

Abstract
The evolution of O2-producing cyanobacteria that use water as terminal reductant transformed Earth's atmosphere to one suitable for the evolution of aerobic metabolism and complex life. The innovation of water oxidation freed photosynthesis to invade new environments and visibly changed the face of the Earth. We offer a new hypothesis for how this process evolved, which identifies two critical roles for carbon dioxide in the Archean period. First, we present a thermodynamic analysis showing that bicarbonate (formed by dissolution of CO2) is a more efficient alternative substrate than water for O2 production by oxygenic phototrophs. This analysis clarifies the origin of the long debated “bicarbonate effect” on photosynthetic O2 production. We propose that bicarbonate was the thermodynamically preferred reductant before water in the evolution of oxygenic photosynthesis. Second, we have examined the speciation of manganese(II) and bicarbonate in water, and find that they form Mn-bicarbonate clusters as the major species under conditions that model the chemistry of the Archean sea. These clusters have been found to be highly efficient precursors for the assembly of the tetramanganese-oxide core of the water-oxidizing enzyme during biogenesis. We show that these clusters can be oxidized at electrochemical potentials that are accessible to anoxygenic phototrophs and thus the most likely building blocks for assembly of the first O2 evolving photoreaction center, most likely originating from green nonsulfur bacteria before the evolution of cyanobacteria.
Keywords: bicarbonate, carbon dioxide, cyanobacteria, evolution, manganese
Oxygen (O2) production by photosynthesis is by far the dominant global process that replenishes atmospheric and oceanic oxygen essential to sustain all aerobic life. Geochemical records of terrestrial oxides indicate that O2 evolution must have taken place in the precursors to cyanobacteria before ca. 2.8 billion years ago and led to the accumulation of O2 in the atmosphere (1, 2). The creation of a photosynthetic apparatus capable of splitting water into O2, protons, and electrons was the pivotal innovation in the evolution of life on Earth. For the first time photosynthesis had an unlimited source of electrons and protons by using water as reductant. By freeing photosynthesis from the availability of reduced chemical substances, the global production of organic carbon could be enormously increased and opened new environments for photosynthesis to occur. This event literally changed the face of the Earth. The accumulation of O2 in the atmosphere led to the biological innovation of aerobic respiration, which harnesses a more powerful metabolic energy source. Because aerobic metabolism generates 18 times more energy (ATP) per metabolic input (hexose sugar) than does anaerobic metabolism, the engine of life became supercharged. This sequence of evolutionary steps enabled the emergence of complex, multicellular, energy-efficient, eukaryotic organisms.
Comparisons of cyanobacteria, green algae, and contemporary plants reveal that the same inorganic core and similar reaction center core proteins have been found at the active site of all O2-producing photosynthetic organisms that have been studied to date (3–5). The available record shows that nature created only a single type of photocatalyst capable of catalyzing this reaction ca. 3 billion years ago, called the photosystem II water-oxidizing complex (PSII-WOC). The stoichiometry of the inorganic components of this core is currently believed to be Mn4OxCa1Cly (6). The absence of evolution of an enzyme's active site over such enormous time scales is unimaginable given the diversity of enzymatic catalysts that nature has invented and improved upon for other reactions in considerably shorter time scales. We believe that one of the main reasons for the lack of catalytic diversity is because the oxidation of water involves a complex, four-electron/four-proton coupled oxidation reaction that is thermodynamically the most challenging multielectron reaction in biology. The overall free energy change for the reaction is 74.6 kcal per mol of O2 (Fig. 1).
Figure 1.

Standard free energy differences and chemical equilibria in pure water and in a two-component system of water and CO2. Energies are in kcal/mol. Data from NIST sources (http://webbook.nist.gov/chemistry) (42).
Three central questions need to be answered concerning the evolutionary process that led to water oxidation in photosynthesis. First, were there transitional electron donors used by the first O2-producing phototrophs before water was adopted as the universal reductant? Second, how did the PSII photochemical apparatus evolve to generate a sufficiently strong one-electron photooxidant as precursor to chlorophyll-a (Chl-a) found in all contemporary PSII organisms? Third, because O2 evolution from water produces no free, partially oxidized, intermediates (i.e., is a concerted four-electron process), what were the evolutionary forms of the inorganic core and how did they acquire the ability to couple the four photochemical steps into a single four-electron substrate oxidation step? We shall come back to a discussion of the inorganic core after first summarizing the somewhat clearer picture of the evolution of PSII reaction centers.
Genomic and Pigment Evidence for a Reaction Center Precursor to Cyanobacterial PSII
All oxygenic photoautotrophs contain two reaction centers, whereas all nonoxygenic photoautotrophs contain only a single reaction center. X-ray and electron diffraction data on PSI (FeS type) and PSII (Fe-quinone type) reaction center complexes and numerous phylogenetic analyses of the core reaction center proteins from cyanobacteria and higher plants reveal that they have several striking structural similarities in common, both with each other and also with type II purple bacterial reaction centers (7, 8). These similarities support the long-standing hypothesis of a common evolutionary origin for all reaction center complexes, despite the large sequence divergence of type I (FeS-type) and type II (Fe-quinone type) reaction center genes (5, 9, 10). Type II reaction centers are the accepted precursor of cyanobacterial PSII. For an overview, see Govindjee and Whitmarsh (11).
Bacteriochlorophyll-a (BChl-a) has a standard potential (E0 = 0.55 Vi) that is quite sufficient for the oxidation of ferrous iron, carbon, and sulfur substrates used as electron donors by nonoxygenic phototrophs. See Fig. 2 for a plot of relevant one-electron potentials for bacterial reaction centers and PSII. The creation of a stronger photooxidant is required to split water into O2, because the reaction has a standard potential of 0.82 V per electron (pH 7). A plausible case has been made for chlorophyll pigment evolution from BChl-a to the much stronger oxidant Chl-a (E0 = 1.12 V; ref. 12) found in oxygenic organisms (10). Blankenship (10) hypothesized that evolution may have occurred via a transitional purple bacterium that first used Chl-d.
Figure 2.

Standard reduction potentials [volts vs. NHE normal hydrogen electrode)] per electron for oxidation of water, aqueous bicarbonate, the 1:2 dimanganese/bicarbonate complex, Mn2(HCO3)4, and the photooxidizable reaction center pigment found in cyanobacteria and higher plants (P680; Chl-a), purple bacteria and green bacteria (P870; BChl-a), and heliobacteria (P798; BChl-g) (31). Energies of the excited states (P*) are in electron volts.
Pigment genes, unlike reaction center genes, are common to all photosynthetic lineages and thus provide an alternative basis for measuring ancestry. Comparison of pigment genes from the green nonsulfur (a.k.a. filamentous) bacterium Chloroflexus aurantiacus, containing the type II reaction center, and the green sulfur bacterium Chlorobium tepidum, containing the type I reaction center, establishes that these organisms are each other's closest relatives and thus may represent the earliest known branching point of the two reaction center types (13). The purple bacteria are now placed as the most ancient of all emerging photosynthetic lineages. Also, Xiong et al. (13) showed that the pigment biosynthetic genes of heliobacteria are the last common ancestor of oxygenic photosynthetic lineages, the cyanobacteria, all of which use Chl-a. Heliobacteria use BChl-g as pigment, the closest evolutionary pigment precursor to Chl-a, and contain a type I reaction center (14). A modified view of the origin of the first oxygenic cyanobacteria is suggested with the Chloroflexus type II reaction center appearing to be the closest type II reaction center protein precursor to cyanobacterial PSII based on pigment genes. Although heliobacteria appear to have the closest pigment precursor to Chl-a, they have the wrong reaction center type for water oxidation. Thus, there is still a missing link to be found that would bring the pigment genes for BChl-g biosynthesis into a Chloroflexus type II reaction center precursor to cyanobacterial PSII. Another provocative hint that C. aurantiacus may be the precursor to cyanobacterial PSII is its preferential binding of manganese in place of iron in the non-heme iron site involved in secondary electron transfer out of the reaction center (15). This suggests that C. aurantiacus is adapted for preferential insertion of manganese. Examination of the inorganic physiology of the green nonsulfur bacteria would thus appear to be the best place to look for new information on the evolution of the catalyst essential for O2 evolution.
Pigment evolution alone cannot account for photosynthetic water oxidation, as the first one-electron potential for oxidation of water to hydroxyl radical is much too high (2.75 V) for any pigment oxidation. Hence, the acquisition of a catalyst is essential to access lower potential, multielectron, oxidation processes, such as the four-electron oxidation of water to O2 (0.82 V). Another solution would be to use a substrate other than water for O2 production.
Was There a Transitional Electron Donor Before Water?
Bacterial photoautotrophs use a variety of substrates that donate electrons one at a time either directly to the BChl photooxidant or indirectly via a protein radical or cytochrome carrier. These include Fe2+, H2S, Sx, formate, oxalate, and others. There are no required intermediary catalysts in bacterial photosynthesis; each turnover of the reaction center leads to one-electron oxidation of the substrate. By contrast, water oxidation in oxygenic photosynthesis is a concerted four-electron process. Thus we can ask was there a transitional multielectron donor before water was adopted as the universal reductant for oxygenic photosynthesis?
Blankenship and Hartman (10) have suggested that hydrogen peroxide (H2O2) may have been a transitional electron donor and that manganese might have been incorporated into PSII by gene fusion of a purple photosynthetic bacterium with a dimanganese catalase enzyme from a nonphotosynthetic bacterial precursor. H2O2 is much easier to oxidize than water (Fig. 2) and thus would be an feasible candidate for as reductant to the bacterial precursor of cyanobacteria. However, this hypothesis now appears to be ruled out for two reasons (6). No obvious sequence homology has been identified between the manganese catalases and the manganese binding domain of the PSII reaction center subunits. Nor is there evidence for an abundant environmental source of hydrogen peroxide in the anaerobic, mildly reducing environment thought to prevail in the Archean period.
In early work, Warburg speculated that CO2 might be the source of oxygen atoms for O2 produced by all oxygenic photosynthesis, contrary to the generally accepted view that water is the source of O2 based on subsequent mass spectrometry experiments performed by Kamen, following the availability of 18O-enriched water (16). Stemler and Govindjee were the first to observe an effect of dissolved CO2 on electron transport (17), and they suggested a possible site for bicarbonate binding on the donor side of PSII involved in stimulating water oxidation. This view was further advocated by Stemler (18) but later refuted by Radmer and Ollinger based on isotopic studies (19). These contradictions, together with the discovery of a low affinity binding site for bicarbonate on the PSII acceptor side, at the non-heme iron site involved in stimulating secondary electron transfer steps out of PSII, shifted attention away from a possible site for bicarbonate functioning on the donor side. An enlightening historical account of this early work is now available (20), with the unfortunate omission of the seminal work by Metzner.
Arguably, Metzner's group gave us the only evidence implicating bicarbonate as a directly oxidizable electron donor to PSII, rather than merely an activator of water oxidation (21, 22). In their final report on this topic, they showed that if 18O-bicarbonate was added to suspensions of algal cells or thylakoids, the photosynthetically generated O2 was transiently enriched in the heavy isotope (23). This enrichment disappeared on a time scale slower than the rate of equilibration of the 18O-bicarbonate with water. The isotope effect is exactly opposite to that expected if water was the primary electron donor to PSII. This observation led to their hypothesis that an unidentified bicarbonate-modified species, designated X(HCO3−), can compete with water as an electron donor to PSII. They were never successful in identifying the X cofactor, nor localizing its site of action within PSII. Their model hypothesized that oxidation of X(HCO3−), might possibly form either the bicarbonate radical (HCO3⋅) or peroxidicarbonic acid (HOOC-O-O-COOH) via dimerization. Neither species was ever observed.
More recently, Klimov's group and their collaborators followed up on these earlier works and found compelling evidence for involvement of bicarbonate in stimulating electron donation from Mn2+ to apo-WOC-PSII, in which the inorganic core was first removed (24–27). Importantly, they found that bicarbonate increases the binding affinity and photooxidation rate of the first two MnII ions, implicating a high affinity pair of MnII ions involved in bicarbonate-dependent electron transfer from MnII (27). They also found definitive evidence for bicarbonate enhancing the rate of light-induced charge separation within the intact holo-WOC-PSII at a site on the donor side of PSII, as well as for stabilizing the holo-WOC-PSII against thermal deactivation (27–29). We speculate that this greater thermal stability could have provided an evolutionary advantage to PSII precursors in the Archean period when ambient temperatures are predicted to have been substantially greater than today (1). The Pushchino group has proposed a direct role for bicarbonate as an intrinsic cofactor involved in stimulation of water oxidation within the WOC. However, direct spectroscopic evidence identifying the location and characteristics of the binding site for bicarbonate in the WOC (rather than alternatively delivering OH−) is still lacking.
Baranov and coworkers (30) provided a new piece of the puzzle by showing that extremely low levels of bicarbonate (<25 μM) also accelerate the rate of binding of Mn2+ ions to apo-WOC-PSII and facilitate their light-driven photooxidation during assembly of the functional Mn4 core during reconstitution of O2 evolution (biogenesis by photoactivation). Two models were favored to account for the data, including the possibility that bicarbonate either binds directly as integral cofactor or delivers hydroxide ion needed for formation of Mn(OH)+ at its binding site within apo-WOC. This result indicates a second role for bicarbonate during biogenesis of the Mn4 core in addition to its role in stimulating the O2 evolution rate in the intact holoenzyme.
In the next section we present an equilibrium thermodynamic argument that has not been previously articulated to explain the “bicarbonate effect” on the donor side of PSII. We shall show that the data as a whole are consistent with bicarbonate serving as a competitive electron donor that can replace water as substrate, rather than merely as an auxiliary cofactor to stimulate water oxidation.
Thermodynamics of Oxygen Production from Water/Bicarbonate in the Archean Period
Fig. 1 lists the relative free energies of the species that form in a simple two-component system comprised of CO2 dissolved in water vs. pure water alone. Because CO2 is a weak acid, it dissolves to form carbonic acid (H2CO3) and, by dissociation, bicarbonate ion and a proton. The concentration of bicarbonate increases with the partial pressure of atmospheric CO2 in an equilibrium system, and this is how nature can control the availability of bicarbonate for O2 production. The equilibrium constants for both dissolution of CO2 and ionization to form bicarbonate are less than unity and thus require the input of free energy (3.9 and 8.6 kcal/mol at pH 7, respectively). This free energy is provided by the increase in partial pressure of CO2. These equilibria allow us to calculate a lower limit to the concentration of bicarbonate dissolved in the oceans during the Archean period, using the range of estimates for the partial pressure of atmospheric CO2 in the Archean (Table 1). The resulting range that is predicted in water is 30 to 30,000 times greater than today. This calculation ignores activity corrections (decreases the increase), buffering from other species (unknown), assumes the same temperature (favors more bicarbonate), and ignores dissolution of mineral sources of bicarbonate (favors more dissolved bicarbonate). Although the range of predicted concentrations is large, even the lowest bicarbonate concentration predicted to have existed in the Archean ocean would have been very substantial; certainly it would have been well in excess of that needed to saturate the bicarbonate binding site that stimulates photosynthetic O2 evolution in all oxygenic organisms studied to date.
Table 1.
CO2, HCO3−, and Mn2+ speciation and concentrations in the Archean ocean
Importantly, the two-component system of CO2/water has a much higher buffer capacity than pure water. This means that for each mole of CO2 that is dissolved in water, only 8.6 kcal/mol is required for dissociation into bicarbonate and proton, whereas dissociation of 1 mol of water to hydroxide and protons requires 21.4 kcal/mol (Fig. 1). Hence, any system that can use bicarbonate as a source for generation of hydroxide will need to input only 8.9 kcal/mol to release it from CO2, vs. 21.4 kcal/mol to ionize pure water. Moreover, if the system can use bicarbonate as a direct surrogate for hydroxide, then no further energy input is needed in the two-component system! This advantage of bicarbonate as a source for hydroxide can be equivalently expressed in terms of the dissociation constants:
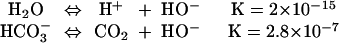 |
In other words, at neutral pH a solution containing 0.1 M bicarbonate ion will contain 106-fold more bicarbonate ion than the concentration of hydroxide ion in pure water.
This brings us to the key issue relevant to the thermodynamics of water oxidation. By use of the thermodynamic cycle summarized in Fig. 1, we can calculate the experimentally unmeasured free energy change for the oxidation of bicarbonate ion to 0.5 mol of O2 and compare this to the directly measured oxidation of water to O2. These energies are 24.8 and 37.3 kcal/mol, respectively. In other words, it is 12.5 kcal/mol easier (34% lower free energy) to produce 0.5 mol of O2 from bicarbonate than from water. This very substantial energetic advantage provides a quantitative thermodynamic rationalization for the observations noted above for bicarbonate stimulation of O2 evolution rate in the intact WOC and also Metzner's observation of the transient incorporation of 18O from 18O-bicarbonate into photosynthetic O2. Thus, bicarbonate is thermodynamically a better electron donor than water for O2 evolution, rather than merely a cofactor that stimulates the enzyme to oxidize water more efficiently. Fig. 1 also implies that the Archean period with its high concentration of dissolved bicarbonate had available a stronger reductant than water for the first inefficient attempts at evolution of an oxygenic reaction center. We hypothesize that bicarbonate, not water, was the transitional electron donor that facilitated the evolution of the bacterial photosynthetic precursor of cyanobacterial PSII. However, the data in Fig. 1 do not explain the bicarbonate stimulation of the binding of manganese ions and their photooxidation by apo-WOC-PSII during biogenesis. We consider this in the next section.
Speciation of Manganese-Bicarbonate Solutions in a Model Archean Ocean
Molecular archaeologists have focused on the genomic record of protein and pigment evolution and have largely ignored the chemical evolution of the Earth as a controlling factor in the availability and speciation of the inorganic components used for assembly of the inorganic core of the WOC. We have obtained several lines of evidence showing that bicarbonate specifically alters the speciation of Mn2+ ions in solution and its redox properties. To illustrate the change in speciation, we show in Fig. 3 representative electrochemical data for the reduction of Mn2+ to Mn0 at a Hg electrode as a function of bicarbonate concentration (pH 8.3) (Y.N.K., A. A. Kazakova, V.V.K., and G.C.D., unpublished work). One observes two transitions corresponding to the formation of two species having different bicarbonate binding constants. From the slopes of these plots (14 mV and 60 mV), one is able to obtain directly the stoichiometry of binding, while the intercepts provide the standard potentials and the formation constants (32). The slopes show that the two species that form between Mn2+ and bicarbonate have stoichiometries equal to 2:1 and 1:2. These correspond to complexes with empirical formulas, MnII2(HCO3)3+ and [MnII(HCO3)2]n. The remaining coordination sites in the first coordination shell will be occupied by water molecules, by analogy to MnIIaq, which coordinates six water molecules in the first shell. The formation constants (KB) that were obtained for these complexes are given in Table 2. By contrast, only the 1:1 complexes form with acetate or formate, and these have a lower affinity (data not shown). These data show that bicarbonate specifically induces formation of manganese clusters having apparent dimanganese composition.
Figure 3.

Representative electrochemical data for the reduction of a solution of MnII (2.5 × 10−4 M MnSO4) to Mn0 at a Hg electrode as a function of the concentration of bicarbonate (pH 8.3, 0.1 M LiCLO4). Voltage scanning at 50 mV s−1. Inset shows the standard reduction potentials and formulas of the MnII species that form.
Table 2.
Manganese(II)-bicarbonate equilibrium speciation in water
Electrochemical oxidation of the MnII clusters to the MnIII state was also performed as a function of bicarbonate concentration and provided the standard potentials (E0) given in Table 2. We found that oxidation of the MnII-bicarbonate clusters in the presence of excess bicarbonate leads to formation of MnIII-bicarbonate clusters at potentials (E0 = 0.61–0.52 V) that are far lower relative to MnIIaq (E0 = 1.18 V). These potential shifts are sufficiently large that they would enable Mn-bicarbonate clusters to function as electron donors to anoxygenic phototrophs (Fig. 2). However, the electrochemical data do not reveal whether bicarbonate complexes to manganese or delivers hydroxide to form the corresponding Mn-hydroxo/oxo species.
Evidence in the literature shows that Mn-bicarbonate solutions catalyze the multielectron dismutation of hydrogen peroxide (H2O2 → O2 + 2H2O), so-called catalase activity (33, 34). Stadtman (33) additionally showed that the rate of Mn-dependent dismutation depended on the bicarbonate concentration to the third power, suggesting the formation of an active species with 1:3 Mn/bicarbonate stoichiometry. We now know from the studies described herein that their experimental conditions lead to formation of MnII-bicarbonate clusters as the active species. It is also known that manganese catalases found in biology contain exclusively dimanganese centers, and that efficient abiotic manganese catalase model complexes contain di- or multimanganese centers. Considering these results, we may attribute the catalase activity of Mn-bicarbonate solutions to the formation of Mn-bicarbonate clusters, possibly MnII2(HCO3)4 produced from MnII2(HCO3)3+. Both complexes are oxidizable by peroxide by forming the Mn2(III,III) oxidation state as intermediate in the two-electron/two-proton-coupled dismutation reaction.
Additional evidence in support of formation of Mn-bicarbonate oligomers in solution comes from variable temperature electron paramagnetic resonance (EPR) experiments on samples that do not contain an electrolyte (LiClO4) but include 60% glycerol as glassing agent. Fig. 4 gives a plot of the EPR intensity for the MnIIaq ion as a function of bicarbonate concentration (six-line spectrum in Inset). This signal is well known to be caused by the monomeric MnIIaq ion. Upon addition of bicarbonate, the MnIIaq signal disappears and is replaced by an unstructured broad signal from a second MnII species (Fig. 4) whose intensity grows and saturates in reciprocal proportion to the loss of MnIIaq. The EPR titration data are compared with the solid line showing the fit to an equilibrium binding reaction: n Mn2+aq + 2n HCO3− ↔ Mn(HCO3)2n. Other stoichiometries gave poorer fits to the data. All titration data were recorded at a fixed time after mixing because the system was found to be time-dependent because of slow precipitation of MnCO3 crystals. The broad EPR signal appears to be attributable to the precursor(s) to solid MnCO3(s). Unlike MnIIaq the bicarbonate-induced MnII EPR signal is detectable only below 70 K, exhibits a non-Curie temperature dependence of signal intensity and a temperature-dependent g-value, and undergoes rapid spin relaxation. These properties differ greatly from isolated MnII ions and indicate that the new species corresponds to an oligomer of MnII ions that interact via both electronic exchange (chemical bonding) and magnetic dipolar interactions (35). Comparisons to simple dimanganese complexes and extended solids indicate that the Mn ions must share one or more bridging ligands, presumably bicarbonate, hydroxide, or carbonate ions. These EPR data are consistent with the electrochemical data in showing that MnIIaq forms a MnII oligomer upon complexation or reaction with bicarbonate rather than remaining monomeric. The composition of the Mn-bicarbonate oligomer is under investigation.
Figure 4.

Titration of the EPR signal intensities as function of bicarbonate concentration for the MnIIaq species (g2 six-line spectrum, Inset) and the broad signal that replaces it upon addition of bicarbonate (broad signal, Inset). The broad signal is detectable only below 70 K, and it exhibits strong spin relaxation and a temperature-dependent linewidth and a g value indicative of formation of a MnIIx-bicarbonate oligomer.
A Proposal for the Origin and Evolution of the Inorganic Core Responsible for Photosynthetic O2 Evolution
The electrochemical data above indicate that dilute solutions of MnII in water above a concentration of 10 mM bicarbonate exist primarily as Mn-bicarbonate (or hydroxide) clusters of 2:1 and 1:2 composition. The measured stability constants for their formation (Table 2) together with estimates for the MnIIaq concentration in the Archean ocean (Table 1) indicate that these MnII clusters would have represented the dominant form of soluble MnII present in the Archean ocean, unlike today where the speciation favors the monomeric aquo ion MnIIaq. The pKa of MnIIaq is 10.5. Hence, at the pH of the contemporary ocean (≈8), the fractional concentration of Mn(OH)+ would be vanishing small (10−2.5 × MnIIaq) if there was no bicarbonate to serve as hydroxide source. In the Archean ocean (pH ≈ 6.5–7), it would have been even smaller (36). Thus, bicarbonate, not free hydroxide, is the major source of hydrolytic species formed from MnIIaq, including Mn(OH)+, in both the contemporary and Archean oceans.
Fig. 2 compares the standard potentials (per electron) for oxidation of water, bicarbonate, and the dimanganese bicarbonate complex Mn2(HCO3)4 vs. the reaction center pigments found in cyanobacteria and higher plants (P680; Chl-a), purple and green nonsulfur bacteria (P870; BChl-a), and green sulfur bacteria and heliobacteria (P840; BChl-a). The Mn-bicarbonate complexes fall much closer to the potential generated by anoxygenic bacterial reaction centers than to the PSII oxygenic reaction center of cyanobacteria. Thus, the Mn-bicarbonate clusters would be readily oxidizable substrates for photosynthetic bacteria, even though free bicarbonate or water are thermodynamically inaccessible as reductants. Additional evidence has accumulated showing that Mn-bicarbonate precursors are highly efficient in the assembly of the tetramanganese-oxo core of PSII during biogenesis (30).
In Fig. 5, we give a hypothesis for the sequence of events that may describe how the inorganic core of the WOC-PSII was first created and evolved from anoxygenic bacteria. We suggest that preformed, abiotic dimanganese-bicarbonate clusters served initially as terminal substrates to primitive anoxygenic phototrophs, such as the green nonsulfur bacteria (Mn-bicarbonate oxidase, stage 1). Mn-bicarbonate clusters would have been feasible, although inefficient, electron donors to photobacteria owing to the mismatch in electrochemical potentials (Fig. 2). In the next stage, two features may have been adopted in the Archean period that characterize the “missing link” in evolutionary development: (i) mutations in the reaction center proteins occurred that favored binding of a tetramanganese-bicarbonate cluster derived from the Mn-bicarbonate clusters present in the environment, and (ii) evolution of a higher potential photooxidant, such as BChl-g, the suggested evolutionary precursor pigment to Chl-a (13). These developments would have enabled bicarbonate to serve as an inefficient terminal substrate for the concerted four-electron oxidation to O2. Hence we call this stage the bicarbonate oxidase stage. The most recent proposed stage of development represents the emergence of cyanobacteria and is denoted as the water oxidase stage. This stage was brought on by the enormous reduction of atmospheric CO2 in the post-Archean period. Although it is unclear how this transition occurred, it would have required the evolution of a stronger inorganic catalyst and a stronger photooxidant to split water efficiently (Fig. 1). We suggest that this developmental stage may correspond to the incorporation of calcium as integral cofactor within the tetramanganese cluster (6). The calcium cofactor boosts the electrochemical potential of the tetramanganese core in the contemporary WOC, thus permitting weaker reductants such as water to serve as terminal substrates. Also, the adoption of a stronger photooxidant such as Chl-a would have greatly increased the quantum efficiency of water oxidation, owing to its considerably higher potential (Fig. 2). It is possible that this final stage of development also included the incorporation of basic amino acid residues in the reaction center protein environment to serve as proton acceptors, thus replacing the lost function of bicarbonate as hydroxide buffer during assembly of the inorganic core.
Figure 5.

(Lower) Proposed evolutionary stages of development of type II bacterial reaction centers towards cyanobacterial (oxygen-evolving) reaction centers in the Archean period. (Upper) Electrochemical potentials of the reaction center photooxidant (P) and terminal substrates (D = formate, oxalate, etc.).
Acknowledgments
This paper is dedicated to Gerald A. Soffer, astrobiology pioneer.
We thank T. Carrell, A. Bocarsly, A. Stemler, and R. Blankenship for proofreading and helpful discussions. Our research was supported by the National Institutes of Health (Grant GM39932), the Russian Foundation for Basic Research (Grant No.9904–48308), and a NATO collaborative linkage grant.
Abbreviations
- BChl-a
bacteriochlorophyll-a
- Chl-a
chlorophyll-a
- EPR
electron paramagnetic resonance
- PSI and PSII
photosystems I and II
- WOC
water-oxidizing complex
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Des Marais D J. Science. 2000;289:1703–1704. [PubMed] [Google Scholar]
- 2.Holland H, Rye R. Am J Sci. 1998;298:621–672. doi: 10.2475/ajs.298.8.621. [DOI] [PubMed] [Google Scholar]
- 3.Debus R J. Biochim Biophys Acta. 1992;1102:269–352. doi: 10.1016/0005-2728(92)90133-m. [DOI] [PubMed] [Google Scholar]
- 4.Debus R. In: Metal Ions in Biological Systems. Sigel H, editor. Vol. 37. New York: Dekker; 2000. pp. 657–710. [PubMed] [Google Scholar]
- 5.Mulkidjanian A Y, Junge W. Photosynth Res. 1997;51:27–42. [Google Scholar]
- 6.Ananyev G, Zaltsman L, Vasko C, Dismukes G C. Biochim Biophys Acta Bioenerg. 2001;1503:52–68. doi: 10.1016/s0005-2728(00)00215-2. [DOI] [PubMed] [Google Scholar]
- 7.Schubert W-D, Klukas O, Saenger W, Witt H-T, Fromme P, Krauss N. J Mol Biol. 1998;280:297–314. doi: 10.1006/jmbi.1998.1824. [DOI] [PubMed] [Google Scholar]
- 8.Barber J, Nield J J, Morris E P, Hankamer B. Trends Biochem Sci. 1999;24:42–45. doi: 10.1016/s0968-0004(98)01348-6. [DOI] [PubMed] [Google Scholar]
- 9.Nitscke W, Rutherford A W. Trends Biochem Sci. 1991;16:241–245. doi: 10.1016/0968-0004(91)90095-d. [DOI] [PubMed] [Google Scholar]
- 10.Blankenship R E, Hartman H. Trends Biochem Sci. 1998;23:94–97. doi: 10.1016/s0968-0004(98)01186-4. [DOI] [PubMed] [Google Scholar]
- 11.Govindjee, Whitmarsh J. In: Concepts in Photobiology: Photosynthesis and Photomorphogenesis. Singhal G S, Renger G R, Sopory S K, Irrgang K-D, Govindjee, editors. New Delhi: Narosa; 1997. pp. 11–51. [Google Scholar]
- 12.Klimov, V. V., Allakhverdiev, S. I., Demeter, S. & Krasnovsky, A. A. (1979) Dokl. Akad. Nauk. USSR (Russian)249.
- 13.Xiong J, Fisher W M, Inoue K, Narakhara M, Bauer C E. Science. 2000;289:1724–1730. doi: 10.1126/science.289.5485.1724. [DOI] [PubMed] [Google Scholar]
- 14.Amesz J. In: The Molecular Biology of Cyanobacteria. Bryant D, editor. Dordrecht, The Netherlands: Kluwer; 1995. pp. 687–697. [Google Scholar]
- 15.Feick R, Shiozawa J A, Ertlmaier A. In: Anoxygenic Photosynthetic Bacteria. Blankenship R E, Madigan M T, Bauer C, editors. Dordrecht, The Netherlands: Kluwer; 1995. pp. 699–708. [Google Scholar]
- 16.Kamen M. Photosynth Res. 1989;21:137–144. doi: 10.1007/BF00037177. [DOI] [PubMed] [Google Scholar]
- 17.Stemler A, Govindjee Plant Physiol. 1973;52:119–123. doi: 10.1104/pp.52.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stemler A. Biochim Biophys Acta. 1980;543:103–112. doi: 10.1016/0005-2728(80)90011-0. [DOI] [PubMed] [Google Scholar]
- 19.Radmer R, Ollinger O. FEBS Lett. 1980;110:57–61. doi: 10.1016/0014-5793(86)80178-8. [DOI] [PubMed] [Google Scholar]
- 20.Van Rensen J J S, Xu C, Govindjee Physiol Plant. 1999;105:585–592. [Google Scholar]
- 21.Metzner H, Gerster R. Photosynthetica. 1976;10:302–306. [Google Scholar]
- 22.Metzner H. In: Photosynthetic Oxygen Evolution. Metzner H E, editor. New York: Academic; 1978. pp. 59–76. [Google Scholar]
- 23.Metzner H, Fischer K, Bazlen O. In: Photosynthesis. Akoynoglou G, editor. II. Philadelphia: Balaban; 1981. pp. 375–387. [Google Scholar]
- 24.Klimov V V, Allakhverdiev S I, Feyziev Y M, Baranov S V. FEBS Lett. 1995;363:251–255. doi: 10.1016/0014-5793(95)00327-6. [DOI] [PubMed] [Google Scholar]
- 25.Klimov V V, Allakhverdiev S I, Baranov S V, Feyziev Y M. Photosynth Res. 1995;46:219–225. doi: 10.1007/BF00020434. [DOI] [PubMed] [Google Scholar]
- 26.Allakhverdiev S I, Yruela I, Picorel R, Klimov V V. Proc Natl Acad Sci USA. 1997;94:5050–5054. doi: 10.1073/pnas.94.10.5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klimov V V, Baranov S V, Allakhverdiev S I. FEBS Lett. 1997;48:243–246. doi: 10.1016/s0014-5793(97)01392-6. [DOI] [PubMed] [Google Scholar]
- 28.Klimov V V, Hulsebosch R, Allakhverdiev S I, Wncencjusz H, van Gorkom H, Hoff A. Biochemistry. 1997;36:16277–16281. doi: 10.1021/bi9717688. [DOI] [PubMed] [Google Scholar]
- 29.Wincencjusz H, Allakhverdiev S I, Klimov V V, van Gorkom H J. Biochim Biophys Acta. 1996;1273:1–3. [Google Scholar]
- 30.Baranov S V, Ananyev G M, Klimov V V, Dismukes G C. Biochemistry. 2000;39:6060–6065. doi: 10.1021/bi992682c. [DOI] [PubMed] [Google Scholar]
- 31.Blankenship R E, Prince R C. Trends Biochem Sci. 1985;10:382–383. [Google Scholar]
- 32.Rieger P H. Electrochemistry. New York: Chapman & Hall; 1990. [Google Scholar]
- 33.Stadtman E R, Berlett P B, Chock P B. Proc Natl Acad Sci USA. 1990;87:384–388. doi: 10.1073/pnas.87.1.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sychev A Y, Isac V G. Russ Chem Rev. 1993;62:279–290. [Google Scholar]
- 35.Khangulov S V, Pessiki P J, Barynin V V, Ash D, Dismukes G C. Biochemistry. 1995;34:2015–2025. doi: 10.1021/bi00006a023. [DOI] [PubMed] [Google Scholar]
- 36.Holland H D. Chemical Evolution of the Atmosphere and Oceans. Princeton: Princeton Univ. Press; 1984. [Google Scholar]
- 37.Kasting J. Nature (London) 1993;364:759–760. [Google Scholar]
- 38.Walker J C G. Origins Life. 1985;16:117–127. doi: 10.1007/BF01809466. [DOI] [PubMed] [Google Scholar]
- 39.Bruhland K. In: Chemical Oceanography. Riley J P, Chester R, editors. Vol. 8. London: Academic; 1983. pp. 147–220. [Google Scholar]
- 40.Smith R M, Martell A E. Critical Stability Constants. New York: Plenum; 1976. [Google Scholar]
- 41.Kozlov Y N, Klimov V V. Membr Cell Biol. 1997;11:115–120. [PubMed] [Google Scholar]
- 42.Snoeyink V, Jenkins D. Water Chemistry. New York: Wiley; 1980. [Google Scholar]