

Abstract
The coagulation and fibrinolytic systems contribute to malignancy by increasing angiogenesis, tumor growth, tumor invasion, and tumor metastasis. Oncogenic transformation increases the expression of tissue factor (TF) that results in local generation of coagulation proteases and activation of protease-activated receptor (PAR)-1 and PAR-2. We compared the PAR-dependent expression of urokinase plasminogen activator (uPA) and plasminogen activator inhibitor (PAI)-1 in 2 murine mammary adencocarcinoma cell lines: metastatic 4T1 cells and nonmetastatic 67NR cells. 4T1 cells expressed TF, PAR-1 and PAR-2 whereas 67NR cells expressed TF and PAR-1. We also silenced PAR-1 or PAR-2 expression in the 4T1 cells. We discovered 2 distinct mechanisms for PAR-dependent expression of uPA and PAI-1. First, we found that factor Xa or thrombin activation of PAR-1 led to a rapid release of stored intracellular uPA into the culture supernatant. Second, thrombin transactivation of a PAR-1/PAR-2 complex resulted in increases in PAI-1 mRNA and protein expression. Cells lacking PAR-2 failed to express PAI-1 in response to thrombin and factor Xa did not activate the PAR-1/PAR-2 complex. Our results reveal how PAR-1 and PAR-2 on tumor cells mediate crosstalk between coagulation and fibrinolysis.
Introduction
Tissue factor (TF) is the cell surface receptor for coagulation factor VII/VIIa (FVII/VIIa). TF is expressed on various cell types and its expression is up-regulated by oncogenic transformation, conferring a procoagulant phenotype to cancer cells.1–4 Tumor cell TF locally activates the coagulation cascade when clotting factors in the blood enter the stroma from leaky tumor vasculature. Indeed, coagulation proteases, such as factor VIIa (FVIIa), factor Xa (FXa), and thrombin, have been shown to contribute to tumor proliferation, migration, and angiogenesis.5–8
The mechanism by which coagulation proteases exert their tumor enhancing effects is, in part, via activation of protease-activated receptor (PAR)-1 and PAR-2.9–12 PARs belong to a family of G-protein coupled receptors that are proteolytically activated by a variety of proteases. FXa and thrombin activate PAR-1, whereas FVIIa and FXa activate PAR-2.9,13,14 In addition, PAR-1 can be activated by matrix metalloproteinase-1, plasmin, and activated protein C bound to endothelial protein C receptor (APC-EPCR).13,15–17 Trypsin, tryptase, kallikreins, and matriptase activate PAR-2 (for review, see Trejo18). Furthermore, PARs can transactivate one another. For instance, thrombin can bind to the N-terminus of PAR-3, which acts as a cofactor that allows the protease to activate PAR-4 and induce intracellular signaling in mouse platelets.19 In human platelets PAR-1 and PAR-4 both signal. In addition, PAR-1 forms a heterodimer with PAR-4 to initiate signaling in response to thrombin.20 Furthermore, O'Brien and colleagues demonstrated, using pharmacological inhibitors of PAR-1 and a mutant PAR-1 incapable of signaling, that thrombin cleaved PAR-1 can transactivate PAR-2 in human endothelial cells and transfected COS-7 cells.21
Urokinase plasminogen activator (uPA) is a serine protease that converts plasminogen to plasmin. Plasminogen activator inhibitor (PAI)-1 is the endogenous inhibitor of uPA that forms a heterotrimeric complex with uPA and the uPA receptor (uPAR).22 Aside from their roles in regulating fibrinolysis, both uPA and PAI-1 promote metastasis.22–25 Furthermore, uPA and PAI-1 are also involved in angiogenesis and endothelial cell migration.26–28 Increased levels of uPA and PAI-1 are found in breast cancer patients, both correlating with poor prognosis and reduced survival.29,30 Proteolytic activation of PAR-2 by FVIIa bound to TF increases uPA, PAI-1, and uPAR mRNA expression in human breast cancer and pancreatic cancer cell lines.31,32 PAR-1 and PAR-2 agonist peptides also increase both uPA and PAI-1 mRNA levels in the MDA-MD-231 human breast cancer cell line.31
The aim of this study was to determine the role of PAR-1 and PAR-2 in the crosstalk between coagulation proteases and the regulation of plasmin generation in breast cancer cells. We used nonmetastatic (67NR) and metastatic (4T1) murine mammary adenocarcinoma cell lines because TF, coagulation proteases, uPA, and PAI-1 have been shown to contribute to metastasis. Here we describe 2 different mechanisms by which FXa and thrombin regulate uPA release and PAI-1 mRNA expression.
Methods
Reagents
Recombinant mouse FVIIa (mFVIIa) was provided by Dr Lars Petersen (Novo Nordisk). Purified human factor X (FX), FXa, and α-thrombin were obtained from Haematologic Technologies Inc. Puromycin dihydrochloride was obtained from Mediatech. Brefeldin A (BFA) was purchased from BD Biosciences. Dimethyl sulfoxide, Triton X-100, DAPI (4′,6-diamidino-2-phenylindole), and dithiothreitol were obtained from Sigma-Aldrich. 4-β-phorbol-12-myristate 13-acetate (PMA) was purchased from Cell Signaling Technologies. Complete protease inhibitor cocktail tablets and phosphatase inhibitor cocktail were purchased from Roche.
Cell culture
67NR and 4T1 mouse mammary adenocarcinoma cell lines were obtained from Dr. Fred Miller (Michigan Cancer Foundation). The 67NR and 4T1 cells were derived from the same spontaneously arising primary tumor and therefore represent the varied metastatic potential that exists in cells within a single tumor.33 Importantly, the 4T1 cell line spontaneously metastasizes to the lymph nodes, lung, liver, bone, and brain and is therefore regarded as a clinically relevant breast tumor model that closely recapitulates human stage IV metastatic disease.34 Conversely, the 67NR cell line is nonmetastatic. Cells were maintained in mimimum essential medium-alpha (Gibco), with 10% fetal bovine serum (Omega Scientific), and 1% penicillin/streptomycin (Sigma-Aldrich). Before treatment with coagulation factors or BFA, cells were grown to confluence in 12-well plates (Corning Inc.) and serum-starved overnight in serum-free media (SFM). The following coagulation proteases or zymogens were diluted in SFM and used to treat the cells: recombinant mFVIIa, human zymogen FX, mFVIIa and FX, human FXa, and human α-thrombin. PMA was diluted in SFM and incubated with the cells for 1 hour. BFA or dimethyl sulfoxide vehicle control were diluted in SFM and cells were pre-incubated for 3 hours before the addition of coagulation proteases or zymogen.
Short hairpin RNA
Lentiviral particles carrying plasmids encoding short hairpin RNA (shRNA) to either mouse PAR-1 or mouse PAR-2 were obtained from the University of North Carolina at Chapel Hill (UNC-CH) shRNA Core Facility. The GFPshRNA control plasmid (Addgene plasmid 12273) was obtained from Addgene, and packaged into lentiviral particles by the UNC-CH shRNA Core Facility.35 Cells were transduced using Viraductin (Cell Biolabs) according to manufacturers protocol with an additional spin inoculation at 1250g for 90 minutes at 22°C before placing the cells in the incubator. Twenty-four hours posttransduction, 12 μg/mL of puromycin was added to the culture media to select stable cell populations containing the lentivirus. Entire populations of cells were selected rather than single cell clones to minimize selecting individual clones that have phenotypically drifted from the parental 4T1 cells because of the lentiviral transduction. Five and 6 separate cell populations were generated for both PAR-1 and PAR-2 silenced 4T1 cells, respectively. Knockdown efficiency was determined by real time polymerase chain reaction (PCR) analysis. The cell populations with the highest silencing efficiency were used for the experiments.
Real time PCR
RNA was isolated from cells using the RNeasy Plus kit (QIAGEN). mRNA was reverse transcribed using the First Strand cDNA Synthesis kit with Oligo-dT primers (Fermentas). Exon-spanning gene specific primers (Table 1) were synthesized by Integrated DNA Technologies. Real time PCR was performed on a Mastercycler Gradient (Eppendorf) using the Maxima SYBR Green qPCR Master Mix (Fermentas). Relative mRNA levels were quantified using the ΔΔCt method normalized to hypoxanthine-guanine phosphoribosyl transferase (HPRT).36
Table 1.
Real-time PCR primers
| Primer | Sequence |
|---|---|
| Mouse TF fwd | 5′-TCA AGC ACG GGA AAG AAA AC-3′ |
| Mouse TF rev | 5′-CAA AAT AGC CCA GGA AGC AG-3′ |
| Mouse PAR-1 fwd | 5′-CAG CCA GAA TCA GAG AGG ACA GA-3′ |
| Mouse PAR-1 rev | 5′-CCA GCA GGA CGC TTT CAT TT-3′ |
| Mouse PAR-2 fwd | 5′-AGC CGG ACC GAG AAC CTT-3′ |
| Mouse PAR-2 rev | 5′-GGA ACC CCT TTC CCA GTG ATT-3′ |
| Mouse uPA fwd | 5′-TTA CTG CAG GAA CCC TGA CAA CCA-3′ |
| Mouse uPA rev | 5′-TGC TAA GAG AGC AGT CAT GCA CCA-3′ |
| Mouse PAI-1 fwd | 5′-TTC AGT GGC CAA TGG AAG ACT CCT-3′ |
| Mouse PAI-1 rev | 5′-AGG GCA GTT CCA CAA CGT CAT ACT-3′ |
| Mouse HPRT fwd | 5′-CTG GTG AAA AGG ACC TCT CG-3′ |
| Mouse HPRT rev | 5′-TGA AGT ACT CAT TAT AGT CAA GGG CA-3′ |
uPA and PAI-1 enzyme-linked immunosorbent assay
Cells were grown in 12-well plates to form confluent monolayers. Cells were then starved overnight in SFM (Gibco). After starvation, new SFM containing coagulation factors were added to the wells. Conditioned media was collected, centrifuged at 5000g for 5 minutes at 4°C to remove cellular debris, and frozen at −20°C. To obtain whole cell lysates, lysis buffer (10nM HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid], 1.5mM MgCl2, 10mM KCl, 0.05% NP-40 [nonyl phenoxypolyethoxylethanol], pH 7.9) containing complete protease inhibitor cocktail was added to the cells. Cells were then scraped, sonicated, and centrifuged at 20 000g for 10 minutes at 4°C to pellet the cellular debris. The supernatant was then removed and stored at −20°C. uPA and PAI-1 enzyme-linked immunosorbent assays (ELISAs) were performed according to the manufacturer's protocol (Molecular Innovations).
uPA immunofluorescence
Cells were grown on chamber slides (BD Biosciences), starved, fixed, and permeablized using BD Cytofix/Cytoperm (BD Biosciences). The cells were then incubated with fluorescein isothiocyanate (green) conjugated uPA antibody (Molecular Innovations) and Alexa Fluor-555 (red) conjugated Golgi marker (GM)130 antibody (BD Biosciences) for 1 hour at 4°C in the dark. Nuclei were counterstained with DAPI (blue). Slides were washed then mounted with HyrdoMount mounting medium (National Diagnostics).
Immunoblotting
Cells were grown to confluence, starved overnight, and treated with either 125nM FXa, 20nM thrombin, or 200nM PMA. Cells were washed with cold phosphate-buffered saline and lysed in lysis buffer (Cell Signaling Technologies) containing 1mM dithiothreitol and phosphatase inhibitor cocktail. Samples were then sonicated and cleared of debris by centrifugation at 20 000 × g for 10 minutes at 4°C. Protein concentration was determined by Bio-Rad protein assay and 50 μg of lysate was combined with Laemmeli sample buffer, boiled, and the proteins were separated on a Novex Tris-Glycine 4% to 12% gradient gel (Invitrogen). Protein was transferred to polyvinylidene fluoride (PVDF) membranes (Millipore) and probed with an antibody against phosphorylated PKCμ at a 1:250 dilution (Cell Signaling Technologies) and an IRDye 800CW conjugated secondary antibody (Rockland) at a 1:10 000 dilution. The membranes were stripped in Restore Stripping Buffer (Thermo Scientific), blocked, and incubated with an antibody against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) at a 1:5000 dilution (Santa Cruz Biotechnology) and an Alexa Fluor 680 conjugated secondary antibody (Invitrogen) at a 1:10 000 dilution. Blots were visualized and quantified using the Odyssey Infrared Imaging System (Licor).
Statistical analysis
All statistical analyses were performed using GraphPad Prism 4 for Mac (GraphPad Software). All data are presented as means ± standard error of the mean (SEM). One-way analysis of variance with a Bonferroni posthoc analysis was performed when indicated. For 2 group comparisons, a 2-tailed Student t test was used. P ≤ .05 was considered statistically significant.
Results
PAR-1 and PAR-2 mediate crosstalk between coagulation and fibrinolysis
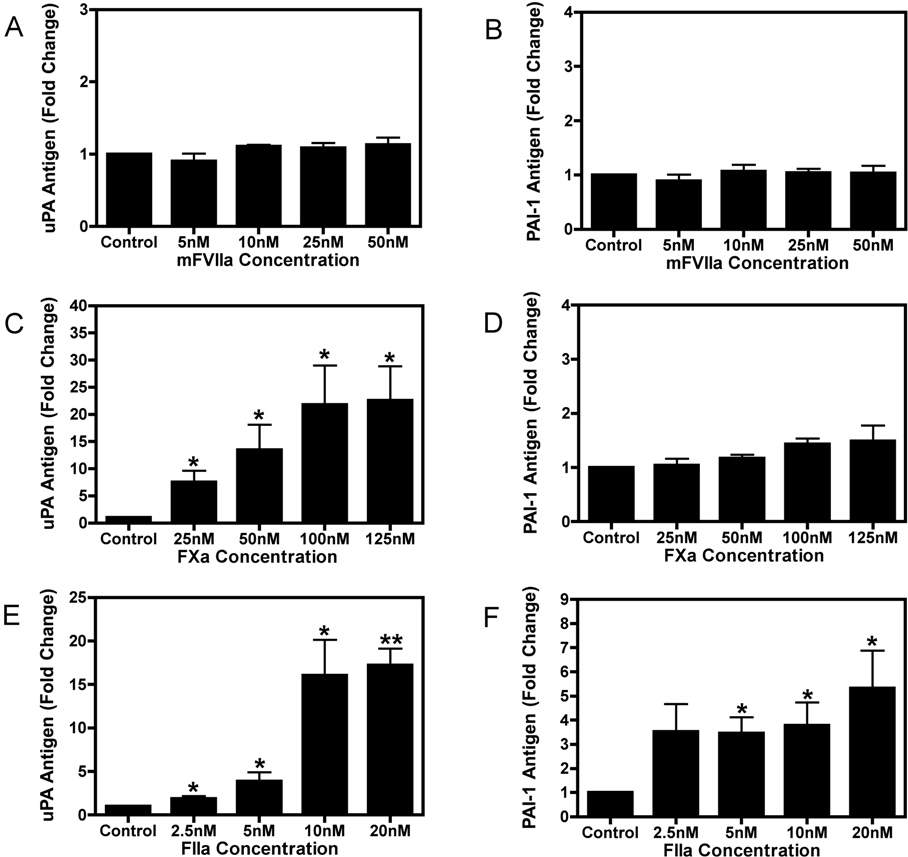
Unstimulated 4T1 and 67NR cells expressed basal levels of TF, PAR-1, uPA, and PAI-1 mRNAs. Importantly, 4T1 cells expressed PAR-2 whereas the 67NR cells lacked detectable PAR-2 mRNA by real time PCR analysis (Figure 1A-B). It was determined using a one-stage clotting assay that 4T1 cells exhibited approximately 3-fold higher levels of TF activity than 67NR cells (data not shown). 4T1 cells were used for mFVIIa, FXa, and thrombin dose-titration experiments. mFVIIa did not increase the levels of either uPA or PAI-1 in the culture supernatant at 24 hours (supplemental Figure 1A-B, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). FXa produced a dose-dependent increase of uPA but not PAI-1 (supplemental Figure 1C-D). Treatment of the cells with thrombin led to dose-dependent increases of both uPA and PAI-1 in the cell culture supernatant (supplemental Figure 1E-F).
Figure 1.

Coagulation proteases increase uPA and PAI-1 expression in the culture supernatant of 4T1 and 67NR breast cancer cell lines. (A-B) Real-time PCR analysis of TF, PAR-1, PAR-2, uPA, and PAI-1 mRNA expression in 4T1 cells (A) and 67NR cells (B). Cells were grown to confluence and starved overnight. Results are shown as mean ± SEM of at least 3 independent experiments. (C-E) Serum starved confluent cell monolayers were incubated for 24 hours with the following coagulation factors: mFVIIa (10nM), FX (130nM), mFVIIa (10nM) and FX (130nM), FXa (125nM), or thrombin (FIIa; 20nM). Levels of uPA in treated 4T1 cells (C) and 67NR cells (D) were determined by ELISA. The amount of PAI-1 released from the treated 4T1 (E) and 67NR (F) cell lines was measured by a PAI-1 ELISA. Results are shown as mean ± SEM of at least 5 independent experiments. *P ≤ .05 and **P ≤ .001 (control versus protease treated).
Stimulation of 4T1 and 67NR cells with either mFVIIa or zymogen FX alone did not increase uPA levels in the culture supernatant (Figure 1C-D). However, incubation of the 4T1 cells with both mFVIIa and FX resulted in high levels of uPA, whereas 67NR cells exhibited a more modest increase in uPA in response to the combination of mFVIIa and FX (Figure 1C-D). The level of uPA generated when cells were incubated with both mFVIIa and FX was similar to the level observed with FXa alone (Figure 1C-D), demonstrating that mFVIIa is enzymatically active. Similarly, thrombin increased uPA levels in the culture supernatant of both cell lines (Figure 1C-D). Interestingly, thrombin was the only protease capable of increasing PAI-1 levels in the culture supernatant of 4T1 cells (Figure 1E). Importantly, incubation of the 67NR cells with any of the proteases failed to increase PAI-1 levels in the cell culture supernatant at 24 hours (Figure 1F). Similar results were obtained by adding mFVIIa, FX, a combination of mFVIIa and FX, FXa, or thrombin to 2 additional cell lines: the PAN02 pancreatic cancer cell line and the 168FARN breast cancer cell line (data not shown).
Absence of PAR-2 is associated with a loss of thrombin induced PAI-1 expression
Our data demonstrated that cells lacking PAR-2 did not express PAI-1 in response to thrombin stimulation. Therefore, we hypothesized that PAR-2 is required for the induction of PAI-1. To test this hypothesis, we silenced PAR-2 expression in 4T1 cells to determine its role in uPA and PAI-1 expression. We also silenced PAR-1 expression in 4T1 cells. The different stably transduced shRNA cell populations are referred to as 4T1GFP, 4T1ΔPAR-1, and 4T1ΔPAR-2. PAR-1 and PAR-2 mRNA levels were knocked down in the 4T1ΔPAR-1 and 4T1ΔPAR-2 by 95% and 80%, respectively (Figure 2A-B). Silencing PAR-1 attenuated the FXa or thrombin-dependent increase of uPA protein (Figure 2C). In contrast, levels of uPA in the culture supernatant of 4T1ΔPAR-2 cells in response to FXa or thrombin did not significantly differ from those observed in the 4T1GFP control cells (Figure 2C). As expected, silencing PAR-1 diminished the thrombin-dependent increase of PAI-1. Importantly, silencing PAR-2 expression also drastically reduced PAI-1 induction in response to thrombin treatment (Figure 2D), supporting our hypothesis that PAR-2 is required for inducible PAI-1 expression.
Figure 2.

Silencing PAR-1 and PAR-2 in 4T1 cells. (A-B) Real-time PCR analysis of PAR-1 (A) and PAR-2 (B) mRNA expression in 4T1ΔPAR-1 and 4T1ΔPAR-2 cells expressed as a percentage of the 4T1GFP control. PAR-1 and PAR-2 levels were normalized to HPRT mRNA. uPA protein (C) and PAI-1 protein (D) were measured by ELISA after 24 hour incubation with FXa (125nM) or thrombin (20nM). Results are shown as mean ± SEM of 3 independent experiments. *P ≤ .05 and **P ≤ .001 (4T1GFP versus 4T1ΔPAR-1), ¶P ≤ .05 and ¶¶P ≤ .001 (4T1GFP versus 4T1ΔPAR-2), §P ≤ .05 and §§P ≤ .001 (4T1ΔPAR-1 versus 4T1ΔPAR-2).
Furthermore, we used Gene Sifter Analysis Edition, Version 3.4 (Geospiza) to mine in vivo gene array data from laser capture microdissected cells from orthotopic 67NR and 4T1 primary tumors. The original data were submitted to the Gene Submission Omnibus database by Lou et al (GSE 11 259).37 We found that 4T1 cells expressed significantly more PAR-2 (33.54-fold) and PAI-1 (9.56-fold) than 67NR cells in vivo. This suggests that the differential PAR-2 expression between the 67NR and 4T1 cells is not a consequence of in vitro culturing conditions and that PAR-2 may indeed be needed for inducible PAI-1 expression in vivo. Taken together, our data indicate that PAR-1 is required for FXa and thrombin mediated increases in uPA whereas both PAR-1 and PAR-2 are required for thrombin-dependent increases in PAI-1.
Transcriptional and posttranslational regulation of PAI-1 and uPA
To understand the mechanism by which PAR activation regulates uPA and PAI-1, we examined the kinetics of uPA and PAI-1 mRNA and protein expression in 4T1 cells treated with FXa or thrombin. uPA mRNA levels remained unchanged in 4T1 cells stimulated with either FXa or thrombin (Figure 3A). This is in stark contrast to uPA protein, which was rapidly increased in the culture supernatant in response to a 1-hour treatment with FXa or thrombin, respectively (Figure 3B). Further increases in uPA were observed throughout the 24-hour period. We also observed uPA release in response to a 1-hour incubation with FXa or thrombin in the PAN02 and 168FARN cell lines (data not shown).
Figure 3.

Timecourse of uPA and PAI-1 mRNA and protein expression in 4T1 cells stimulated with FXa or thrombin. Levels of uPA mRNA (A) and protein (B) were determined by real-time PCR and ELISA, respectively. PAI-1 mRNA (C) and protein (D) induction were also measured using real-time PCR and ELISA. uPA and PAI-1 mRNA levels were normalized to HPRT mRNA. Results are shown as mean ± SEM of at least 3 independent experiments. *P ≤ .05 and **P ≤ .001 (control versus FXa treated). §P ≤ .05 and §§P ≤ .001 (control versus thrombin treated).
Incubation of 4T1 cells with FXa did not significantly increase PAI-1 mRNA or protein levels (Figure 3C-D). In contrast, thrombin treatment of 4T1 cells increased PAI-1 mRNA levels, reaching a maximum induction of 2.2-fold before returning to baseline by 24 hours (Figure 3C). An accumulation of PAI-1 protein in the culture supernatant was observed starting at 6 hours in thrombin treated 4T1 cells (Figure 3D).
4T1 cells contain a store of intracellular uPA associated with the Golgi
Analysis of uPA by direct immunofluorescence in SFM treated 4T1 cells under basal conditions revealed that the protease was stored in the perinuclear region of the cell (Figure 4A). This uPA staining colocalized with the Golgi marker GM130, suggesting that intracellular uPA was associated with this secretory organelle. After treating the 4T1 cells for 30 minutes with either FXa or thrombin, we observed a marked decrease in the intensity of uPA staining (Figure 4A). Next, we measured levels of uPA in whole cell lysates and culture supernatants of 4T1 cells with or without FXa or thrombin treatment. After a 1-hour incubation with FXa or thrombin, cellular uPA levels diminished whereas levels in the culture supernatant increased (Figure 4B-C).
Figure 4.

Stimulation of 4T1 cells with FXa or thrombin induces uPA secretion. (A) Intracellular uPA staining. Subconfluent 4T1 cells were starved overnight then incubated with SFM, FXa, or thrombin for 30 minutes. The cells were then fixed and permeablized on glass chamber slides. Cells were incubated with both uPA-fluorescein isothiocyanate (green) and GM130-Alexa Fluor555 (red) antibodies, then counterstained with DAPI (blue). Slides were viewed on an Olympus BX51WI fluorescence microscope fitted with an Olympus DP70 cooled digital color camera. Total magnification is 400 × (10 × ocular; 40 × objective). DP Controller version 2.2.1.227 software was used for image acquisition. GraphicConverter X V5.4 was used to compile images. (B-C) Serum starved 4T1 cells were incubated with FXa (125nM) or thrombin (20nM) for 1 hour. Cellular (B) and culture supernatant (C) uPA expressed as percentage of total uPA in 4T1 cells. Results are shown as mean ± SEM of 3 independent experiments. *P ≤ .05 (control versus FXa treated) and §P ≤ .05 (control versus thrombin treated). (D) Cells were treated with BFA (10 μg/mL) or vehicle control then stimulated with FXa (125nM) or thrombin (20nM) for 1 hour. uPA was quantified in cell culture supernatant. **P ≤ .05 (control versus FXa treated) and §P ≤ .05 (control versus thrombin treated). (E) uPA levels in the culture supernatant of 4T1 cells treated with 100nM or 200nM PMA for 1 hour. *P ≤ .05 and **P ≤ .001 (control versus PMA treated). Results are shown as mean ± SEM of 3 independent experiments.
Next, we used BFA to inhibit budding of newly formed secretory vesicles from the trans-Golgi network. FXa or thrombin mediated release of uPA was decreased by approximately 50% when cells were pre-treated with BFA (Figure 4D). In addition, we directly activated the secretory pathway by incubating 4T1 cells with PMA. Exposure of 4T1 cells to PMA for 1 hour induced uPA release in a dose-dependent manner (Figure 4E).
FXa and thrombin activate the secretory signaling pathway in 4T1 cells
Protein kinase C-μ (PKCμ) is an intracellular signaling protein involved in secretion from the Golgi. Importantly, we detected phosphorylated PKCμ in 4T1 cells treated with FXa, thrombin, or PMA for 5, 10, or 15 minutes (Figure 5A), which suggests that PKCμ is involved in uPA release. Our previous results demonstrate that activation of PAR-1 results in the rapid release of uPA in 4T1 cells. Therefore, we hypothesized that cleavage of PAR-1 induces PKCμ phosphorylation. 4T1GFP, 4T1ΔPAR-1, and 4T1ΔPAR-2 cells were treated with SFM, FXa, thrombin, or PMA for 5 minutes, and the cell lysates were used to detect phosphorylation of PKCμ. 4T1ΔPAR-1 cells exhibited significantly decreased levels of phosphorylated PKCμ when treated with either FXa or thrombin in comparison to the 4T1GFP and 4T1ΔPAR-2 cells (Figure 5B), thereby supporting our hypothesis that cleavage of PAR-1 activates the secretory pathway.
Figure 5.

Coagulation proteases activate the secretory pathway in the 4T1 cell line. (A) 4T1 cells were treated for the indicated amount of time with SFM, FXa, thrombin, or 200nM PMA. Blots were probed with antibodies for p-PKCμ and GAPDH. Data shown is representative of 3 independent experiments. (B) 4T1GFP, 4T1ΔPAR-1, and 4T1ΔPAR-2 cells were treated with SFM, FXa, thrombin, or PMA for 5 minutes. Blots were probed with antibodies for p-PKCμ and GAPDH. Phosphorylated PKCμ was quantified by dividing the background-corrected p-PKCμ signal intensity by the background corrected GAPDH signal intensity. The blot shown is representative of at least 3 independent experiments. Quantification results are shown as mean ± SEM of at least 3 independent experiments. *P ≤ .05 (4T1GFP versus 4T1ΔPAR-1) and #P ≤ .05 (4T1ΔPAR-1 versus 4T1ΔPAR-2).
Discussion
Our results show that FXa or thrombin mediated activation of PAR-1 induces a rapid release of uPA in murine mammary adenocarcinoma cells (Figure 6). In contrast to previous reports using human cells lines, we did not find evidence for the induction of uPA mRNA expression in the murine tumor lines used in this study.31,32,38 Cellular uPA colocalized with the Golgi. In addition, inhibiting secretion with BFA decreased uPA protein release. BFA collapses the trans-Golgi network, thereby preventing trafficking of immature and newly synthesized secretory vesicles.39 The portion of uPA not inhibited by BFA treatment presumably represents uPA that is already within mature secretory vesicles. Furthermore, uPA was released by PMA activation of the secretory pathway. Activated PKCμ translocates to the Golgi apparatus where it is involved in vesicular trafficking from the trans-Golgi network to the plasma membrane.40 Phosphorylation of the 2 activation loops of PKCμ resulting from FXa or thrombin treatment is indicative of activation of this secretory pathway. Taken together, these data demonstrate a novel mechanism of uPA regulation downstream of cellular activation by FXa or thrombin.
Figure 6.

Proposed model of how coagulation protease activation of PAR-1 and PAR-2 regulates uPA and PAI-1 expression in mouse breast cancer cell lines. PAR-1 and PAR-2 can exist as individual receptors or in a complex. Cells, such as the 67NR cell line (left panel), express PAR-1 only whereas 4T1 cells express PAR-1 and a PAR-1/PAR-2 complex (right panel). The configuration of PAR-1 and PAR-2 determines the expression of uPA and PAI-1 in response to different coagulation proteases.
Both PAR-1 and PAR-2 are required for PAI-1 mRNA and protein expression because silencing of either receptor in 4T1 cells abolished the increase of PAI-1 in the culture supernatant. This result is consistent with the absence of inducible PAI-1 expression in breast tumor lines lacking PAR-2, such as 67NR cells. This codependency suggests that PAR-1 and PAR-2 are complexed together and that cleaved PAR-1 transactivates PAR-2 to induce PAI-1 expression. Interestingly, thrombin, but not FXa, activated the PAR-1/PAR-2 complex. These data are consistent with the thrombin-dependent activation of a PAR-1/PAR-2 complex previously described by O'Brien et al.21 Thrombin activated PAR-1 and transactivated PAR-2 have been shown to use separate intracellular signaling pathways in an endothelial cell model of sepsis using pharmacological inhibitors and siRNA against PAR-1 or PAR-2.41 This may explain the differential regulation of uPA secretion and PAI-1 mRNA expression by PAR-1 and a PAR-1/PAR-2 complex, respectively, in tumor cells.
Our model suggests that there is differential signaling in tumor cells containing either PAR-1 alone or both PAR-1 and PAR-2 (Figure 6). In cells expressing only PAR-1, FXa or thrombin activate the receptor resulting in uPA secretion. Thrombin activates PAR-1 in a membrane-independent manner whereas FXa is anchored to the membrane. This membrane tethering may be achieved when FXa is complexed with TF-FVIIa or when FXa is bound to the membrane. In cells that express both PAR-1 and PAR-2, we found evidence of a PAR-1/PAR-2 complex. In these cells, we propose that PAR-1, the more abundantly expressed receptor, can exist as a lone receptor or as part of the PAR-1/PAR-2 complex, whereas all of the PAR-2 is sequestered into the complex. Importantly, unlike PAR-1 alone, the PAR-1/PAR-2 complex is activated by thrombin but not by FXa. This thrombin-dependent cleavage of PAR-1 transactivates PAR-2 resulting in increased PAI-1 expression. Previous studies have shown that receptors, such as TF and PARs, are often clustered together in specialized membrane microdomains, namely caveolae and lipid rafts, to enhance signaling.42 Presumably, PAR-1 and the PAR-1/PAR-2 complex reside in different membrane domains explaining why only a trans-acting protease, such as thrombin, is capable of activating both sets of PARs on the cell surface.
We were unable to observe a TF-FVIIa-PAR-2 dependent regulation of either uPA or PAI-1 in the 4T1 cell line. In contrast, TF-FVIIa-PAR2 signaling has been described using the human breast cancer cell line MDA-MB-231.31,43 Using Oncomine (Compendia Biosciences), we mined gene array data (GSE 2603) and found that the mRNA levels of TF and PAR-2 were significantly greater than that of PAR-1 in MDA-MB-231 tumor xenografts.44 We found similar results by quantitative real-time PCR using cultured MDA-MB-231 cells (data not shown). TF-FVIIa-PAR-2 signaling was also observed in baby hamster kidney cells transfected with TF and human endothelial cells transfected with TF and PAR-2.45 Taken together, this suggests that the levels of TF and PAR-2 expressed on the 4T1 cells are not high enough to support for TF-FVIIa-PAR-2 signaling.
PARs enable cells to detect, and therefore respond to proteases present in the local environment. Tumor metastasis and angiogenesis, whether lymphatic or hematogenous, requires a variety of matrix remodeling proteases, including matrix metalloproteinase-1 and plasmin.46,47 These proteases, in addition to mast cell tryptase, and tissue kallikreins, are present within the breast tumor stroma and are known to activate PAR-1 or PAR-2.18,48,49 Recently, it has been demonstrated that oncogenes increase TF, PAR-1, and PAR-2 expression.2 We hypothesize that the local generation of FXa and thrombin on the surface of tumor cells, in addition to the aforementioned proteases present in the tumor stroma, may activate PAR-1 or PAR-2, leading to increased release/expression of uPA and PAI-1. Both uPA and PAI-1 have established roles in matrix degradation, tumor motility, and angiogenesis.22 Our study explains how the coagulation system may use PAR-1 and PAR-2 to promote malignancy via increased generation of plasmin.
Supplementary Material
Acknowledgments
We would like to thank Dr Lars Petersen for the recombinant mouse FVIIa.
This work was supported by an F31-NRSA fellowship from the National Cancer Institute to T.A.M. (1F31CA142162-01) and grants from the National Institutes of Health to N.M. (R01-HL095096). R.P. is supported by a grant from the American Heart Association (AHA-09BGIA2150078). F.C.C. is supported by grants from the Susan G. Komen Breast Cancer Foundation (BCTR0503475 and BCTR45206).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: T.A.M. designed the experiments, performed the experiments, interpreted the data, and wrote the manuscript; R.P. aided in experimental design, data interpretation, and critically read and edited the manuscript; K.L.R. provided invaluable technical assistance and critically read the manuscript; F.C.C. aided in data interpretation and critically read the manuscript; and N.M. designed the experiments, interpreted the data, edited the manuscript, and contributed to the overall design of the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Nigel Mackman, Division of Hematology/Oncology, 917 Mary Ellen Jones Bldg, Campus Box 7035, Chapel Hill, NC 27599-7035; e-mail: nigel_mackman@med.unc.edu.
References
- 1.Haas SL, Jesnowski R, Steiner M, et al. Expression of tissue factor in pancreatic adenocarcinoma is associated with activation of coagulation. World J Gastroenterol. 2006;12(30):4843–4849. doi: 10.3748/wjg.v12.i30.4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Magnus N, Garnier D, Rak J. Oncogenic epidermal growth factor receptor upregulates multiple elements of the tissue factor signaling pathway in human glioma cells. Blood. 2010;116(5):815–818. doi: 10.1182/blood-2009-10-250639. [DOI] [PubMed] [Google Scholar]
- 3.Rong Y, Belozerov VE, Tucker-Burden C, et al. Epidermal growth factor receptor and PTEN modulate tissue factor expression in glioblastoma through JunD/activator protein-1 transcriptional activity. Cancer Res. 2009;69(6):2540–2549. doi: 10.1158/0008-5472.CAN-08-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu JL, May L, Lhotak V, et al. Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood. 2005;105(4):1734–1741. doi: 10.1182/blood-2004-05-2042. [DOI] [PubMed] [Google Scholar]
- 5.Hembrough TA, Swartz GM, Papathanassiu A, et al. Tissue factor/factor VIIa inhibitors block angiogenesis and tumor growth through a nonhemostatic mechanism. Cancer Res. 2003;63(11):2997–3000. [PubMed] [Google Scholar]
- 6.Jiang X, Bailly MA, Panetti TS, et al. Formation of tissue factor-factor VIIa-factor Xa complex promotes cellular signaling and migration of human breast cancer cells. J Thromb Haemost. 2004;2(1):93–101. doi: 10.1111/j.1538-7836.2004.00545.x. [DOI] [PubMed] [Google Scholar]
- 7.Hu L, Ibrahim S, Liu C, et al. Thrombin induces tumor cell cycle activation and spontaneous growth by down-regulation of p27Kip1, in association with the up-regulation of Skp2 and MiR-222. Cancer Res. 2009;69(8):3374–3381. doi: 10.1158/0008-5472.CAN-08-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kasthuri RS, Taubman MB, Mackman N. Role of tissue factor in cancer. J Clin Oncol. 2009;27(29):4834–4838. doi: 10.1200/JCO.2009.22.6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci U S A. 2000;97(10):5255–5260. doi: 10.1073/pnas.97.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Versteeg HH, Schaffner F, Kerver M, et al. Protease-activated receptor (PAR) 2, but not PAR1, signaling promotes the development of mammary adenocarcinoma in polyoma middle T mice. Cancer Res. 2008;68(17):7219–7227. doi: 10.1158/0008-5472.CAN-08-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Booden MA, Eckert LB, Der CJ, Trejo J. Persistent signaling by dysregulated thrombin receptor trafficking promotes breast carcinoma cell invasion. Mol Cell Biol. 2004;24(5):1990–1999. doi: 10.1128/MCB.24.5.1990-1999.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang E, Boire A, Agarwal A, et al. Blockade of PAR1 signaling with cell-penetrating pepducins inhibits Akt survival pathways in breast cancer cells and suppresses tumor survival and metastasis. Cancer Res. 2009;69(15):6223–6231. doi: 10.1158/0008-5472.CAN-09-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ludeman MJ, Kataoka H, Srinivasan Y, et al. PAR1 cleavage and signaling in response to activated protein C and thrombin. J Biol Chem. 2005;280(13):13122–13128. doi: 10.1074/jbc.M410381200. [DOI] [PubMed] [Google Scholar]
- 14.Bhattacharjee G, Ahamed J, Pawlinski R, et al. Factor Xa binding to annexin 2 mediates signal transduction via protease-activated receptor 1. Circ Res. 2008;102(4):457–464. doi: 10.1161/CIRCRESAHA.107.167759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boire A, Covic L, Agarwal A, et al. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120(3):303–313. doi: 10.1016/j.cell.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 16.Mannaioni G, Orr AG, Hamill CE, et al. Plasmin potentiates synaptic N-methyl-D-aspartate receptor function in hippocampal neurons through activation of protease-activated receptor-1. J Biol Chem. 2008;283(29):20600–20611. doi: 10.1074/jbc.M803015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296(5574):1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 18.Russo A, Soh UJK, Trejo J. Proteases display biased agonism at protease-activated receptors: location matters! Mol Interv. 2009;9(2):87–96. doi: 10.1124/mi.9.2.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakanishi-Matsui M, Zheng YW, Sulciner DJ, et al. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 2000;404(6778):609–613. doi: 10.1038/35007085. [DOI] [PubMed] [Google Scholar]
- 20.Leger AJ, Jacques SL, Badar J, et al. Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation. 2006;113(9):1244–1254. doi: 10.1161/CIRCULATIONAHA.105.587758. [DOI] [PubMed] [Google Scholar]
- 21.O'Brien PJ, Prevost N, Molino M, et al. Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1. J Biol Chem. 2000;275(18):13502–13509. doi: 10.1074/jbc.275.18.13502. [DOI] [PubMed] [Google Scholar]
- 22.McMahon B, Kwaan HC. The plasminogen activator system and cancer. Pathophysiol Haemost Thromb. 2008;36(3–4):184–194. doi: 10.1159/000175156. [DOI] [PubMed] [Google Scholar]
- 23.Maillard CM, Bouquet C, Petitjean MM, et al. Reduction of brain metastases in plasminogen activator inhibitor-1-deficient mice with transgenic ocular tumors. Carcinogenesis. 2008;29(11):2236–2242. doi: 10.1093/carcin/bgn204. [DOI] [PubMed] [Google Scholar]
- 24.Henneke I, Greschus S, Savai R, et al. Inhibition of urokinase activity reduces primary tumor growth and metastasis formation in a murine lung carcinoma model. Am J Respir Crit Care Med. 2010;181(6):611–619. doi: 10.1164/rccm.200903-0342OC. [DOI] [PubMed] [Google Scholar]
- 25.Pakneshan P, Szyf M, Farias-Eisner R, Rabbani SA. Reversal of the hypomethylation status of urokinase (uPA) promoter blocks breast cancer growth and metastasis. J Biol Chem. 2004;279(30):31735–31744. doi: 10.1074/jbc.M401669200. [DOI] [PubMed] [Google Scholar]
- 26.Sandberg T, Casslén B, Gustavsson B, Benraad TJ. Human endothelial cell migration is stimulated by urokinase plasminogen activator:plasminogen activator inhibitor 1 complex released from endometrial stromal cells stimulated with transforming growth factor beta1; possible mechanism for paracrine stimulation of endometrial angiogenesis. Biol Reprod. 1998;59(4):759–767. doi: 10.1095/biolreprod59.4.759. [DOI] [PubMed] [Google Scholar]
- 27.Binder BR, Mihaly J, Prager GW. uPAR-uPA-PAI-1 interactions and signaling: a vascular biologist's view. Thromb Haemost. 2007;97(3):336–342. [PubMed] [Google Scholar]
- 28.Lacroix R, Sabatier F, Mialhe A, et al. Activation of plasminogen into plasmin at the surface of endothelial microparticles: a mechanism that modulates angiogenic properties of endothelial progenitor cells in vitro. Blood. 2007;110(7):2432–2439. doi: 10.1182/blood-2007-02-069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hildenbrand R, Schaaf A, Dorn-Beineke A, et al. Tumor stroma is the predominant uPA-, uPAR-, PAI-1-expressing tissue in human breast cancer: prognostic impact. Histol Histopathol. 2009;24(7):869–877. doi: 10.14670/HH-24.869. [DOI] [PubMed] [Google Scholar]
- 30.De Cremoux P, Grandin L, Diéras V, et al. Urokinase-type plasminogen activator and plasminogen-activator-inhibitor type 1 predict metastases in good prognosis breast cancer patients. Anticancer Res. 2009;29(5):1475–1482. [PubMed] [Google Scholar]
- 31.Albrektsen T, Sørensen BB, Hjortø GM, et al. Transcriptional program induced by factor VIIa-tissue factor, PAR1 and PAR2 in MDA-MB-231 cells. J Thromb Haemost. 2007;5(8):1588–1597. doi: 10.1111/j.1538-7836.2007.02603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taniguchi T, Kakkar AK, Tuddenham EG, Williamson RC, Lemoine NR. Enhanced expression of urokinase receptor induced through the tissue factor-factor VIIa pathway in human pancreatic cancer. Cancer Res. 1998;58(19):4461–4467. [PubMed] [Google Scholar]
- 33.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52(6):1399–1405. [PubMed] [Google Scholar]
- 34.Lelekakis M, Moseley JM, Martin TJ, et al. A novel orthotopic model of breast cancer metastasis to bone. Clin Exp Metastasis. 1999;17(2):163–170. doi: 10.1023/a:1006689719505. [DOI] [PubMed] [Google Scholar]
- 35.Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 36.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 37.Lou Y, Preobrazhenska O, auf dem Keller U, et al. Epithelial-mesenchymal transition (EMT) is not sufficient for spontaneous murine breast cancer metastasis. Dev Dyn. 2008;237(10):2755–2768. doi: 10.1002/dvdy.21658. [DOI] [PubMed] [Google Scholar]
- 38.Yoshida E, Verrusio EN, Mihara H, Oh D, Kwaan HC. Enhancement of the expression of urokinase-type plasminogen activator from PC-3 human prostate cancer cells by thrombin. Cancer Res. 1994;54(12):3300–3304. [PubMed] [Google Scholar]
- 39.Orci L, Tagaya M, Amherdt M, et al. Brefeldin A, a drug that blocks secretion, prevents the assembly of non-clathrin-coated buds on Golgi cisternae. Cell. 1991;64(6):1183–1195. doi: 10.1016/0092-8674(91)90273-2. [DOI] [PubMed] [Google Scholar]
- 40.Yeaman C, Ayala MI, Wright JR, et al. Protein kinase D regulates basolateral membrane protein exit from trans-Golgi network. Nat Cell Biol. 2004;6(2):106–112. doi: 10.1038/ncb1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaneider NC, Leger AJ, Agarwal A, et al. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol. 2007;8(12):1303–1312. doi: 10.1038/ni1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Awasthi V, Mandal SK, Papanna V, Rao LVM, Pendurthi UR. Modulation of tissue factor-factor VIIa signaling by lipid rafts and caveolae. Arterioscler Thromb Vasc Biol. 2007;27(6):1447–1455. doi: 10.1161/ATVBAHA.107.143438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morris DR, Ding Y, Ricks TK, et al. Protease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res. 2006;66(1):307–314. doi: 10.1158/0008-5472.CAN-05-1735. [DOI] [PubMed] [Google Scholar]
- 44.Minn AJ, Gupta GP, Siegel PM, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436(7050):518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Versteeg HH, Borensztajn KS, Kerver ME, et al. TF:FVIIa-specific activation of CREB upregulates proapoptotic proteins via protease-activated receptor-2. J Thromb Haemost. 2008;6(9):1550–1557. doi: 10.1111/j.1538-7836.2008.03091.x. [DOI] [PubMed] [Google Scholar]
- 46.Bohn OL, Nasir I, Brufsky A, et al. Biomarker profile in breast carcinomas presenting with bone metastasis. Int J Clin Exp Pathol. 2009;3(2):139–146. [PMC free article] [PubMed] [Google Scholar]
- 47.Clavel C, Chavanel G, Birembaut P. Detection of the plasmin system in human mammary pathology using immunofluorescence. Cancer Res. 1986;46(11):5743–5747. [PubMed] [Google Scholar]
- 48.Xiang M, Gu Y, Zhao F, et al. Mast cell tryptase promotes breast cancer migration and invasion. Oncol Rep. 2010;23(3):615–619. doi: 10.3892/or_00000676. [DOI] [PubMed] [Google Scholar]
- 49.Papachristopoulou G, Avgeris M, Scorilas A. Expression analysis and study of KLK4 in benign and malignant breast tumours. Thromb Haemost. 2009;101(2):381–387. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}