

Abstract
The most frequent MLL-gene rearrangement found in leukemia is a reciprocal translocation with AF4 on chromosome 4 resulting in the formation of the MLL-AF4 and the AF4-MLL fusion genes. The oncogenic role of MLL-AF4 is documented but the significance of the reciprocal product - AF4-MLL in leukemia is less clear. In the human leukemia cell lines – RS4;11 and SEMK2-M1, both of which express MLL-AF4 and AF4-MLL, we knocked down the expression of AF4-MLL using siRNA. Loss of AF4-MLL had no effect on the growth of either RS4;11 or SEMK2-M1 cells. Furthermore, in SEMK2-M1 cells there were no changes in cell cycle or apoptosis with loss of AF4-MLL. In contrast, knockdown of MLL-AF4 significantly inhibited growth of both RS4;11 and SEMK2-M1. Additionally, in SEMK2-M1 cells, loss of MLL-AF4 led to G2/M cell cycle arrest and increased apoptosis. Overall, these results demonstrate that in t(4;11) leukemia, the MLL-AF4 fusion protein is critical for leukemia cell proliferation and survival while the AF4-MLL fusion product is dispensable.
Keywords: MLL, AF4, t(4;11), leukemia, 11q23
Introduction
The MLL gene on chromosome 11q23 is rearranged most often in infant leukemia and in secondary leukemia. The most common rearrangement is a reciprocal translocation between MLL and a partner gene resulting in the formation of a fusion gene composed of the amino terminus domains of MLL and carboxy terminus domains of the partner gene. More than 70 genes have been identified as participating in translocations with MLL in leukemia, although the most commonly encountered partners are AF4, AF9 and ENL(1). The normal MLL protein is composed of 3969 amino acids and undergoes proteolysis by Taspase1 to yield two fragments that dimerize to form the functional unit(2). MLL is required for normal hematopoiesis and for the expression of the 5′ HOX-A genes (HOXA5, HOXA7, HOXA9, HOXA10)(3–4). Transcriptional activation of the HOX gene cluster is principally mediated by the SET domain of MLL, located at the carboxy terminus(5). In leukemia-associated translocations, the der11 product composed of the amino terminus of MLL is expressed universally while the reciprocal product [e.g. der4 in the case of t(4;11)] containing the carboxy terminus (and the SET domain) is not detected in up to 20% of the cases(6). Several experimental models of MLL-rearranged leukemia have been described using various fusion partners, utilizing either retroviral expression or transgenic knock-in methods(7–10). In all these models, expression of the der11 MLL-fusion gene induces growth deregulation in hematopoietic cells and majority of them lead to leukemia development in mice. These results suggest that the reciprocal fusion, composed of the carboxy terminus of MLL is not required for the pathogenesis of leukemia. On the other hand, all the above experimental models suffer from a long latency in the complete transformation to leukemia, suggesting that the der11 MLL-fusion by itself is insufficient for transformation and that additional factors are required. It is thus possible that the reciprocal fusion might be the missing factor that is required in the rapid transformation seen in leukemia patients. Recent studies suggest that leukemia may be initiated by retroviral expression in mouse hematopoietic cells of the reciprocal MLL protein – AF-MLL, the der4 product in t(4;11) leukemia(11). We sought to test the requirement of the reciprocal fusion protein in human t(4;11) leukemia – the der4 fusion AF4-MLL. We used RNA-interference to specifically knockdown the AF4-MLL fusion protein in two human t(4;11) cell lines. Our results demonstrate that inhibition of the der4 fusion gene has little effect on the leukemia cells, suggesting that the reciprocal fusion is non-essential in established leukemias.
Materials and Methods
Cell lines
Human leukemia cell lines, RS4;11, SEMK2-M1 (a subclone of the original SEMK2 cell line) and HPB-NULL maintained in RPMI-1640 with 10%FBS as described previously(12–13).
siRNA
siRNA duplexes were designed using Dharmacon’s online siDesign algorithm (Darmacon, Lafayette, CO). Scrambled control consisted of pooled non-targeting duplexes. All siRNAs were reconstituted in Reconstitution Buffer. For delivery, 4 × 106 cells in 200 µL OPTI-MEM containing 750 or 1000 nm siRNA were elctroporated to 250 V for 25 ms using a BTX Electro Square Porator ECM830 (BTX, Holliston, MA). Cells were then washed with serum free media and placed back in regular culture conditions at a concentration of 5 × 105 cells/mL.
Cell growth and cell cycle analysis
Cells were stained with Trypan blue and live cells were counted in triplicate. Cell cycle analysis was performed as before.(14)
RT-PCR
RNA extraction, reverse transcription and real time quantitative RT-PCR using SYBR green detection was performed as before.(14)
Western blotting
To determine the MLL-AF4-specific siRNA knockdown efficiencies, three million SEMK2-M1 cells were harvested after electroporation as described and washed with cold PBS. Nuclear proteins were extracted by a NE-PER Nuclear and cytoplasmic extraction kit (Pierce, Rockford, IL). The proteins were separated in 4% SDS-PAGE, transferred to nitrocellulose membranes and probed with antibodies against AF4(15) or MLLN (Millipore, Billerica, MA). HRP-conjugated secondary antibodies and ECL (Pierce, Rockford, IL) were used for detection.
Results
Design of siRNA duplexes against the der4 fusion in t(4;11) cell lines
To evaluate the role AF4-MLL fusion gene in leukemia, we chose to study RS4;11 and SEMK2-M1 - two t(4;11) cell lines that express both the der11 and the der4 fusion genes.(16) By sequencing of PCR products, we first confirmed the previously published findings that the der4 fusion (AF4-MLL) gene results in two alternatively spliced transcripts and that these transcripts are identical in RS4;11 and SEMK2-M1 cells.(16) The major transcript, termed der4a, is formed by the splicing of AF4-exon3 to MLL-exon10 while in the minor transcript – der4b, AF4-exon3 is spliced to MLL-exon11 (Supplementary Figure 1). However, we were unable to detect a protein product for AF4-MLL using various antibodies directed against either the amino-terminus of AF4 or the carboxy terminus of MLL. Notably, there are no published Western blot results showing successful detection of AF4-MLL or any other reciprocal fusion protein. We then screened several siRNA duplexes directed against the junction regions of the two AF4-MLL transcripts and selected the ones that showed most efficient knockdown in RS4;11 and SEMK2-M1 cells compared to scrambled controls (Supplementary Figure 2). In repeated experiments, we found that the siRNA constructs der4a#2 and der4b#6 efficiently knocked down their respective targets der4a and der4b in both RS4;11 and SEMK2-M1 compared to scrambled siRNA or mock electroporation. Results of one representative experiment are shown in Figure 1, where der4a#2 and der4b#6 treatment knocked down their respective targets by 70% and 60% in RS4;11 cells. Surprisingly, siRNA der4b#6 (henceforth referred to as D4B6) resulted in knockdown of both der4a and der4b transcripts in both SEMK2-M1 and RS4;11. At 24 hours after transfection with D4B6, der4a and der4b mRNA levels were significantly reduced in SEMK2-M1 (by an average 62% and 59% respectively, N=3) and RS4;11 (by average 54% for both de4a and der4b) compared to scrambled siRNA pool or mock electroporation (Figure 1). The reasons for this cross-reactivity are not obvious but common sequences in the 5′ ends of the two siRNA constructs might be involved as none of the other constructs we screened showed this feature (Supplementary Figure 3). Furthermore, combining the two siRNAs had no additive inhibitory effect on the expression of AF4-MLL transcripts (Figure 1). In both RS4;11 and SEMK2-M1, D4B6 treatment had no effect on the levels of the corresponding wild type mRNAs - MLL and AF4, and of the der11 fusion product MLL-AF4. Additionally, D4B6 treatment had no effect on the levels of MLL and of AF4 in the HPB-NULL cell line that does not harbor t(4;11) (Figure 1). As positive controls for our siRNA knockdown strategy, we treated both the SEMK2-M1 and RS4;11 cells with the previously described MLL-AF4 siRNAs – MARS for RS4;11 and MA6 for SEMK2-M1.(17) In contrast to the der4 transcripts, the der11 transcripts are different in the two cell lines, requiring the use of different siRNAs to knockdown these fusions.(16) In agreement with the previously described results, at 24 hours post transfection with the corresponding siRNA, MLL-AF4 mRNA levels were significantly reduced in both SEMK2-M1 (average 63% knockdown, N=3) and RS4;11 (average 50% knockdown) compared to scrambled siRNA pool or mock electroporation (Figure 1). Knockdown of MLL-AF4 protein was verified by Western blot analysis using anti-AF4 and anti-MLL antibodies, with little change evident in the levels of the wild type proteins AF4 and MLL (Figure 1). Treatment of SEMK2-M1, RS4;11 or HPB-NULL cells with MARS or MA6 had no effect on the levels of AF4, MLL or the der4 mRNAs (for SEMK2-M1 and RS4;11 only), confirming the specificity of the siRNA constructs for their respective targets (Figure 1). Overall, these results demonstrate that we achieved relatively specific knockdown of AF4-MLL and MLL-AF4 in both SEMK2-M1 and RS4;11 cells.
Figure 1. siRNAs againt MLL-AF4 and AF4-MLL specifically knockdown their respective targets.
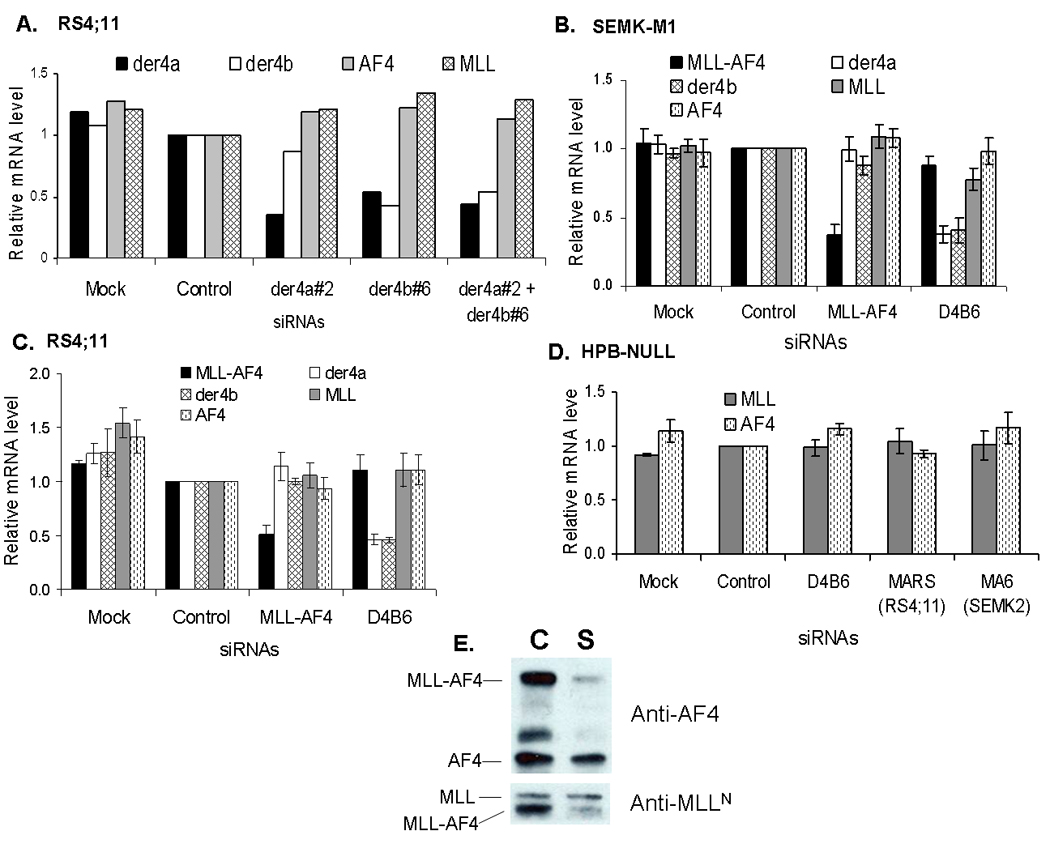
Human leukemia cell lines were electroporated with various siRNA duplexes as described and gene expression measured by real time quantitative RT-PCR at 24 hours. Bar graphs show relative expression of AF4-MLL (der4a and der4b), MLL-AF4, AF4 and MLL in RS4;11 (A, C) and SEMK2-M1 (B) cells while HPB-NULL (D) served as a control cell line since it lacks the MLL-translocation. Relative expression levels were derived using beta-actin as internal control and normalized to levels in the scrambled control condition. Panel A shows data from one representative experiment in RS4;11 while for the remaining graphs data represent mean ± std. err of 3 separate experiments. Panel E shows Western blot results for MLL-AF4 knockdown in SEMK2-M1 probed with anti-AF4 (upper panel) or anti-MLLN (lower panel) antibodies. Lane labels on top represent the siRNA treatments – control (C) or MA6 (S) - the SEMK2-specific MLL-AF4 siRNA. Protein bands are labeled on the left and the antibodies used in each blot are on the right.
MLL-AF4 is essential for cell growth while AF4-MLL is dispensable
We next analyzed the effects of AF4-MLL inhibition on in vitro cell growth. Cells were electroporated with siRNA duplexes and viable cell numbers were counted daily for 4 days. Specific knockdown of target mRNA was confirmed by real-time quantitative RT-PCR as described above. In repeated experiments, growth of AF4-MLL-knocked-down (D4B6 treated) SEMK2-M1 and RS4;11 cells was identical to that of cells treated with scrambled pool or no siRNA (Figure 2). The results were the same with the der4a#2 siRNA (Supplementary Figure 4). While these results indicate that AF4-MLL knockdown had no significant effects on cell growth, the kinetics of AF4-MLL synthesis and degradation are unknown. Given our inability to detect AF4-MLL protein, to ensure sustained inhibition of the der4 fusion, we performed sequential electroporations with siRNAs. In these experiments, we electroporated SEMK2-M1 cells on days 0, 2, 4 and 7 with mock (no siRNA), scrambled pool, a mismatched siRNA (siMM), MA6 (anti-MLL-AF4) or D4B6 siRNA.(17) Quantitative RT-PCR analysis showed >50% knockdown of der4a, der4b and of MLL-AF4 with the corresponding siRNA treatments compared to controls (data not shown). Over a 9 day observation period (from day 0 to 48 hours after the fourth electroporation), cells with knockdown of der4 mRNA showed growth characteristics identical to those that were treated with siMM, scrambled pool or mock siRNA (electroporation only).
Figure 2. Knockdown of AF4-MLL has no effect on cell growth.
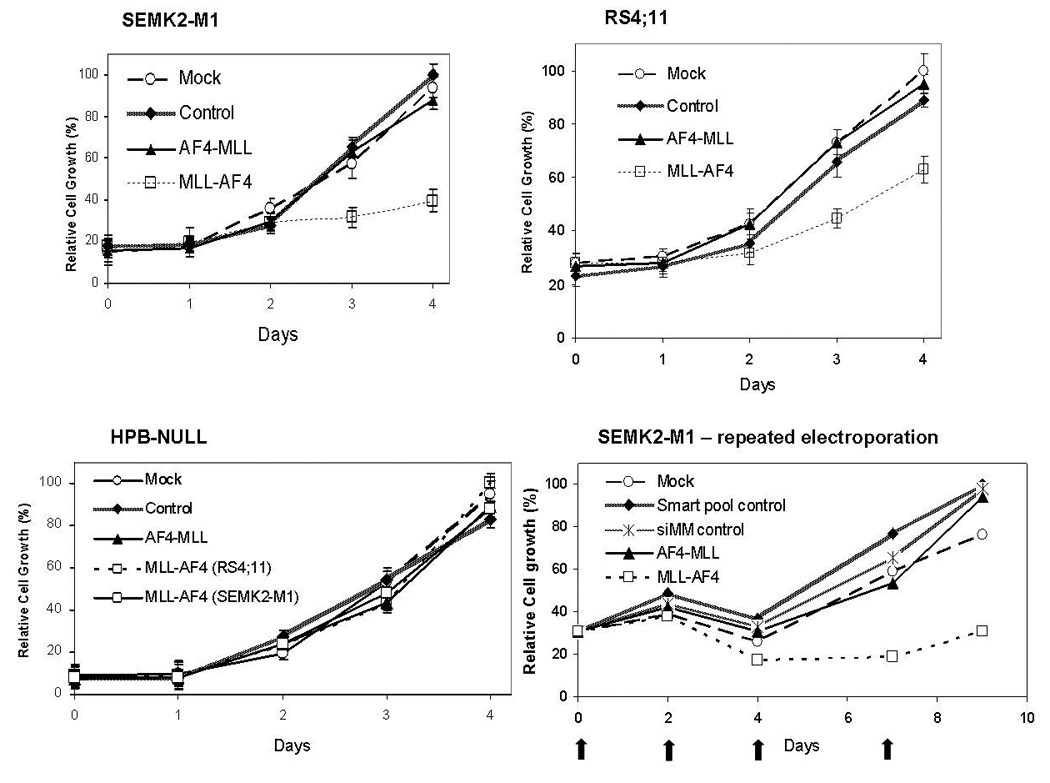
Leukemia cell lines SEMK2-M1, RS4;11 and HPB-NULL were electroporated with various siRNAs as described and viable cells were counted by Trypan blue exclusion. Daily cell counts were normalized to those on day 4 which were considered 100%. Graphs below depict growth curves for each cell line with time since electroporation indicated on the x-axis. Data represent mean ± std. err. of 3 separate experiments. In the SEMK2-M1 repeat electroporation experiment, SEMK2-M1 cells were electroporated sequentially on days 0, 2, 4 and 7 (indicated by arrows on the x-axis) observed for a total of 9 days. Viable cells were counted on the days of electroporation and normalized to counts on day 9 (100%).
In contrast to the above results, with MLL-AF4 knockdown, we observed a significant inhibition of cell growth in both SEMK2-M1 and RS4;11 cell lines. As shown in Figure 2, at 96 hours post single electroporation, growth of SEMK2-M1 cells with MLL-AF4 knockdown was reduced to 39.6 ± 5.3 % that of scrambled controls while for RS4;11, MLL-AF4 knock-down reduced cell growth to 62.9 ± 5.0% that of controls (mean ± std. err., N=3, p < 0.01, t-test). Growth of the t(4;11)-negative cell line HPB-NULL was unaffected by electroporation with either the AF4-MLL siRNA D4B6 or the MLL-AF4 siRNAs MARS and MA6 (Figure 2). Similarly, with repeated doses of MA6, we observed a significant and persistent growth reduction in SEMK2-M1 (MA6 treated cell numbers 46.7 % that of scrambled pool or siMM treated controls, Figure 2).
To discern the effects of AF4-MLL knockdown on cell proliferation that may not have been detected by the cell growth assay described above, we performed cell cycle analysis on SEMK2-M1 cells. Using flow cytometric analysis of DNA content, we measured the proportions of cells in the various phases of the cell cycle every 24 hours after siRNA treatments for a total of 4 days. As shown in Figure 3, the proportions of cells in G0/G1, S and G2/M phases of the cell cycle at the different time points for the AF4-MLL knocked-down cells were identical to those for cells treated with scrambled pool or mock electroporation. The sub-diploid (<2N) fraction which marks apoptotic cells was also identical among the AF4-MLL knockdown and control conditions. On the other hand, MLL-AF4 knockdown resulted in significant changes in cell cycling and survival of SEMK2-M1 cells. At 48 hours post electroporation, we observed a significant increase in the proportion of cells in the G2/M phase with MLL-AF4 knockdown (24.5 ± 1.0 %) compared to scrambled control (15.7 ± 1.4%, mean ± std. err., N=3, p<0.05, t-test). Concomitantly, we saw a decrease in the proportion of cells in G0/G1 with MLL-AF4 knockdown (45.6 ± 0.7%) compared to scrambled control (53.2 ± 0.8%, p<0.05). This G2/M cell cycle arrest gradually resolved by 96 hours post electroporation (Figure 3). Further, with MLL-AF4 knockdown we observed an increase in the <2N fraction starting at 48 hours post electroporation, peaking at 96 hours (53.8 ± 3.4% with MLL-AF4 knockdown compared to 21.8 ± 0.4% for scrambled controls, mean ± std. err., N=3, p < 0.01, t-test, Figure 3). These results demonstrate that MLL-AF4 knockdown induced G2/M cell cycle arrest and apoptosis while loss of AF4-MLL had no significant effect on proliferation or survival of SEMK2-M1 cells.
Figure 3. Knockdown of AF4-MLL has no effect on cell cycling or cell survival.
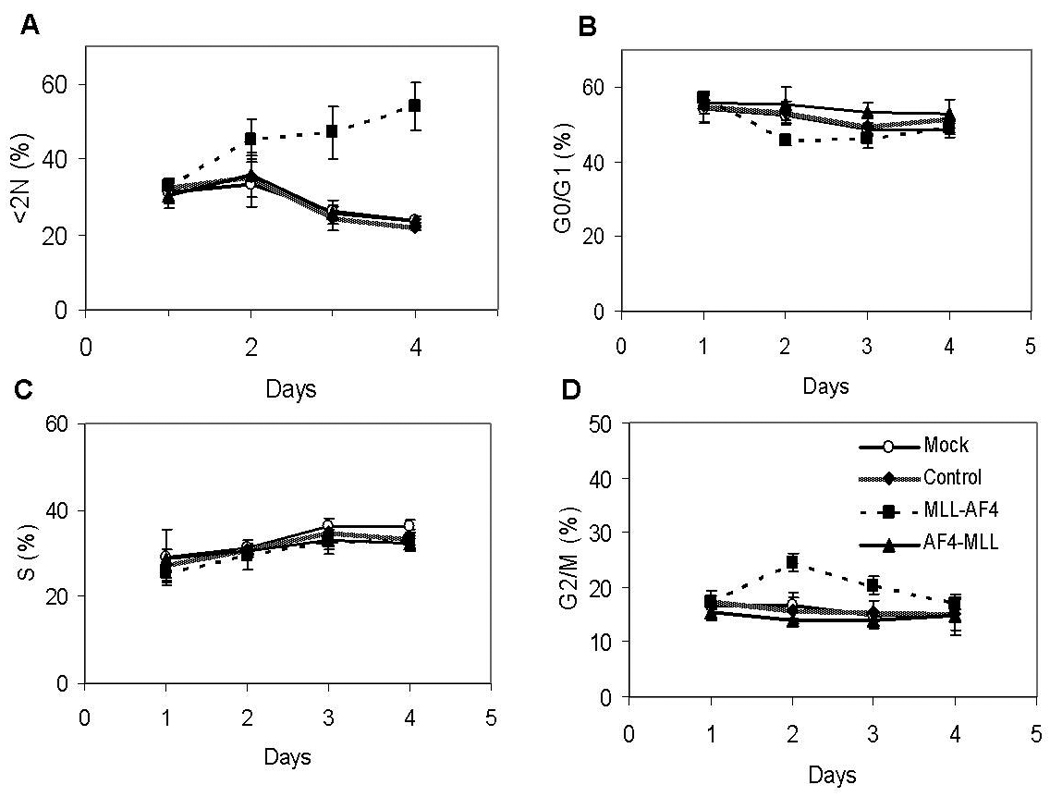
SEMK2-M1 cells were electroporated with various siRNA duplexes as described. Cell aliquots were harvested every 24 hours for a total of 4 days and DNA content analyzed by flow cytometry. Line graphs represent the proportions of cells in the subdiploid fraction [<2N, (A)] and various phases of the cell cycle [G0/G1 (B), S (C) and G2/M (D)] with the x-axis representing time in days. Depicted are mean ± std. err. of three separate experiments.
Discussion
Translocations involving the MLL gene in leukemia are uniquely characterized by the expression of both the MLL-fusion and the reciprocal fusion product in majority of the cases. The lack of detectable reciprocal fusions in up to 20% of cases may be due to complex rearrangements involving multiple genes that have been recently identified(18). Given that the der11 MLL-fusion protein is expressed in all 11q23 rearranged leukemias, all experimental models have been based on expressing this MLL-fusion gene and the data from these models demonstrate that the MLL-fusion gene indeed promotes transformation of the host cell. However, the role of the reciprocal gene has not been evaluated in detail. Our results indicate that in established t(4;11) leukemias the reciprocal product – AF4-MLL is not essential for leukemia cell growth or survival. We observed no significant effects of siRNA mediated knockdown of AF4-MLL in both RS4;11 and SEMK2-M1 cell lines. The lack of an antibody to detect AF4-MLL precludes our ability to evaluate the knockdown of the protein, but our real-time quantitative RT-PCR data show that we achieved consistent knockdown of the mRNA by >50%. Moreover, sequential electroporations with the AF4-MLL siRNA failed to show any effect on SEMK2-M1 cell growth while under identical conditions, MLL-AF4 knockdown resulted in significant growth inhibition. In a recent publication, Bursen et al report that murine Lin−/Sca1+ bone marrow cells were transformed by retrovirally expressed AF4-MLL, resulting in the development of acute leukemia in a third of the mice transplanted with AF4-MLL or AF4-MLL + MLL-AF4 expressing cells(11). While at first our results appear to contradict these published results, several considerations highlight that we cannot compare the study by Bursen et al to ours. Firstly, the two studies address different questions. In their study, Bursen et al show that leukemia may be initiated by retrovirally expressed AF4-MLL, while our results demonstrate that established t(4;11) leukemias are no longer dependent on AF4-MLL for continued growth. It is possible that events downstream of the translocation that culminate in the development of leukemia render the reciprocal fusion redundant. Additionally, the cellular origin of MLL-rearranged leukemia remains unknown. The Lin−/Sca1+ bone marrow fraction used by Bursen et al is a heterogeneous population largely composed of proliferating progenitor cells. Recently published results suggest that t(4;11) leukemias might originate in relatively more primitive cells that are pre-hematopoietic in their lineage commitment (20). Thus, the patient-derived leukemia cell lines used in our study might differ significantly in their developmental origins compared to the cells transformed by retroviral expression of AF4-MLL in the study by Bursen et al. However, it is worth noting that in contrast to the results of Bursen et al, other groups have successfully generated leukemia in murine models driven by the MLL-AF4 fusion alone (i.e. without the reciprocal fusion) either via a conditional knock-in approach or via retroviral expression(10, 21). Finally, MLL-rearranged leukemias are characterized by increased H3K79 methylation at specific loci that correlate with increased expression of the corresponding genes(10). Recent data indicate that the carboxy terminus of AF4 plays an important role in recruiting the H3K79 methyltransferase DOT1L to these loci of increased transcription, implicating the der11 MLL-AF4 as the major oncogene in t(4;11) leukemias.
Overall, our results show that leukemias with MLL-translocations remain dependent on the der11 MLL-fusion protein for continued growth while the reciprocal fusion appears to be non-essential. Therapies targeting the MLL-fusion protein itself, its interacting proteins, or its downstream mediators may thus be beneficial for treating patients with these leukemias.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Eguchi M, Eguchi-Ishimae M, Greaves M. Molecular pathogenesis of MLL-associated leukemias. Int J Hematol. 2005 Jul;82(1):9–20. doi: 10.1532/IJH97.05042. [DOI] [PubMed] [Google Scholar]
- 2.Hsieh JJ, Cheng EH, Korsmeyer SJ. Taspase1: a threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell. 2003 Oct 31;115(3):293–303. doi: 10.1016/s0092-8674(03)00816-x. [DOI] [PubMed] [Google Scholar]
- 3.Ernst P, Fisher JK, Avery W, Wade S, Foy D, Korsmeyer SJ. Definitive hematopoiesis requires the mixed-lineage leukemia gene. Dev Cell. 2004 Mar;6(3):437–443. doi: 10.1016/s1534-5807(04)00061-9. [DOI] [PubMed] [Google Scholar]
- 4.Hanson RD, Hess JL, Yu BD, Ernst P, van Lohuizen M, Berns A, et al. Mammalian Trithorax and polycomb-group homologues are antagonistic regulators of homeotic development. Proc Natl Acad Sci U S A. 1999 Dec 7;96(25):14372–14377. doi: 10.1073/pnas.96.25.14372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002 Nov;10(5):1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 6.Downing J, Head D, Raimondi S, Carroll A, Curcio-Brint A, Motroni T, et al. The der(11)-encoded MLL/AF-4 fusion transcript is consistently detected in t(4;11)(q21;q23)-containing acute lymphoblastic leukemia. Blood. 1994 January 15;83(2):330–335. 1994. [PubMed] [Google Scholar]
- 7.Chen W, Li Q, Hudson WA, Kumar A, Kirchhof N, Kersey JH. A murine Mll-AF4 knock-in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood. 2006 Jul 15;108(2):669–677. doi: 10.1182/blood-2005-08-3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corral J, Lavenir I, Impey H, Warren AJ, Forster A, Larson TA, et al. An Mll AF9 Fusion Gene Made by Homologous Recombination Causes Acute Leukemia in Chimeric Mice: A Method to Create Fusion Oncogenes. 1996;85(6):853–861. doi: 10.1016/s0092-8674(00)81269-6. [DOI] [PubMed] [Google Scholar]
- 9.Lavau C, Szilvassy SJ, Slany R, Cleary ML. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J. 1997 Jul 16;16(14):4226–4237. doi: 10.1093/emboj/16.14.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008 Nov 4;14(5):355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bursen A, Schwabe K, Ruster B, Henschler R, Ruthardt M, Dingermann T, et al. The AF4bulletMLL fusion protein is capable of inducing ALL in mice without requirement of MLLbulletAF4. Blood. Mar 1; doi: 10.1182/blood-2009-06-229542. [DOI] [PubMed] [Google Scholar]
- 12.Yao Q, Nishiuchi R, Li Q, Kumar AR, Hudson WA, Kersey JH. FLT3 expressing leukemias are selectively sensitive to inhibitors of the molecular chaperone heat shock protein 90 through destabilization of signal transduction-associated kinases. Clin Cancer Res. 2003 Oct 1;9(12):4483–4493. [PubMed] [Google Scholar]
- 13.Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, Den Boer ML, et al. Inhibition of FLT3 in MLL: Validation of a therapeutic target identified by gene expression based classification. Cancer Cell. 2003;3(2):173–183. doi: 10.1016/s1535-6108(03)00003-5. [DOI] [PubMed] [Google Scholar]
- 14.Kumar AR, Li Q, Hudson WA, Chen W, Sam T, Yao Q, et al. A role for MEIS1 in MLL-fusion gene leukemia. Blood. 2009 Feb 19;113(8):1756–1758. doi: 10.1182/blood-2008-06-163287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Q, Frestedt JL, Kersey JH. AF4 encodes a ubiquitous protein that in both native and MLL-AF4 fusion types localizes to subnuclear compartments. Blood. 1998 Nov 15;92(10):3841–3847. [PubMed] [Google Scholar]
- 16.Marschalek R, Greil J, Lochner K, Nilson I, Siegler G, Zweckbronner I, et al. Molecular analysis of the chromosomal breakpoint and fusion transcripts in the acute lymphoblastic SEM cell line with chromosomal translocation t(4;11) Br J Haematol. 1995 Jun;90(2):308–320. doi: 10.1111/j.1365-2141.1995.tb05151.x. [DOI] [PubMed] [Google Scholar]
- 17.Thomas M, Gessner A, Vornlocher HP, Hadwiger P, Greil J, Heidenreich O. Targeting MLL-AF4 with short interfering RNAs inhibits clonogenicity and engraftment of t(4;11)-positive human leukemic cells. Blood. 2005 Nov 15;106(10):3559–3566. doi: 10.1182/blood-2005-03-1283. [DOI] [PubMed] [Google Scholar]
- 18.Kowarz E, Burmeister T, Lo Nigro L, Jansen MWJC, Delabesse E, Klingebiel T, et al. Complex MLL rearrangements in t(4;11) leukemia patients with absent AF4.MLL fusion allele. Leukemia: Official Journal Of The Leukemia Society Of America, Leukemia Research Fund, UK. 2007;21(6):1232–1238. doi: 10.1038/sj.leu.2404686. [DOI] [PubMed] [Google Scholar]
- 19.Chen W, Kumar AR, Hudson WA, Li Q, Wu B, Staggs RA, et al. Malignant transformation initiated by Mll-AF9: gene dosage and critical target cells. Cancer Cell. 2008 May;13(5):432–440. doi: 10.1016/j.ccr.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menendez P, Catalina P, Rodriguez R, Melen GJ, Bueno C, Arriero M, et al. Bone marrow mesenchymal stem cells from infants with MLL-AF4+ acute leukemia harbor and express the MLL-AF4 fusion gene. J Exp Med. 2009 Dec 21;206(13):3131–3141. doi: 10.1084/jem.20091050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z, Smith KS, Murphy M, Piloto O, Somervaille TCP, Cleary ML. Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature. 2008;455(7217):1205–1209. doi: 10.1038/nature07284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.