1743–1748. doi: 10.1038/sj.bjp.0704630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol Rev. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- Macrez N, Mironneau C, Carricaburu V, Quignard JF, Babich A, Czupalla C, et al. Phosphoinositide 3-kinase isoforms selectively couple receptors to vascular L-type Ca2+ channels. Circ Res. 2001;89:692–699. doi: 10.1161/hh2001.097864. [DOI] [PubMed] [Google Scholar]
- Maier U, Babich A, Nürnberg B. Roles of non-catalytic subunits in Gbeta-gamma induced activation of class I phosphoinositide 3-kinase isoforms beta and gamma. J Biol Chem. 1999;274:29311–29317. doi: 10.1074/jbc.274.41.29311. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Kobayashi T, Kamata K. Alterations in vascular endothelial function in the aorta and mesenteric artery in type II diabetic rats. Can J Physiol Pharmacol. 2004;82:175–182. doi: 10.1139/y04-002. [DOI] [PubMed] [Google Scholar]
- Mayhan WG, Irvine SD, Sharpe GM. Constrictor responses of resistance arterioles during diabetes mellitus. Diabetes Res Clin Pract. 1999;44:147–156. doi: 10.1016/s0168-8227(99)00031-5. [DOI] [PubMed] [Google Scholar]
- Miao L, Dai Y, Zhang J. Mechanism of RhoA/Rho kinase activation in endothelin-1-induced contraction in rabbit basilar artery. Am J Physiol Heart Circ Physiol. 2002;283:H983–H989. doi: 10.1152/ajpheart.00141.2002. [DOI] [PubMed] [Google Scholar]
- Morello F, Perino A, Hirsch E. Phosphoinositide 3-kinase signalling in the vascular system. Cardiovasc Res. 2009;82:261–271. doi: 10.1093/cvr/cvn325. [DOI] [PubMed] [Google Scholar]
- Murakami M, Yamamura H, Suzuki T, Kang MG, Ohya S, Murakami A, et al. Modified cardiovascular L-type channels in mice lacking the voltage-dependent Ca2+ channel beta3 subunit. J Biol Chem. 2003;278:43261–43267. doi: 10.1074/jbc.M211380200. [DOI] [PubMed] [Google Scholar]
- Murga C, Fukuhara S, Gutkind JS. A novel role for phosphatidylinositol 3-kinase in signaling from G protein-coupled receptors to Akt. J Biol Chem. 2000;275:12069–12073. doi: 10.1074/jbc.275.16.12069. [DOI] [PubMed] [Google Scholar]
- Navedo MF, Takeda Y, Nieves-Cintrón M, Molkentin JD, Santana LF. Elevated Ca2+ sparklet activity during acute hyperglycemia and diabetes in cerebral arterial smooth muscle cells. Am J Physiol Cell Physiol. 2010;298:C211–C220. doi: 10.1152/ajpcell.00267.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott CA, Poy MN, Najjar SM, Watts SW. Phosphoinositide 3-kinase mediates enhanced spontaneous and agonist-induced contraction in aorta of deoxycorticosterone acetate-salt hypertensive rats. Circ Res. 2002;91:360–369. doi: 10.1161/01.res.0000030861.13850.f1. [DOI] [PubMed] [Google Scholar]
- Northcott CA, Hayflick JS, Watts SW. PI3-kinase upregulation and involvement in spontaneous tone in arteries from DOCA-salt rats: is p110delta the culprit? Hypertension. 2004;43:885–890. doi: 10.1161/01.HYP.0000118518.20331.e8. [DOI] [PubMed] [Google Scholar]
- Northcott CA, Hayflick J, Watts SW. Upregulated function of phosphatidylinositol-3-kinase in genetically hypertensive rats: a moderator of arterial hypercontractility. Clin Exp Pharmacol Physiol. 2005;32:851–858. doi: 10.1111/j.1440-1681.2010.04276.x. [DOI] [PubMed] [Google Scholar]
- Nuzum DS, Pharm D. Macrovascular complications of diabetes mellitus. J Pharm Pract. 2009;22:135–148. [Google Scholar]
- Okon EB, Szado T, Laher I, Mcmanus B, Van Breemen C. Augmented contractile response of vascular smooth muscle in diabetic mouse model. J Vasc Res. 2003;40:520–530. doi: 10.1159/000075238. [DOI] [PubMed] [Google Scholar]
- Orasanu G, Plutzky J. The pathologic continuum of diabetic vascular disease. J Am Coll Cardiol. 2009;53:35–42. doi: 10.1016/j.jacc.2008.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper GM. Enhanced, unaltered and impaired nitric oxide-mediated endothelium-dependent relaxation in experimental diabetes mellitus: importance of disease duration. Diabetologia. 1999;42:204–213. doi: 10.1007/s001250051140. [DOI] [PubMed] [Google Scholar]
- Pieper GM, Mei DA, Lamgemstroer P, O'rourke ST. Bioassay of endothelium-derived relaxing factor in diabetic rat aorta. Am J Physiol. 1992;263:676–680. doi: 10.1152/ajpheart.1992.263.3.H676. [DOI] [PubMed] [Google Scholar]
- Pieper GM, Siebeneich W, Moore Hilton G, Roza AM. Reversal by L-arginine of a dysfunctional arginine/nitric oxide pathway in the endothelium of the genetic diabetic BB rat. Diabetologia. 1997;40:910–915. doi: 10.1007/s001250050767. [DOI] [PubMed] [Google Scholar]
- Piercy VG, Taylor SA. Comparison of spasmogenic and relaxant responses in aortae from C57/BL/KsJ diabetic mice with those from their non-diabetic littermates. Pharmacology. 1998;56:267–275. doi: 10.1159/000028208. [DOI] [PubMed] [Google Scholar]
- Quignard JF, Mironneau J, Carricaburu V, Fournier B, Babich A, Nurnberg B, et al. Phosphoinositide 3-Kinase gamma mediates angiotensin II-induced stimulation of L-type calcium channels in vascular myocytes. J Biol Chem. 2001;276:32545–32551. doi: 10.1074/jbc.M102582200. [DOI] [PubMed] [Google Scholar]
- Rabelo LA, Côrtes SF, Alvarez-Leite JI, Lemos VS. Endothelium-dysfunction in LDL receptor knockout mice: a role for H2O2. Br J Pharmacol. 2003;7:1215–1220. doi: 10.1038/sj.bjp.0705164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhu C, Dick K, Tino WT, Staunton DE. Selective role of PI3Kδ in neutrophil inflammatory responses. Biochem Biophys Res Commun. 2003a;308:764–769. doi: 10.1016/s0006-291x(03)01480-3. [DOI] [PubMed] [Google Scholar]
- Sadhu C, Masinovsky B, Dick K, Sowell CG, Staunton DE. Essential role of phosphoinositide 3-kinase {delta} in neutrophil directional movement. J Immunol. 2003b;170:2647–2654. doi: 10.4049/jimmunol.170.5.2647. [DOI] [PubMed] [Google Scholar]
- Taylor PD, Oon BB, Thomas CR, Poston L. Prevention by insulin treatment of endothelial dysfunction but not enhanced noradrenaline-induced contractility in mesenteric resistance arteries from streptozotocin-induced diabetic rats. Br J Pharmacol. 1994;111:35–41. doi: 10.1111/j.1476-5381.1994.tb14020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchione C, Patrucco E, Marino G, Barberis L, Poulet R, Aretini A, et al. Protection from angiotensin II-mediated vasculotoxic and hypertensive response in mice lacking PI3Kγ. Nat Med. 2005;201:1217–1228. doi: 10.1084/jem.20040995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viard P, Exner T, Maier U, Mironneau J, Nurnberg B, Macrez N. Gbetagamma dimers stimulate vascular L-type Ca2+ channels via phosphoinositide 3-kinase. FASEB J. 1999;13:685–694. doi: 10.1096/fasebj.13.6.685. [DOI] [PubMed] [Google Scholar]
- Viard P, Butcher AJ, Halet G, Davies A, Nurnberg B, Heblich F. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat Neurosci. 2004;7:939–946. doi: 10.1038/nn1300. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yoshioka K, Azam MA, Takuwa N, Sakurada S, Kayaba Y, et al. Class II phosphoinositide 3-kinase α-isoform regulates Rho, myosin phosphatase and contraction in vascular smooth muscle. Biochem J. 2006;394:581–592. doi: 10.1042/BJ20051471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witczak CA, Wamhoff BR, Sturek M. Exercise training prevents Ca2+ dysregulation in coronary smooth muscle from diabetic dyslipidemic yucatan swine. J Appl Physiol. 2006;101:752–762. doi: 10.1152/japplphysiol.00235.2006. [DOI] [PubMed] [Google Scholar]