

Abstract
Cytochrome P450 CYP125A1 of Mycobacterium tuberculosis, a potential therapeutic target for tuberculosis in humans, initiates degradation of the aliphatic chain of host cholesterol and is essential for establishing M. tuberculosis infection in a mouse model of disease. We explored the interactions of CYP125A1 with a reverse type I inhibitor by x-ray structure analysis and UV-vis spectroscopy. Compound LP10 (α-[(4-methylcyclohexyl)carbonyl amino]-N-4-pyridinyl-1H-indole-3-propanamide), previously identified as a potent type II inhibitor of Trypanosoma cruzi CYP51, shifts CYP125A1 to a water-coordinated low spin state upon binding with low micromolar affinity. When LP10 is present in the active site, the crystal structure and spectral characteristics both demonstrate changes in lipophilic and electronic properties favoring coordination of the iron axial water ligand. These results provide an insight into the structural requirements for developing selective CYP125A1 inhibitors.
Keywords: P450, CYP125, Mycobacterium tuberculosis, Cholesterol degradation, CYP125-inhibitor complex, x-ray structure
The catalytic functions elucidated for four Mycobacterium tuberculosis P450 enzymes [1-6] and physiological functions demonstrated for two of them, CYP121A1 and CYP125A1 [3-6], testify to their essential roles in M. tuberculosis physiology, and highlight their potential as targets for an urgently needed next generation of anti-tuberculosis drugs. A re-emergent pathogen, M. tuberculosis has become resistant to combinations of standard and reserve medications.
The utilization of host cholesterol, mediated in part by the igr operon (Rv3540c-Rv3545c), is essential for M. tuberculosis infectivity and persistence and is predicted to play a role in lipid metabolism [7-11]. As an element of the igr operon, CYP125A1 initiates degradation of the cholesterol aliphatic chain by means of C-27 monooxygenase activity [4–6, 12]. Knockout of the cyp125a1 gene alters lipid composition and causes accumulation of the intermediate cholest-4-en-3-one, which may be one of the physiological substrates of CYP125A1 [4, 6]. X-ray structures of CYP125A1 determined both in the ligand-free form and soaked with androstenedione or econazole [5], and also as a complex co-crystallized with the substrate cholest-4-en-3-one [6], revealed a narrow substrate binding site defining the selective ω-hydroxylation activity of CYP125A1 [13]. Identification of this feature gives a starting point for the rational design of inhibitors targeting this protein.
Cytochrome P450 enzymes respond to ligand binding with changes in the metal center electronic state, indicated by a red- or blue-shift of the heme iron Soret band. Soret shifts in cytochrome P450 spectra are typically caused either by expulsion of the iron distal water ligand or by direct coordination of the heme iron to an aromatic or aliphatic nitrogen atom of the incoming small molecule. Spectral changes associated with these interactions are referred to in the literature as type I or type II respectively [14]. These spectral characteristics have been routinely utilized to identify P450 inhibitors or substrates, including in high throughput assays [15, 16]. Both types of complexes are structurally well characterized, with many examples deposited in the Protein Data Bank (PDB) (http://www.rcsb.org/pdb/home/home.do). Binding with spectral characteristics referred to as “reverse type I” or “modified type II” [17] has also been observed in many instances with different P450 enzymes [18-20]. Spectrally, reverse type I mode is manifested by a shift of the Soret band to 420 nm, reflecting a transition from the high- to low-spin form. Initially it was suggested that reverse type I spectra result from coordination of the hydroxyl group of the incoming ligand to the heme iron [17, 21, 22]. However, it has been observed that molecules that do not carry a hydroxyl group also elicit reverse type I spectra upon binding to P450, suggesting that the heme-ligand interactions may be mediated by a water molecule [20]. Structurally, water molecule-mediated binding is represented by the P450cam-2-phenylimidazole [23] and CYP121-fluconazole [24] complexes, both of which retain an axial water ligand bound to the heme iron upon binding of the inhibitor. Fluconazole in the CYP121-fluconazole complex H-bonds with the iron axial water and partially shifts the equilibrium toward the low-spin form [24]. In both cases, steric constraints imposed by the protein active site prevent formation of the energetically more favorable direct coordination bond between the heme iron and the lone pair of electrons on the aromatic nitrogen atom of the inhibitor.
Inhibitors of the azole class developed by the pharmaceutical industry to target CYP51 in sterol biosynthetic pathways of pathogenic microorganisms have demonstrated anti-mycobacterial efficacy, both in vitro [25-30] and in suppressing M. tuberculosis infection in the mouse model of the disease [30, 31]. Azole inhibitors are therefore routinely tested for binding against all recombinant heterologously expressed M. tuberculosis CYPs [32, 33]. The narrow substrate binding site of CYP125A1 enforces an unusual binding behavior on antifungal azoles, which would otherwise coordinate to the heme iron via the aromatic nitrogen atom to produce a type II shift of the Soret band. Both fluconazole and ketoconazole have been shown to produce partial type II shifts in the range of 20-35% coordination [5], whereas econazole induced a nearly complete high-spin transition [34].
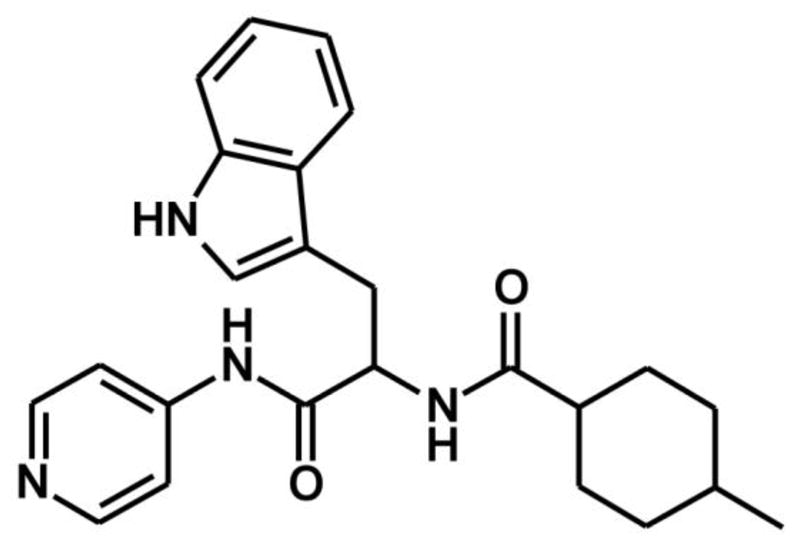
Here, we report that the non-azole compound, α-[(4-methylcyclohexyl)carbonyl amino]-N-4-pyridinyl-1H-indole-3-propanamide (Fig. 1), previously identified as a potent type II inhibitor of Trypanosoma cruzi CYP51 and referred as LP10 [35, 36], binds in a different manner to CYP125A1. Upon binding with LP10, the spin equilibrium of the CYP125A1 heme iron, which is a mixture of high-spin and low-spin states in the ligand-free form [4-6], shifts entirely to the hexa-coordinated low-spin state. To understand the structural bases of the interactions of LP10 with CYP125A1 and elucidate the origin of the reverse type I spectra, we determined the crystal structure of the CYP125A1-LP10 complex.
Fig. 1. Chemical structure of α-[(4-methylcyclohexyl)carbonyl amino]-N-4-pyridinyl-1H-indole-3-propanamide, LP10.
Material and methods
Reagents
Compound LP10 (α-[(4-methylcyclohexyl)carbonyl amino]-N-4-pyridinyl-1H-indole-3-propanamide) was purchased as a racemic mixture from ChemDiv (# C155-0123) (San Diego, California). Cholest-4-en-3-one, spinach ferredoxin, spinach ferredoxin-NADP+-reductase, bovine liver catalase, glucose-6-phosphate dehydrogenase, and glucose-6-phosphate were purchased from Sigma-Aldrich (St. Louis, MO).
Cloning, expression and purification of CYP125A1
The wild-type cyp125A1 gene was cloned from M. tuberculosis H37Rv genomic DNA as described previously [34]. To improve crystallization, a CYP125A1C429L protein construct was made in which the N-terminus was truncated at Val17, Cys429 was substituted with a leucine, and a His6-tag was engineered at the N-terminus upstream of Val17 via modification of the pCWori expression vector [37]. PCR amplification of the truncated cyp125A1 gene with the amino acid substitution was carried out using the forward primer 5′-ACAGCCCTGCATATGCCCAGCCCCAATCTGCC-3′ and the reverse primer 5′-CCAAGCTTCTAGATTAGTGAGCAACCGGGCATC-3′. Letters in boldface indicate an engineered NdeI restriction cloning site in the forward primer, including the initiation codon ATG which replaced Val17 with methionine. The underlined letters in the reverse primer indicate a HindIII restriction cloning site. The amino acid substitution was performed by site-directed mutagenesis following the QuikChange protocol (Stratagene). The truncated cyp125A1 gene was used as a template to generate C429L mutation using the set of complementary primers 5′-GACTACACCGGTAGACTCCCGGTTGCTCACTAA-3′ and 5′-TTAGTGAGCAACCGGGAGTCTACCGGTGTAGTC-3′. The DNA construct was confirmed by sequencing. The truncated CYP125A1C429L mutant was expressed in Escherichia coli DH5α cells co-transformed with pCWori/cyp125C429L and pGro7 plasmids (Takara) encoding for CYP125A1C429L and the E. coli GroEL/GroES chaperones, respectively. Cells were grown at 37°C in Terrific broth (TB) medium containing 100 μg/ml of ampicillin, 40 μg/ml of chloramphenicol and 0.2% (w/v) of L-arabinose. When OD590 reached 0.8, expression of CYP125A1C429L was induced with 500 μM isopropyl β-D-1-thiogalactopyranoside (IPTG) in the presence of 0.25 mM σ-aminolevulinic acid. The temperature was decreased from 37 °C to 25 °C and the culture continued for 36 h. The protein was purified as described previously [38]. Upon CO binding, the sodium dithionate-reduced protein exhibited a typical spectrum with maximum at 450 nm and significant absorbance at 420 nm, indicating heterogeneity in heme coordination by the cysteine axial ligand. The concentration of the 450 nm form was determined based on an extinction coefficient of 91000 M−1cm−1 [39] from the difference in absorbance between 450 and 490 nm, while the total protein concentration was estimated by heme content quantified using the pyridine hemochrome method [40].
Spectroscopic binding assays
All ligand binding assays were performed at room temperature by spectrophotometric titration in 50 mM potassium phosphate buffer (pH 7.4) containing 0.1 mM EDTA using a Cary UV-visible scanning spectrophotometer (Varian). Stock solutions of LP10 were prepared in DMSO. To account for the absorbance of the test compound, 1 ml of protein (2.5 μM) in buffer was placed in the first chamber of two split cuvettes and 1 ml of buffer was placed in the second chamber. After background scanning, equal volumes (0.25 to 1.0 μl) of ligand solution were titrated into both the second chamber of the reference cuvette containing only buffer and the first chamber of the sample cuvette containing the protein. The same volume of solvent was added into the alternate chambers to correct for organic solvent effects. Difference spectra were recorded from 300 to 500 nm. To determine the KD values, titration data points were fitted to Equation 1 using Kaleidagraph software (Synergy). In this equation, Aobs is the absorption shift determined at any ligand concentration; Amax is the maximal absorption shift obtained at saturation; KD is the dissociation constant for the inhibitor-enzyme complex; S is the ligand concentration; Et is the total enzyme concentration.
| Equation 1 |
CYP125A1 catalytic activity assay
Enzymatic reactions (0.5 ml) were carried out both for the wild-type and the CYP125A1C429L mutant at 25 °C in glass tubes containing 50 mM potassium phosphate, pH 7.5, supplemented with 0.05% (v/v) Tween-20. The enzyme (1.5 μM) was pre-incubated for 2 h at 25 °C with the substrate, cholest-4-en-3-one. Reactions were initiated by adding spinach ferredoxin, spinach ferredoxin-NADP+ reductase, catalase, magnesium chloride and an NADPH-regenerating system consisting of glucose-6-phosphate dehydrogenase and glucose-6-phosphate as described elsewhere [2]. The CYP125A1 specific activity was determined at saturating substrate concentrations. Inhibition of enzymatic activity was tested by the addition of increasing concentrations (0–200 μM) of LP10. NADP+ was omitted in the negative control. The enzymatic reactions and negative controls were quenched by adding 1 volume of acetonitrile in 0.1% formic acid and centrifuging at 10,000 rpm for 3 min. A 100-μl aliquot from each reaction was injected into the HPLC column and monitored at 240 nm. The HPLC system was an Agilent Technologies 1200 Series equipped with a photodiode array detector connected to a Polaris C8 column (250 × 4.6 mm i.d. stainless steel, 5 μm particles, Varian). Substrates and reaction products were separated using an isocratic mobile phase consisting of H2O:acetonitrile:methanol (25:25:50) with 0.1% (v/v) formic acid at a flow rate of 1 ml/min.
Crystallization, data collection and crystal structure determination
All crystals reported in this work were obtained from the engineered truncated CYP125A1C429L form. The initial screening of crystallization conditions was performed with commercial high-throughput screening kits (Hampton Research), a nanoliter drop-setting Mosquito robot (TTP; Labtech) operating with 96-well plates, and a hanging-drop crystallization protocol. The protein was from a 1 mM frozen stock in 20 mM Tris (pH 7.5), 0.5 mM EDTA and 0.25 M NaCl. The 50 mM LP10 stock solution was prepared in acetonitrile. Prior to co-crystallization the protein was diluted to 0.2 mM by mixing with 10 mM Tris HCl (pH 7.5) alone or supplemented with 0.3 mM LP10. Crystals of the CYP125C249L-LP10 complex grew at 4 °C in a solution of 2.0 M ammonium sulfate, 0.1 M bis-Tris (pH 5.5) and 0.5 M NaCl in 4 μl hanging drops in 24-well plates. Two distinct crystal forms were obtained for the ligand-free CYP125A1C429L at 4 °C: one in a solution of 0.1 M ammonium acetate, 0.1 M Bis-Tris (pH 5.5) and 17% PEG 10000 (2X5L), and another in a solution of 2.0 M ammonium sulfate and 0.1 M Tris HCl (pH 8.5) (2XN8). The crystals were harvested by first plunging them into a drop of reservoir solution supplemented with 20% (v/v) glycerol for cryo-protection, then flash freezing them in liquid nitrogen. Diffraction data were collected at 100–110 K at Beamline 8.3.1, Advanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA. Data indexing, integration and scaling were conducted with the ELVES automated software suite [41]. Crystal structures were determined by molecular replacement using the atomic coordinates of CYP125A1 (3IVY) as a search model. Automated model building using ARP/wARP [42] placed the majority of the polypeptide chain in the asymmetric unit. The remaining residues were built manually with COOT [43], alternated with positional refinement using REFMAC5 [44]. Refinement of 2X5L converged with R and Rfree of 15.0% and 20.1% respectively, and Ramachandran statistics of 95.6% residues in preferred regions, 4.1% in allowed regions, and 0.3% (1 residue, Thr311) outliers. Refinement of 2XN8 converged with R and Rfree of 14.1% and 20.0%, respectively, and Ramachandran statistics of 95.6% residues in preferred regions and 4.4% in allowed regions. Refinement of the CYP125A1C429L-LP10 complex converged with R and Rfree of 12.2% and 17.7%, respectively, and Ramachandran statistics of 95.7% residues in preferred regions, 4.0% in allowed regions, and 0.3% (1 residue, Thr311) outliers. Data collection and refinement statistics are provided in Table 1.
TABLE 1.
Data collection and refinement statistics.
| Protein | CYP125C429L | CYP125C429L | CYP125C429L |
|---|---|---|---|
| Ligand | none | none | LP10 |
| PDB ID | 2X5L | 2XN8 | 2XC3 |
| Data collection | |||
| Space group | P212121 | P212121 | P212121 |
| Cell dimensions | |||
| a, b, c (Å) | 54.5, 74.6, 102.2 | 54.4, 76.3, 90.4 | 54.4, 76.4, 90.4 |
| Molecules in AU | 1 | 1 | 1 |
| Wavelength | 1.1159 | 1.1159 | 1.1159 |
| Resolution (Å) | 1.48 | 1.64 | 1.50 |
| Rsym or Rmerge (%) | 7.4 (55.2)1 | 14.3 (37.0) | 4.4 (13.7) |
| I / σI | 9.5 (1.5) | 9.0 (2.2) | 26.0 (6.5) |
| Completeness (%) | 94.9 (71.6) | 99.0 (94.3) | 91.9 (63.0) |
| Redundancy | 4.1 (1.9) | 6.1 (3.3) | 7.1 (4.3) |
| Refinement | |||
| No. reflections | 63143 | 43947 | 53185 |
| Rwork / Rfree (%) | 15.0/20.1 | 14.1/21.0 | 12.2/17.7 |
| No. atoms | |||
| Protein | 3327 | 3236 | 3271 |
| Heme | 43 | 43 | 43 |
| Ligand | none | 30 | |
| Water | 714 | 464 | 584 |
| Mean B value | 18.6 | 16.0 | 16.0 |
| B-factors | |||
| Protein | 16.0 | 14.2 | 13.2 |
| Heme | 9.0 | 9.1 | 8.9 |
| Ligand | none | none | 21.3 |
| Water | 31.2 | 29.0 | 32.0 |
| R.m.s deviations | |||
| Bond lengths (Å) | 0.024 | 0.023 | 0.024 |
| Bond angles (°) | 2.0 | 1.83 | 1.86 |
Values in parentheses are for highest-resolution shell.
RESULTS AND DISCUSSION
Design of the truncated CYP125A1C429L mutant
Full-length CYP125A1 was not easily amenable to crystallization in our hands, and we thus introduced sequence modifications to improve crystallization properties. First, reiterating an earlier successful strategy to reduce the heterogeneity of recombinant M. tuberculosis CYP51 [45], Cys429 was replaced with leucine. Second, following the observation of Capyk et al. that heterologous expression from full-length cyp125A1 gene in Rhodococcus jostii RHA1 results in production of two CYP125A1 variants of different sizes, suggesting Val17 as a second potential start codon [4], we truncated the N- terminus at Val17. Third, to facilitate purification, a His6-tag was introduced at the N-terminus. After confirming that these modifications affected neither the binding nor the catalytic properties of the enzyme, the modified form was used for further studies.
Analysis of LP10 binding by UV-vis spectroscopy
Probing the CYP125A1 active site, both in the wild-type and in the CYP125A1C429L mutant, with compounds identified as type II inhibitors in other P450 enzymes, we singled out LP10 (Fig. 1) as the instigator of atypical spectral behavior of the CYP125A1-LP10 complex. Binding of LP10 to CYP125A1 stabilized the low-spin state of the heme iron, completely shifting the spin equilibrium to the low-spin form characterized by a Soret band at 420 nm and α and β bands at 569 and 536 nm respectively (thick trace, Fig. 2). For comparison, titration with imidazole resulted in the formation of a more red-shifted type II complex characterized by a Soret band at 425-426 nm (dashed trace). Thus, to obtain the KD value, binding LP10 to CYP125A1C429L was monitored via the reverse type I shift of the Soret band (Fig 3A). The titration plot was best fitted to Equation 1, with KD of 1.68 ± 0.08 μM.
Fig. 2. UV-visible absorption spectra of M. tuberculosis CYP125A1C429L.

The Soret (A) and enlarged visible (B) regions are shown. Spectra of the resting ferric (thin line), ferric LP10 (thick line), and ferric imidazole (dashed line) forms were recorded at the total hemoprotein concentration of 6.45 μM in 50 mM potassium phosphate buffer (pH 7.4) containing 0.1 mM EDTA at 23 °C.
Fig. 3. Binding of LP10 to M. tuberculosis CYP125A1C429L.

A. Concentration dependence of LP10 binding deduced from difference absorption changes obtained from titration of protein with increasing concentrations of inhibitor. A representative set of the non-corrected difference spectra obtained using the two-chamber cuvettes is shown in the inset. B, Inhibition of CYP125A1 enzymatic activity by LP10. Concentration of the substrate cholest-4-en-3-one used for the inhibition assay was 50 μM.
LP10’s affinity is an order of magnitude lower than that of cholesterol or cholest-4-en-3- one (although some variability in values is reported in the literature [4, 6]), and thus LP10 inhibited the catalytic conversion of cholest-4-en-2-one inefficiently. With concentrations of CYP125A1 at 50 μM and LP10 at 200 μM, only 30% inhibition was achieved (Fig. 3B).
X-ray structure of the CYP125A1C429L-LP10 complex
The modifications introduced in the CYP125A1 sequence were essential to obtain LP10 co-crystals. With respect to the CYP125A1C429L ligand-free forms, binding of LP10 caused virtually no conformational changes. Despite the fact that an LP10 racemic mixture was used for co-crystallization, an individual (R)-diastereomer with a trans-configuration of the methylcyclohexyl moiety was found in the active site. LP10 failed to penetrate deeply into the narrow tunnel accommodating the aliphatic chain of cholest-4-en-3-one [6]. Instead, the branched LP10 molecule filled the space between the B, G and I helices (highlighted in purple in Fig. 4A) normally occupied by the tetracyclic steroid nucleus, and the empty adjacent pocket observed in the CYP125A1-cholest-4-en-3-one complex. The pyridinyl ring of LP10 pointed toward the heme, whereas the methylcyclohexyl and indole moieties were oriented toward the protein surface. Two water molecules mediated protein-ligand interactions via the amide nitrogen atoms of LP10 (Fig. 4B). The closest contacts of the indole ring were within 4 Å of Phe100, Val111, Gln112 and Lys214. The indole aromatic nitrogen atom of LP10 H-bonded to the carbonyl oxygen of Asp108. The methylcyclohexyl group resided in a more hydrophobic environment. The methyl group on the cyclohexane ring made extensive hydrophobic interactions within 5 Å of the Val111, Phe114, Phe260, Ile221, Ser217 and Lys214 residues, with the terminal oxygen and nitrogen atoms of Ser217 and Lys214, respectively, pointing away from the inhibitor (Fig. 4C). Removal of this single methyl group from the LP10 molecule resulted in an approximately seven-fold drop in binding affinity (data not shown). In contrast to the CYP125A1-cholest-4-en-3-one complex, Lys214 adopted alternate conformations instead of forming a salt bridge with Asp108.
Fig. 4. Structure of the CYP125A1C429L-LP10 complex.

A. Overall ribbon structure of the CYP125A1C429L-LP10 complex (2XC3) with B, G and I helices highlighted in purple. LP10 (yellow), heme (orange) and the iron axial water ligand (red) are shown in stick mode. The pyridinyl ring of LP10 points toward heme. The indole and methylcyclohexyl moieties point toward the viewer between the B and G helices. B. A view of LP10 emphasizing the H-bonding interactions in the active site. Selected residues within 4 Å of LP10 are in purple. Alternate conformations of Lys214 interact with both indole and methylcyclohexyl moieties of LP10. C. Interactions of the methylcyclohexyl moiety within 5 Å of the methyl group. D. Catalytic chamber filled with three water molecules. In B, C and D, fragments of the 2Fo-Fc electron density map (calculated with LP10 coordinates omitted from the input file) are shown for heme, LP10, selected amino acid residues (blue mesh) and water molecules (pink mesh). Black lines highlight H-bonding interactions.
Spatial constraints prevent LP10 from penetrating sufficiently into the active site to directly coordinate to the heme iron. A cluster of three water molecules engaged in an H-bonding network was trapped in the catalytic chamber between the heme iron and the pyridinyl moiety of the inhibitor. The LP10 pyridinyl ring H-bonds to one of the water molecules in the cluster (distance 2.75 Å), while the other one, closest to the heme iron, serves as its axial ligand (coordination bond length 2.17 Å) (Fig. 4D). As usual, the carbonyl group of the conserved alanine 268 in the I helix provides an H-bond to the axial water ligand (distance 2.6 Å).
X-ray structure of CYP125A1C429L in the ferric resting state
The truncated CYP125A1C429 was also easily crystallized in two distinct ligand-free forms, both very similar two each other (r.m.s.d=0.38 Å for Cα carbon atoms) and to previously reported ligand-free full-length CYP125A1 structures [5], suggesting that neither the truncation nor cysteine substitution affected the overall protein structure. The main difference between the all four substrate-free forms is in the binding of the iron axial water ligand, a behavior which sheds light on the unusual spectral characteristics of CYP125A1.
Based on the UV-vis spectroscopy, CYP125A1 tends to convert to the penta-coordinated high-spin state even in the absence of substrate. The proportion of the high-spin state in a CYP125A1 preparation has been reported to reach 70-80% [4, 5]. This spectral ambiguity accords with the uncertainty in the position of the distal water as deduced from the x-ray structures of substrate-free CYP125A1 determined in this work and elsewhere [5]. In 2X5L, a water axial ligand was modeled in the elongated electron density map adjacent to the heme iron (Fig. 5). The shape of the electron density was best approximated by two water molecules with a partial occupancy of 0.5 and separated by 1.46 Å, suggesting oscillation of the water ligand between ligated (within 2.24 Å of the heme iron) and non-ligated positions. Oscillation of the water ligand may switch the coordination bond on-and-off, which would explain the mixed characteristics of the CYP125A1 spectrum. In 2XN8, the distal water molecule was 2.65 Å away from the iron and off the heme orthogonal, suggesting a weak coordination bond. The displacement of the iron atom from the porphyrin plane toward the proximal Cys377 thiolate ligand is in accord with partial occupancy of the coordination site. In the CYP125A1-cholest-4-en-3-one complex [6], the iron atom moves even further out of the porphyrin plane toward Cys377. In both crystal structures reported by McLean et al., the distal water molecule coordinates to the heme iron, and ambiguity in the Val267 conformation has been reported [5], although we did not observe such ambiguity. In both ligand-free structures in this work, Val267 is involved in conventional α-helix H-bonding interactions, but the water molecule is largely off-coordination. Thus, the mixed spectral characteristics of CYP125A1 correlate well with the on-and-off binding equilibrium of the distal water, which may be synchronized with the Val267 conformational transitions exposing its carbonyl group to the oxygen binding site. We speculate that the increased hydrophobicity of the CYP125A1 oxygen binding site may ultimately account for poor coordination of the axial water to the heme iron and its tendency to dissociate. Indeed, the CYP125A1 catalytic site is extremely hydrophobic, being made up of Leu117, Ala268, Val313, Phe316, and the methyl group of Thr272, all of which are within 6 Å of the distal water (Fig. 5). This speculation accords with our prior observations on CYP130, where a single glycine to alanine substitution in the conserved position in the oxygen binding site increased hydrophobicity enough to facilitate release of the axial water [46].
Fig. 5. Structure of CYP125A1C429L in the ferric resting state.
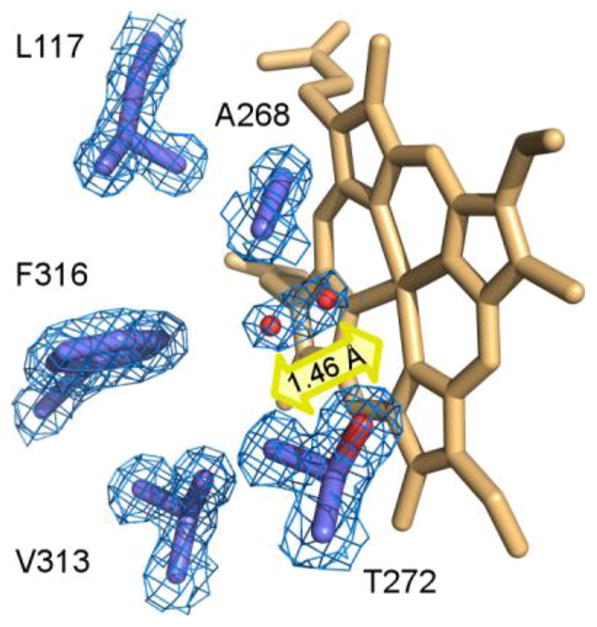
Catalytic chamber of CYP125A1C429L (2X5L) surrounded by residues within 6 Å of the heme iron axial water ligand (purple sticks). Two water molecules (red spheres) with partial occupancy of 0.5 are placed into elongated electron density (blue mesh) 1.46 Å away from each other. The line connecting both molecules runs at a 45° angle to the heme plane. A fragment of the 2Fo-Fc map was calculated with the water coordinates omitted from the input file. Heme is in orange. The yellow arrow suggests an oscillation of the water ligand between ligating and non-ligating positions.
In summary, we have identified an inhibitory scaffold that binds CYP125A1 with low micromolar affinity without utilizing the heme iron’s capacity to coordinate to the aromatic nitrogen atom. However, this affinity is not high enough to efficiently inhibit cholest-4-en-3-one hydroxylation. The structure of the CYP125A1-LP10 complex suggests that extension of the LP10 scaffold into the buried lipophilic tunnel toward the Fe center is necessary for tight recognition by CYP125A1. This is an often-used strategy in structure-based drug design because under optimal conditions correct placement of a single methyl group can increase affinity up to tenfold [47]. Both crystal structure and spectral characteristics demonstrate that changes in the lipophilic and electronic properties caused by LP10 in the active site force the water ligand into coordination to the heme iron, resulting in reverse type I spectral characteristics. This is in contrast to the ferric resting state, where probable oscillation of the iron axial water ligand between the coordinated and non-coordinated positions manifests itself in a mixed low-spin/high-spin spectrum. Thus, the conventional sequence of events is reversed in the CYP125A1-LP10 complex, with the incoming inhibitor stabilizing rather than expelling the axial water, and enhancing rather than diminishing the low-spin character of the binding spectrum.
Acknowledgments
We thank Mr. Potter Wickware for critical reading of the manuscript, the staff members of beamline 8.3.1, James Holton, George Meigs and Jane Tanamachi, at the Advanced Light Source at Lawrence Berkeley National Laboratory, for assistance with data collection. This work was supported by National Institutes of Health RO1 Grants GM078553 (to L.M.P.), GM25515 and AI74824 (to P.O.M.). The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
Data deposition note: The atomic coordinates and structure factors (codes 2X5L, 2XN8 and 2XC3) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bellamine A, Mangla AT, Nes WD, Waterman MR. Characterization and catalytic properties of the sterol 14alpha-demethylase from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 1999;96:8937–8942. doi: 10.1073/pnas.96.16.8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnston J, Kells PM, Podust LM, Ortiz de Montellano PR. Biochemical and structural characterization of CYP124, a methyl-branched lipid w-hydroxylase from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2009;106:20687–20692. doi: 10.1073/pnas.0907398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belin P, Le Du MH, Fielding A, Lequin O, Jacquet M, Charbonnier J-B, Lecoq A, Thai R, Courcon M, Masson C, Dugave C, Genet R, Pernodet J-L, Gondry M. Identification and structural basis of the reaction catalyzed by CYP121, an essential cytochrome P450 in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2009;106:7426–7431. doi: 10.1073/pnas.0812191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Capyk JK, Kalscheuer R, Stewart GR, Liu J, Kwon H, Zhao R, Okamoto S, Jacobs WR, Jr, Eltis LD, Mohn WW. Mycobacterial cytochrome P450 125 (CYP125) catalyzes the terminal hydroxylation of C27 steroids. J Biol Chem. 2009;284:35534–35542. doi: 10.1074/jbc.M109.072132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLean KJ, Lafite P, Levy C, Cheesman MR, Mast N, Pikuleva IA, Leys D, Munro AW. The Structure of Mycobacterium tuberculosis CYP125: molecular basis for cholesterol binding in a P450 needed for host infection. J Biol Chem. 2009;284:35524–35533. doi: 10.1074/jbc.M109.032706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ouellet H, Guan S, Johnston JB, Chow ED, Kells PM, Burlingame AL, Cox JS, Podust LM, de Montellano PR. Mycobacterium tuberculosis CYP125A1, a steroid C27 monooxygenase that detoxifies intracellularly generated cholest-4-en-3-one. Mol Microbiol. 2010;77:730–742. doi: 10.1111/j.1365-2958.2010.07243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang JC, Harik NS, Liao RP, Sherman DR. Identification of Mycobacterial genes that alter growth and pathology in macrophages and in mice. J Infect Dis. 2007;196:788–795. doi: 10.1086/520089. [DOI] [PubMed] [Google Scholar]
- 8.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang JC, Miner MD, Pandey AK, Gill WP, Harik NS, Sassetti CM, Sherman DR. igr Genes and Mycobacterium tuberculosis cholesterol metabolism. J Bacteriol. 2009;191:5232–5239. doi: 10.1128/JB.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yam KC, D’Angelo I, Kalscheuer R, Zhu H, Wang JX, Snieckus V, Ly LH, Converse PJ, Jacobs WR, Jr, Strynadka N, Eltis LD. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 2009;5:e1000344. doi: 10.1371/journal.ppat.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu Y, van der Geize R, Besra GS, Gurcha SS, Liu A, Rohde M, Singh M, Coates A. 3-Ketosteroid 9alpha-hydroxylase is an essential factor in the pathogenesis of Mycobacterium tuberculosis. Mol Microbiol. 2010;75:107–121. doi: 10.1111/j.1365-2958.2009.06957.x. [DOI] [PubMed] [Google Scholar]
- 12.Rosloniec KZ, Wilbrink MH, Capyk JK, Mohn WW, Ostendorf M, van der Geize R, Dijkhuizen L, Eltis LD. Cytochrome P450 125 (CYP125) catalyses C26-hydroxylation to initiate sterol side-chain degradation in Rhodococcus jostii RHA1. Mol Microbiol. 2009;74:1031–1043. doi: 10.1111/j.1365-2958.2009.06915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnston JB, Ouellet H, Podust LM, Ortiz de Montellano PR. Structural control of cytochrome P450-catalyzed w-hydroxylation. Arch Biochem Biophys. 2010 doi: 10.1016/j.abb.2010.08.011. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schenkman JB, Remmer H, Estabrook RW. Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol Pharmacol. 1967;3:113–123. [PubMed] [Google Scholar]
- 15.Podust LM, von Kries JP, Nasser Eddine A, Kim Y, Yermalitskaya LV, Kuehne R, Ouellet H, Warrier T, Altekoster M, Lee J-S, Rademann J, Oschkinat H, Kaufmann SHE, Waterman MR. Small molecule scaffolds for CYP51 inhibitors identified by high-throughput screening and defined by x-ray crystallography. Antimicrob Agents Chemother. 2007;51:3915–3923. doi: 10.1128/AAC.00311-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.von Kries JP, Warrier T, Podust LM. Identification of small-molecule scaffolds for p450 inhibitors. Curr Protoc Microbiol. 2010;Chapter 17(Unit17):14. doi: 10.1002/9780471729259.mc1704s16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schenkman JB, Cinti DL, Orrenius S, Moldeus P, Kraschnitz R. The nature of the reverse type I (modified type II) spectral change in liver microsomes. Biochemistry. 1972;11:4243–4251. doi: 10.1021/bi00773a008. [DOI] [PubMed] [Google Scholar]
- 18.Wilson BJ, Orrenius S. A study of the modified type II spectral change produced by the interaction of agroclavine with cytochrome P-450. Biochim Biophys Acta. 1972;261:94–101. doi: 10.1016/0304-4165(72)90318-2. [DOI] [PubMed] [Google Scholar]
- 19.Vaz AD, Coon MJ, Peegel H, Menon KM. Substituted pyridines: nonsteroidal inhibitors of human placental aromatase cytochrome P-450. Drug Metab Dispos. 1992;20:108–112. [PubMed] [Google Scholar]
- 20.Shimada T, Tanaka K, Takenaka S, Foroozesh MK, Murayama N, Yamazaki H, Guengerich FP, Komori M. Reverse type I binding spectra of human cytochrome P450 1B1 induced by flavonoid, stilbene, pyrene, naphthalene, phenanthrene, and biphenyl derivatives that inhibit catalytic activity: a structure-function relationship study. Chem Res Toxicol. 2009;22:1325–1333. doi: 10.1021/tx900127s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshida Y, Kumaoka H. Studies on the substrate-induced spectral change of cytochrome P-450 in liver microsomes. J Biochem. 1975;78:55–68. [PubMed] [Google Scholar]
- 22.Kumaki K, Sato M, Kon H, Nebert DW. Correlation of type I, type II, and reverse type I difference spectra with absolute changes in spin state of hepatic microsomal cytochrome P-450 iron from five mammalian species. J Biol Chem. 1978;253:1048–1058. [PubMed] [Google Scholar]
- 23.Poulos TL, Howard AJ. Crystal structures of metyrapone- and phenylimidazole-inhibited complexes of cytochrome P-450cam. Biochemistry. 1987;26:8165–8174. doi: 10.1021/bi00399a022. [DOI] [PubMed] [Google Scholar]
- 24.Seward HE, Roujeinikova A, McLean KJ, Munro AW, Leys D. Crystal structure of the Mycobacterium tuberculosis P450 CYP121-fluconazole complex reveals new azole drug-P450 binding mode. J Biol Chem. 2006;281:39437–39443. doi: 10.1074/jbc.M607665200. [DOI] [PubMed] [Google Scholar]
- 25.McLean KJ, Marshall KR, Richmond A, Hunter IS, Fowler K, Kieser T, Gurcha SS, Besra GS, Munro AW. Azole antifungals are potent inhibitors of cytochrome P450 mono-oxygenases and bacterial growth in mycobacteria and streptomycetes. Microbiology. 2002;148:2937–2949. doi: 10.1099/00221287-148-10-2937. [DOI] [PubMed] [Google Scholar]
- 26.Ahmad Z, Sharma S, Khuller GK. In vitro and ex vivo antimycobacterial potential of azole drugs against Mycobacterium tuberculosis H37Rv. FEMS Microbiology Letters. 2005;251:19–22. doi: 10.1016/j.femsle.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 27.Ahmad Z, Sharma S, Khuller GK, Singh P, Faujdar J, Katoch VM. Antimycobacterial activity of econazole against multidrug-resistant strains of Mycobacterium tuberculosis. Int J Antimicrob Agents. 2006;28:543–544. doi: 10.1016/j.ijantimicag.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 28.Ahmad Z, Sharma S, Khuller GK. The potential of azole antifungals against latent/persistent tuberculosis. FEMS Microbiology Letters. 2006;258:200–203. doi: 10.1111/j.1574-6968.2006.00224.x. [DOI] [PubMed] [Google Scholar]
- 29.Banfi E, Scialino G, Zampieri D, Mamolo MG, Vio L, Ferrone M, Fermeglia M, Paneni MS, Pricl S. Antifungal and antimycobacterial activity of new imidazole and triazole derivatives, A combined experimental and computational approach. J Antimicrob Chemother. 2006;58:76–84. doi: 10.1093/jac/dkl182. [DOI] [PubMed] [Google Scholar]
- 30.Byrne ST, Denkin SM, Gu P, Nuermberger E, Zhang Y. Activity of ketoconazole against Mycobacterium tuberculosis in vitro and in the mouse model. J Med Microbiol. 2007;56:1047–1051. doi: 10.1099/jmm.0.47058-0. [DOI] [PubMed] [Google Scholar]
- 31.Ahmad Z, Sharma S, Khuller GK. Azole antifungals as novel chemotherapeutic agents against murine tuberculosis. FEMS Microbiology Letters. 2006;261:181–186. doi: 10.1111/j.1574-6968.2006.00350.x. [DOI] [PubMed] [Google Scholar]
- 32.McLean KJ, Munro AW. Structural biology and biochemistry of cytochrome P450 systems in Mycobacterium tuberculosis. Drug Metab Rev. 2008;40:427–446. doi: 10.1080/03602530802186389. [DOI] [PubMed] [Google Scholar]
- 33.Ouellet H, Johnston JB, Ortiz de Montellano PR. The Mycobacterium tuberculosis cytochrome P450 system. Archives of Biochemistry and Biophysics. 2010;493:82–95. doi: 10.1016/j.abb.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ouellet H, Lang J, Couture M, Ortiz de Montellano PR. Reaction of Mycobacterium tuberculosis cytochrome P450 enzymes with nitric oxide. Biochemistry. 2009;48:863–872. doi: 10.1021/bi801595t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C-K, Doyle PS, Yermalitskaya LV, Mackey ZB, Ang KKH, McKerrow JH, Podust LM. Trypanosoma cruzi CYP51 inhibitor derived from a Mycobacterium tuberculosis screen hit. PLoS Negl Trop Dis. 2009;3:e372. doi: 10.1371/journal.pntd.0000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doyle PS, Chen C-K, Johnston JB, Hopkins SD, Leung SSF, Jacobson MP, Engel JC, McKerrow JH, Podust LM. A non-azole CYP51 inhibitor cures Chagas Disease in a mouse model of acute infection. Antimicrob Agents Chemother. 2010 doi: 10.1128/AAC.00281-10. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barnes HJ, Arlotto MP, Waterman MR. Expression and enzymatic activity of recombinant cytochrome P450 17 alpha-hydroxylase in Escherichia coli. Proc Natl Acad Sci U S A. 1991;88:5597–5601. doi: 10.1073/pnas.88.13.5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ouellet H, Podust LM, de Montellano PR. Mycobacterium tuberculosis CYP130: crystal structure, biophysical characterization, and interactions with antifungal azole drugs. J Biol Chem. 2008;283:5069–5080. doi: 10.1074/jbc.M708734200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. II. Solubilization, purification, and properties. J Biol Chem. 1964;239:2379–2385. [PubMed] [Google Scholar]
- 40.Appleby CA. Purification of Rhizobium cytochromes P-450. Methods Enzymol. 1978;52:157–166. doi: 10.1016/s0076-6879(78)52018-1. [DOI] [PubMed] [Google Scholar]
- 41.Holton J, Alber T. Automated protein crystal structure determination using ELVES. Proc Natl Acad Sci U S A. 2004;101:1537–1542. doi: 10.1073/pnas.0306241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 44.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 45.Podust LM, Yermalitskaya LV, Lepesheva GI, Podust VN, Dalmasso EA, Waterman MR. Estriol bound and ligand-free structures of sterol 14a-demethylase. Structure. 2004;12:1937–1945. doi: 10.1016/j.str.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 46.Podust LM, Ouellet H, von Kries JP, Ortiz de Montellano PR. Interaction of Mycobacterium tuberculosis CYP130 with heterocyclic arylamines. J Biol Chem. 2009;284:25211–25219. doi: 10.1074/jbc.M109.017632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]