

Abstract
Tandem methods for the catalytic asymmetric preparation of enantioenriched β-hydroxy (E)-enamines and cyclopropylamines are presented. The diastereoselective hydrogenation of enantioenriched (E)-trisubstituted hydroxy enamines to generate 1,2-disubstituted 1,3-amino alcohols is also outlined. These methods are initiated by highly regioselective hydroboration of N-tosyl substituted ynamides with diethylborane to generate β-amino alkenyl boranes. In situ boron to zinc transmetalation generates β-amino alkenyl zinc reagents. These functionalized vinylzinc intermediates were subsequently added to aldehydes in the presence of catalyst derived from an enantioenriched amino alcohol (morpholino isoborneol, MIB). The catalyst promotes highly enantioselective C–C bond-formation to provide β-hydroxy enamines in good isolated yields (68–86%) with 54–98% enantioselectivity. The intermediate zinc β-alkoxy enamines can be subjected to a tandem cyclopropanation to afford amino cyclopropyl carbinols with three continuous stereocenters in a one-pot procedure with good yields (72–82%), enantioselectivities of 76–94% and diastereomeric ratios >20:1. Diastereoselective hydrogenation of isolated enantioenriched β-hydroxy enamines over Pd/C furnished syn-1,2-disubstituted-1,3-amino alcohols with high yields (82–90%) and moderate to excellent diastereoselectivities. These methods were used in an efficient preparation of the enantioenriched precursor to PRC200-SS derivatives, which are potent serotonin-norepinephrine-dopamine reuptake inhibitors.
1. Introduction
β-Hydroxy enamines are present in several natural and unnatural products1–5 and are valuable synthetic intermediates.6–8 The absence of efficient catalytic enantioselective methods for their preparation, however, impedes their wider utilization in synthesis and drug discovery. Methods for the synthesis of racemic β-hydroxy enamines have been introduced. Meyer has recently developed a five step synthesis of β-hydroxy enamines from ynamides.7,8 Derivatization followed by Wittig rearrangement aforded amino alcohols, highlighting the utility of β-hydroxy enamines. The only one-step method for the synthesis of β-hydroxy enamines is Urabe's reaction of ynamide-titanium complexes with aldehydes (Scheme 1).6, 9, 10 Yields as high as 94% were achieved with moderate to excellent diastereoselectivities (2:1 to <20:1) and good substrate scope. The method, however, requires the use of enantioenriched sulfonamide-based auxiliaries that were synthesized in four steps, one of which employed another chiral auxiliary.11
Scheme 1.
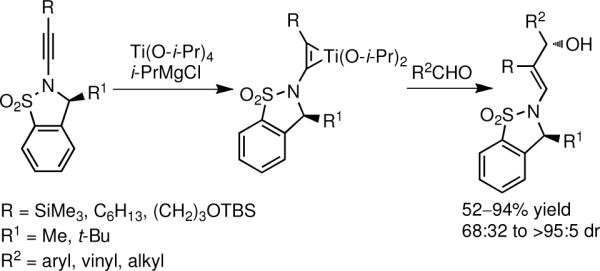
Urabe's Addition of Ynamide-Titanium Complexes to Aldehydes to form β-Hydroxy Enamines
Given the challenges associated with the development of a catalytic enantioselective version of this reaction, we considered an alternative strategy for the stereoselective synthesis of β-hydroxy enamines. Ynamides12,13 were chosen as precursors because they can be easily prepared in one step from terminal alkynes, amine derivatives, and copper catalysts in the presence of oxygen following the method of Stahl and co-workers.14 As outlined in Scheme 2, we envisioned hydroboration of ynamides to generate β-amino vinyl boranes, boron to zinc transmetalation of the resulting vinyl group and catalytic enantioselective carbonyl addition of the β-amino vinyl group to aldehydes to afford zinc alkoxides. Simple protonation of these intermediates is expected to provide β-hydroxy enamines as single double bond isomers with high ee (Scheme 2). For the choice of catalyst for asymmetric addition of β-amino vinyl zinc reagents to aldehydes, we anticipated using enantioenriched amino alcohol derivatives, which we15–19 and others20–29 have successfully employed in the asymmetric addition of vinylzinc reagents to aldehydes.
Scheme 2.

One-Pot Generation of (E)-Trisubstituted β-Hydroxy Enamines
In addition to their intrinsic value, β-hydroxy enamines are also potentially important synthetic intermediates for further elaboration via alkoxide and hydroxyl directed diastereoselective reactions.30 Alkoxide directed cyclopropanation of β-alkoxide enamines could provide access to amino cyclopropyl alcohols (Scheme 3A). Enantioenriched amino cyclopropanes are pharmacophores that are present in numerous natural products and synthetic materials.31–33 As a result, their synthesis has attracted significant attention.34–54 β-Hydroxy enamines are anticipated to be useful precursors in the synthesis of enantioenriched 1,2-disubstituted 1,3-amino alcohols (Scheme 3B), a class of compounds that has garnered much interest for their biological activity.55–58
Scheme 3.

Application of β-Hydroxy Enamine Derivatives in the Synthesis of Amino Cyclopropyl Carbinols (A) and 1,3-Amino Alcohols (B).
Herein we disclose the first one-pot catalytic asymmetric synthesis of β-hydroxy enamines starting from readily available ynamides. Using the approach outlined in Scheme 2, β-hydroxy enamines can be accessed with good to excellent enantioselectivity, good to high yields, and as single double bond isomers. The intermediate β-alkoxide enamine can be subjected to a tandem diastereoselective cyclopropanation to afford amino cyclopropyl carbinols with high enantio- and diastereoselectivity. We have also developed an efficient diastereoselective hydrogenation of β-hydroxy enamines leading to 1,2-disubstituted 1,3-amino alcohols with high enantio- and diastereoselectivities. This method has been applied to the synthesis of PRC200-SS and it's derivatives, which are potent serotonin-norepinephrine-dopamine reuptake inhibitors.
2. Experimental Section
Representative procedures and characterization of the products are described herein. Full experimental details and characterization of all compounds are provided in the Supporting Information.
General Methods
Reactions involving dialkylzinc reagents were performed under nitrogen using standard Schlenk or vacuum-line techniques in oven-dried glassware. Hydrogenations were carried out in magnetically stirred glass test tubes placed in a Parr high pressure hydrogenator. Chemicals were purchased from Aldrich or Acros unless otherwise specified, and solvents were purchased from Fisher Scientific. Toluene and hexanes were dried through alumina columns. Aldehydes were distilled prior to use and stored under nitrogen. Solutions of dimethylzinc and diethylzinc (2M in toluene) were prepared and stored in a Vacuum Atmospheres drybox. Ynamides were synthesized according to Stahl's procedure.14 N-Benzyl-N-ethynyl-4-methylbenzenesulfonamide was synthesized by method of Bruckner.59 Silica gel (230–400 mesh, Silicycle) was used for air-flashed chromatography. Deactivated silica gel was prepared by addition of 17 mL of Et3N to 1 L of silica gel. The progress of reactions was monitored by thin-layer chromatography (TLC) on Whatman precoated silica-gel F-254 plates and visualized by ultraviolet light or ceric ammonium molybdate stain. 1H NMR and 13C{1H} NMR spectra were obtained on Brüker 300, 360, 400 or 500 MHz Fourier transform spectrometers at the University of Pennsylvania NMR facility. Chemical shifts are reported in units of parts per million downfield from tetramethylsilane and coupling constants are reported in hertz. The infrared spectra were obtained using a PerkinElmer 1600 series spectrometer. HRMS data were obtained on a Waters LC-TOF mass spectrometer (model LCT-XE Premier) using electrospray ionization in positive or negative mode, depending on analyte. Melting points were determined on a Unimelt Thomas-Hoover melting point apparatus and are uncorrected.
Caution! Dialkylzinc reagents are pyrophoric. Care and proper laboratory attire must be used when handling them.
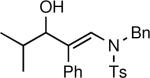
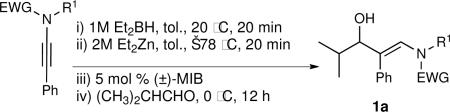
General Procedure A. Asymmetric Amino Vinylation of Aldehydes with β-Amino Vinyl Zinc Reagents: (R,E)-N-Benzyl-N-(3-hydroxy-4-methyl-2-phenylpent-1-enyl)-4-methylbenzenesulfonamide (1a)
A 10 mL Schlenk flask was charged with a solution of N-benzyl-N-(p-toluenesulfonyl)-2-phenylethynylamine (1.0 mL, 0.25 M in toluene, 0.25 mmol) and a solution of diethylborane (0.25 mL, 1.0 M in toluene, 0.25 mmol) was added dropwise at room temperature. The resulting solution was stirred at room temperature for 20 min. The reaction flask was then cooled to −78 °C, Et2Zn (0.25 mL, 2.0 M in toluene, 0.5 mmol) was added, and the reaction mixture was stirred for 20 min. (−)-MIB (3.0 mg, 0.012 mmol, 5 mol %) was added followed by dropwise addition of isobutyraldehyde (24 μL, 0.3 mmol) at −78 °C. The reaction flask was placed in a −30 °C cold bath and allowed to warm to 0 °C over several hours. The solution was stirred at 0 °C until vinyl addition was complete by TLC (typically 12 h). The reaction was then quenched by addition of brine (2 mL). The organic and aqueous layers were separated, and the aqueous layer was extracted with 3 × 20 mL of diethyl ether. The combined organic layers were then washed with 50 mL of water and dried over MgSO4. The filtrate was concentrated in vacuo and the residue was chromatographed on deactivated silica gel (10% ethyl acetate in hexanes) to afford 107 mg (82% yield) of 1a as an amorphous solid. [α]D20: −19.5 (c = 1.0, CHCl3); 1H NMR (CDCl3, 360 MHz): δ 0.82 (d, 3H, J = 7.0 Hz), 1.01 (d, 3H, J = 7.0 Hz), 1.57 (sept., 1H, J = 7.0 Hz), 2.07 (s, 3H), 3.84 (d, 1H, J = 6.4 Hz), 4.27 (dd, 2H, J = 21.3 Hz, 15.0 Hz), 6.54 (s, 1H), 6.94–7.02 (m, 4H), 7.09–7.19 (m, 7H), 7.29 (s, 1H), 7.84–7.90 (m, 2H); 13C{1H} NMR (CDCl3, 75 MHz): δ 27.65, 28.66, 35.45, 44.95, 77.47, 127.52, 127.66, 127.89, 128.81, 129.09, 129.62, 129.91, 134.50, 136.20, 136.32, 156.2; IR (neat): 3537 (OH), 2961, 2870, 1723, 1643, 1598, 1494, 1455, 1343, 1162, 1092, 1026, 814 cm−1; HRMS-CI: m/z 458.1766 [(M + Na)+; calculated for C26H29NO3SNa: 458.1766].
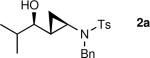
General Procedure B. Asymmetric Amino Vinylation of Aldehydes/Diastereoselective Cyclopropanation: N-Benzyl-N-((1R,2R)-2-((R)-1-hydroxy-2-methylpropyl)cyclopropyl)-4-methylbenzenesulfonamide (2a)
A 10 mL Schlenk flask was charged with a solution of N-benzyl-N-ethynyl-4-methylbenzenesulfonamide (1.0 mL, 0.25 M in toluene, 0.25 mmol) and a solution of diethylborane (0.25 mL, 1.0 M in toluene, 0.25 mmol) was added dropwise at room temperature. The resulting solution was stirred at room temperature for 20 min. The reaction flask was then cooled to −78 °C, Et2Zn (0.25 mL, 2.0 M in toluene, 0.5 mmol) was added, and the reaction mixture was stirred for 20 min. (−)-MIB (3.0 mg, 0.012 mmol, 5 mol %) was added followed by dropwise addition of isobutyraldehyde (24 μL, 0.3 mmol) at −78 °C. The reaction flask was placed in a −30 °C IPA/dry ice cold bath and allowed to warm to 0 °C over 12 h. The solution was stirred at 0 °C until vinyl addition was complete by TLC (typically 12 h). The volatile materials, including Et3B byproduct, were removed in vacuo at 0 °C. Hexanes (2 mL) was added, and the volatile materials were again removed under reduced pressure. This step was repeated two more times to ensure the complete removal of Et3B. A solution of Et2Zn (0.63 mL, 2.0 M in toluene, 1.25 mmol) and neat CF3CH2OH (91 μL, 1.25 mmol) were added slowly at 0 °C and the Schlenk flask was wrapped in aluminum foil to exclude light. The resulting mixture was stirred at 0 °C for 5 min and diiodomethane (101 μL, 1.25 mmol) was added. The stirring was continued at 0 °C for 40 h, after which the reaction mixture was quenched with brine (2 mL). The organic and aqueous layers were separated and the aqueous layer was extracted with 3 × 20 mL of diethyl ether. The combined organic layers were then washed with 50 mL of water and dried over MgSO4. The filtrate was concentrated under reduced pressure and the residue was chromatographed on deactivated silica gel (10% ethyl acetate in hexanes) to afford 59 mg (80% yield) of 2a as an yellow oil. [α]D20: −28.1 (c = 0.5, CHCl3); 1H NMR (CDCl3, 360 MHz): δ 0.68 (dd, 1H, J = 14.32 Hz, 6.95 Hz), 0.74 (d, 3H, J = 6.7 Hz), 0.77 (d, 3H, J = 6.7 Hz), 1.00 (d, 1H, J = 5.2 Hz), 1.09 (m, 1H), 1.52 (m, 1H), 2.00 (m, 1H), 2.42 (s, 3H), 3.16 (q, 1H, J = 4.6 Hz), 4.18 (d, 1H, J = 14.6 Hz), 4.38 (d, 1H, J = 14.6 Hz), 7.20–7.38 (m, 7H), 7.67–7.76 (m, 2H); 13C{1H} NMR (CDCl3, 75 MHz): δ 9.90, 17.22, 18.94, 21.77, 23.69, 34.11, 34.44, 54.96, 74.52, 127.78, 127.86, 128.61, 128.65, 129.84, 135.68, 137.40, 143.67; IR (neat): 3538 (OH), 2960, 2873, 1598, 1495, 1455, 1341, 1163, 1093, 927, 815 cm−1; HRMS-CI: m/z 396.1604 [(M + Na)+; calculated for C21H27NO3SNa: 396.1609].
General Procedure C. Diastereoselective Hydrogenation of β-Hydroxy Enamines with Aliphatic Substituents in 3-Position: N-Benzyl-N-(3-hydroxy-4-methyl-2-phenylpentyl)-4-methylbenzenesulfonamide (3a)
In a 10 × 75 mm glass test tube was dissolved (E)-N-benzyl-N-(3-hydroxy-4-methyl-2-phenylpent-1-enyl)-4-methylbenzenesulfonamide (44 mg, 0.1 mmol) in methanol (4 mL) at room temperature. The space above the solution was purged with nitrogen to remove most of the air. 10% Palladium on carbon (8 mg, 7 mol %) was added and the test tube was placed in a Parr hydrogenator. Good stirring was confirmed before closing apparatus. After flushing three times with hydrogen, the system was pressured with 9.65 MPa (1400 psi) of hydrogen and the reaction was stirred for 12 h at room temperature. After opening the apparatus, the palladium catalyst was removed via filtration through a plug of Celite. The filtrate was concentrated under reduced pressure and the residue was chromatographed on silica gel (10% ethyl acetate in hexanes) to afford 39 mg (90% yield) of 3a as an amorphous solid. 1H NMR (CDCl3, 360 MHz): δ 0.36 (d, 3H, J = 6.6 Hz), 0.86 (d, 3H, J = 6.6 Hz), 1.16 (sept., 1H, J = 7.2 Hz), 2.41 (s, 3H), 2.73 (dd, 1H, J = 14.3 Hz, 4.4 Hz), 3.08 (d, 1H, J = 5.4 Hz), 3.48 (m, 1H), 3.74 (d, 1H, J = 14.0 Hz), 3.96 (dd, 1H, J = 14.8 Hz, 11.7 Hz), 4.73 (d, 1H, J = 14.0 Hz), 7.06–7.13 (m, 2H), 7.14–7.22 (m, 3H), 7.28–7.44 (m, 7H), 7.68–7.76 (m, 2H); 13C{1H} NMR (CDCl3, 75 MHz): δ 18.86, 20.21, 21.75, 31.15, 46.69, 53.51, 55.23, 75.25, 126.97, 127.43, 128.39, 128.48, 129.05, 129.24, 129.66, 130.11, 136.70, 139.05, 139.21, 143.80; IR (neat): 3532 (OH), 2924, 1599, 1494, 1330, 1156, 1094, 925, 814 cm−1; HRMS-CI: m/z 438.2111 [(M + H)+; calculated for C26H32NO3S: 438.2103].
General Procedure D. Diastereoselective Hydrogenation of β-Hydroxy Enamines with Aromatic Substituents in 3-Position: N-Benzyl-N-(3-hydroxy-2,3-diphenylpropyl)-4-methylbenzenesulfonamide (3d)
In a 10 × 75 mm glass test tube was dissolved (E)-N-benzyl-N-(3-hydroxy-2,3-diphenylprop-1-enyl)-4-methylbenzenesulfonamide (47 mg, 0.1 mmol) in ethyl acetate (3 mL) at room temperature. 10% Palladium on carbon (8 mg, 7 mol %) was added and the test tube was placed in a Parr hydrogenator at room temperature. Good stirring was confirmed before closing the apparatus. After flushing three times with hydrogen, the system was pressured with 9.65 MPa (1400 psi) of hydrogen and the reaction was stirred for 12 h at room temperature. After opening the apparatus, the palladium catalyst was removed via filtration through a plug of Celite. The filtrate was concentrated in vacuo and the residue was chromatographed on silica gel (10% ethyl acetate in hexanes) to afford 43 mg (92% yield) of 3d as an amorphous solid. 1H NMR (CDCl3, 360 MHz): δ 2.42 (s, 3H), 2.65 (m, 1H), 2.79 (d, 1H, J = 4.3 Hz), 2.99 (dd, 1H, J = 14.3 Hz, 6.1 Hz), 3.88 (d, 1H, J = 14.5 Hz), 4.55 (d, 1H, J = 14.5 Hz), 4.96 (t, 1H, J = 4.3 Hz), 6.59–6.70 (m, 4H), 6.82–6.89 (m, 2H), 7.04–7.13 (m, 3H), 7.25–7.36 (m, 8H), 7.61–7.66 (m, 2H). 13C{1H} NMR (CDCl3, 75 MHz): δ 21.78, 52.15, 54.66, 55.36, 72.28, 127.09, 127.32, 127.53, 128.16, 128.33, 129.03, 129.10, 129.62, 130.05, 134.51, 136.12, 136.79, 138.21, 143.83, 158.61. IR (neat): 3519 (OH), 2919, 1611, 1513, 1454, 1331, 1246, 1156, 1103, 1034, 928 cm−1. HRMS-CI: m/z 494.1760 [(M + Na)+; calculated for C29H29NO3SNa: 494.1766].
3. Results and Discussion
3.1. Asymmetric Synthesis of β-Hydroxy Enamines
Our interest in enantiomerically enriched β-hydroxy enamines with stereodefined double bonds stems from their potential utility in medicinal chemistry and as synthetic intermediates. We therefore setout to develop a highly enantioselective, practical and efficient one-pot method for their generation.
3.1.1. Optimization of Enantioselective β-Amino Vinylation of Aldehydes
For the synthesis of enantioenriched β-hydroxy enamines, we envisaged use of Oppolzer's method20 for the key C–C bond-forming step. Based on Srebnik's observation60 that alkenyl boranes undergo reversible transmetalation with dialkylzinc reagents to generate vinylzinc intermediates, Oppolzer20 developed a catalytic asymmetric synthesis of allylic alcohols involving hydroboration of alkynes, transmetalation of the vinyl group to zinc, and enantioselective addition to aldehydes to afford enantioenriched allylic alcohols (Scheme 4). We,15–19 and others,20–29 have used this method to make allylic alcohols, and we have applied it to the synthesis of α- and β-amino acid derivatives,61, 62 epoxy alcohols,16, 17, 63 and cyclopropyl and vinyl cyclopropyl alcohols.19, 64, 65 This method works well with terminal and internal alkynes and we have shown that ethoxy ethynyl ether can also be employed.18, 66 It was not clear at the outset whether the uncatalyzed hydroboration of internal ynamides would proceed with high regioselectivity. Only hydroboration of terminal ynamides was previously reported with good regioselectivity.67, 68
Scheme 4.

Oppolzer's Alkenylation of Aldehydes
Our synthesis of β-hydroxy enamines involves application of Oppolzer's procedure to ynamides, which are readily available using Stahl's copper catalyzed oxidative coupling of alkynes with amines.14 As outlined in Table 1, several phenyl-substituted ynamides were synthesized using amines with electron withdrawing groups (EWG) on the nitrogen. The presence of the EWG is important for the synthesis and stabilization of ynamides and the resulting β-hydroxy enamines.
Table 1.
Examination of various Electron Withdrawing Groups on the Ynamide
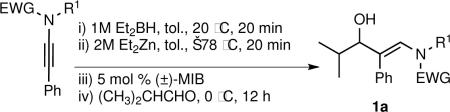
| entry | ynamide | (E)-trisubstituted hydroxy enamide | isolated yield (%) |
|---|---|---|---|
| 1 |
|
|
25 |
| 2 |
|
|
0 |
| 3 |
|
|
0 |
| 4 |
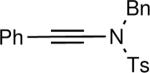
|
|
82 |
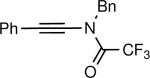
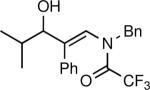
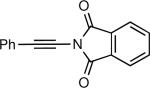
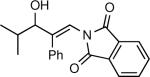
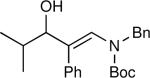
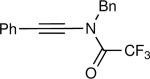
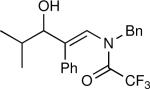
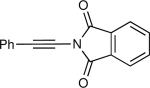
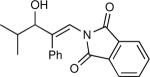
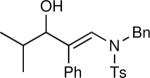
The synthesis of β-hydroxy enamines begins with hydroboration of ynamides with diethyborane and transmetalation of vinylborane with Et2Zn to generate the β-amino vinyl zinc intermediates. The aldehyde alkenylation was performed in the presence of catalyst derived from Nugent's isoborneol-based amino alcohol ligand MIB.69 As illustrated in Table 1, yields of the addition product are strongly dependent on the nature of the EWG. Although the Boc group resulted in formation of the desired addition product, the isolated yield did not exceed 25% (entry 1). More electron withdrawing carbonyl groups on the nitrogen such as trifluoro acetyl or imide did not result in formation of the β-hydroxy enamine product (entries 2–3), perhaps because the borohydride added to the carbonyl group. In contrast, the more robust tosyl group led to formation of the β-hydroxy enamine in 82% yield (entry 4). The tosyl group serves a dual role in the chemistry, both enabling the generation of the product in good yield and preventing the elimination of β-hydroxy enamine. Decomposition via elimination eventually affords α,β-unsaturated aldehydes after reaction with water.
To perform the analogous asymmetric addition, we used catalyst derived from enantiomerically pure (−)-MIB (Table 2), which is readily prepared in three steps or can be purchased.70 Following the general procedure in Table 1, entry 4, several hydroborating agents were examined (Table 2). After the hydroboration and addition of the dialkylzinc reagent, (−)-MIB (5 mol %) and isobutyraldehyde were added. When BH3•SMe2 was used in the hydroboration, only trace addition product was observed (entry 1). Use of 9-BBN resulted in formation of the β-hydroxy enamine in 44% yield with 82% ee (entry 2). Screening combinations of diethyl- and dicyclohexyl borane with diethyl- and dimethylzinc (entries 3–5) led to identification of diethylborane and diethylzinc as the optimal combination, with formation of the β-hydroxy enamine in 82% yield and 92% ee. It is noteworthy that under the reaction conditions the addition of Et2Zn to the aldehyde is very slow compared to the reaction of the vinyl zinc species and less then 5% of ethyl addition product was observed. The optimized conditions in Table 2 were then used to determine the substrate scope, as outlined in the following section.
Table 2.
Optimization of Yield and Enantioselectivity in the Synthesis of β-Hydroxy Enamines
| entry | ynamide | (E)-trisubstituted hydroxy enamide | isolated yield (%) |
|---|---|---|---|
| 1 |
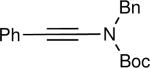
|
|
25 |
| 2 |
|
|
0 |
| 3 |
|
|
0 |
| 4 |
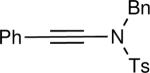
|
|
82 |
3.1.2. Substrate Scope of the Synthesis of β-Hydroxy Enamines
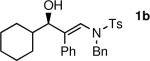
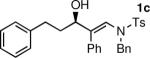
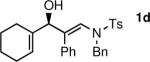
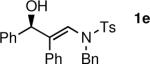
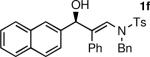
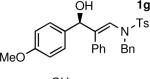
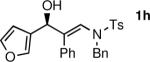
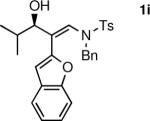
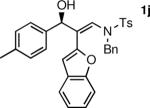
To determine the scope and limitations of our one-pot synthesis of enantioenriched β-hydroxy enamines, a series of aldehydes and ynamides were tested using the conditions in Table 2 (entry 4). As shown in Table 3, the asymmetric vinylation reaction is compatible with a range of ynamides and aldehydes. Initially, ynamides derived from phenyl acetylene were employed (entries 1–8). With this coupling partner, aliphatic aldehydes with α-branching underwent additions with enantioselectivities >90% and yields of 73–82% (entries 1 and 2). With dihydrocinnamaldehyde, which lacks α-branching, a significant drop in enantioselectivity to 54% was observed (entry 3). The α,β-unsaturated aldehyde cyclohexenecarboxaldehyde afforded β-hydroxy enamine in 92% enantioselectivity with 70% yield (entry 4). This product also contains an allylic alcohol, which is a useful functional group for further elaboration.30 Aromatic aldehydes proved to be excellent substrates undergoing the reaction with enantioselectivities ranging from 91–98% and yields >80% (entries 5–7). The heteroaromatic 3-furan carboxaldehyde resulted in formation of the product with 89% enantioselectivity in 68% yield (entry 8). Ynamides with a 2-benzofuranyl substituent also exhibited excellent enantioselectivities when used in the asymmetric addition to isobutyraldehyde (91% ee, entry 9) and 4-methylbenzaldehyde (98% ee, entry 10). Yields >80% were achieved in both cases. Comparison of the enantioselectivities with phenyl- (entries 1–8) and 2-benzofuranyl-substituted ynamides (entries 9 and 10) suggests that the addition is not strongly dependent on the nature of the aryl group.
Table 3.
Substrate Scope of the Asymmetric One-Pot Generation of (E)-Trisubstituted β-Hydroxy Enamines

| entry | β-hydroxy enamide | isolated yield (%) ee (%)a | |
|---|---|---|---|
| 1 |
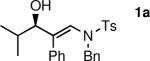
|
82 | 92 |
| 2 |
|
73 | 91 |
| 3 |
|
78 | 54 |
| 4 |
|
70 | 92 |
| 5 |
|
85 | 91 |
| 6 |
|
81 | 98 |
| 7 |
|
86 | 96 |
| 8 |
|
68 | 89 |
| 9 |
|
82 | 91 |
| 10 |
|
85 | 98 |
| 11 |
|
81 | 93 |
| 12 |
|
85 | 96 |
| 13 |
|
80 | 93 |
| 14 |
|
77 | 94 |
| 15 |
|
67 | 93 |
| 16 |
|
81 | 76 |
| 17 |
|
79 | 83 |
| 18 |
|
25 | 97 |
Enantioselectivities were determined by HP
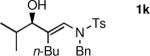
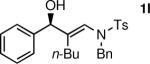
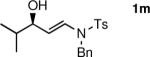
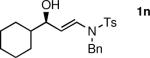
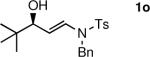
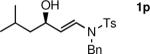
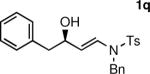
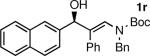
The n-Bu-substituted ynamide underwent addition with representative aliphatic and aromatic aldehydes with high levels of enantioselectivity. Thus, with isobutyraldehyde and benzaldehyde, the addition products were obtained with 93% and 96% enantioselectivity, respectively (entries 11–12). The yields were >80% in both cases. The parent ynamide (R=H) was also a useful coupling partner as shown in entries 13–17. With aliphatic aldehydes isobutyraldehyde and cyclohexanecarboxaldehyde, yields were 80% and 77%, respectively, and enantioselectivities were ≥93% (entries 13–14). Bulky pivaldehyde reacted with high enantioselectivity (93%), albeit decreased yield (67%, entry 15). Not surprisingly, aliphatic aldehydes lacking α-branching were difficult substrates with (–)-MIB (entries 16–17), as with most amino alcohol catalysts.71–82 Isovaleraldehyde and phenyl acetaldehyde gave 76% and 83% enantioselectivities, respectively. Presumably the advent of more enantioselective catalysts will enable additions to these challenging substrates to occur with higher stereoselectivities. As indicated in Table 1, entry 1, the Boc protected ynamide gave low yield. Nonetheless, the product formed on addition to 2-naphthaldehyde in the presence of (–)-MIB had an ee of 97% (Table 3, entry 18).
The absolute stereochemistry was determined for a derivative of 1b by single crystal X-ray diffraction (see the Supporting Information and section 3.2). Accordingly, the (–)-MIB-based catalyst promoted the formation of (R)-hydroxy enamines, consistent with transition state proposed by Noyori83, 84 for the asymmetric alkylation of aldehydes with dialkyl zinc reagents and the amino alcohol-based catalyst derived from DIAB. The stereochemistry of the remaining entries in Table 3 was assigned by analogy. It is noteworthy that coordination of the heteroatoms, such as the nitrogens present in the ynamides and oxygens of the sulfonamides and benzofuryl groups (entries 9 and 10) to zinc was not problematic in the enantioselective vinyl addition step. Finally, only (E)-isomers of the β-hydroxy enamines were observed in all cases by 1H NMR spectroscopy, indicating that isomerization of the double bond does not occur during the transmetalation, addition, work-up or purification steps. As will be seen in subsequent sections, the conservation of the double bond geometry of the enamine is essential for diastereoselective elaboration of these products.
In order for a method to be useful, scalability must be demonstrated. As shown in eq 1, the phenyl substituted ynamide and isobutyraldehyde were used in the catalytic asymmetric β-amimo vinylation to afford 1.5 g of β-hydroxy enamine 1a (92% ee).
 |
(1) |
3.2. Tandem Asymmetric β-Amino-Vinylation of Aldehydes/Diastereoselective Cyclopropanation
Cyclopropyl amines continue to attract interest due to their abundance in natural products and biologically active compounds,31 including several commercial medications.85–93 As a result, many methods have been developed to synthesize the amino cyclopropyl moiety.34–54 Direct methods to effectively synthesize functionalized enantio- and diastereoenriched cyclopropyl amines remain challenging, however. Existing routes to prepare amino cyclopropyl carbinols suffer from either low yields94 (Scheme 5, A), low diastereoselectivities95, 96 (Scheme 5, B) or are not enantioselective.97–99
Scheme 5.

Barrett's (A) and de Meijere's (B) Syntheses of Amino Cyclopropyl Carbinols
Because amino cyclopropyl carbinols can serve as precursors to compounds of biological relevance,52,100–102 their direct, enantio- and diastereoselective synthesis is desirable. Our approach to the catalytic enantio- and diastereoselective synthesis of amino cyclopropyl carbinols is based on our prior efforts involving the diastereoselective directed cyclopropanation of allylic alcohols.19, 64, 65 At the outset of this study, however, it was not clear how the nitrogen of the enamine would impact the reactivity of the double bond toward cyclopropanation or if it would affect the diastereoselectivity. A search of the literature revealed very few examples of Simmons-Smith cyclopropanations of enamines.50,103 Nonetheless, we attempted the tandem asymmetric amino vinylation/diastereoselective cyclopropanation. Following our synthesis of β-hydroxy enamines, hydroboration of ynamides, transmetalation, and asymmetric addition to aldehydes generated zinc β-alkoxy enamine intermediates (Scheme 6). The alkoxide complex was then subjected to a directed Simmons–Smith cyclopropanation using a carbenoid introduced by Shi.104, 105
Scheme 6.

Asymmetric and Diastereoselective One-Pot Generation of Amino Cyclopropyl Carbinols
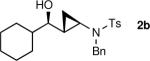
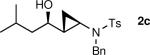
The generation of the Shi modified carbenoid involves reaction of CF3CH2OH with diethylzinc to form the monoalkoxide complex, CF3CH2OZnEt. Reaction of this intermediate with diiodomethane resulted in formation of the Shi carbenoid, CF3CH2OZnCH2I. Upon combining CF3CH2OZnCH2I with zinc β-alkoxy enamine intermediates, we were pleased to see formation of the desired cyclopropyl amines as a single diastereomer, although the yields were moderate. As we have observed with other systems,19,64,65 the cyclopropanation proceeds in low to moderate yield in the presence of the triethylborane, a byproduct formed in the transmetalation of the β-amino vinyl group from boron to zinc (Scheme 6). To achieve high yields the triethyborane was removed from the reaction mixture before the cyclopropanation step. Removal of the volatile triethylborane can be accomplished by subjecting the reaction mixture to reduced pressure. The best results were obtained when hexanes was added to the residue and the volatile materials were removed under reduced pressure. This procedure was repeated two more times. Under these conditions, aliphatic aldehydes (entries 1–5, Table 4) underwent the addition/cyclopropanation sequence with the terminal ynamide in 72–82% isolated yields, 76–94% enantioselectivities and excellent diastereoselectivities (>20:1 in all cases).
Table 4.
Enantio- and Diastereoselective One-Pot Generation of Amino Cyclopropyl Carbinols

| entry | product | isolated yield (%) | ee (%)a | drb |
|---|---|---|---|---|
| 1 |
|
80 | 93 | >20:1 |
| 2 |
|
76 | 94 | >20:1 |
| 3 |
|
78 | 76 | >20:1 |
| 4 |
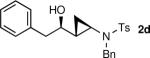
|
78 | 83 | >20:1 |
| 5 |
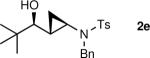
|
72 | 93 | >20:1 |
| 6 |
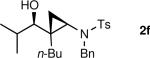
|
28c | 93 | >20:1 |
Ee determined by HPLC of the intermediate enamines.
Diastereoselectivity determined by 1H NMR analysis of crude reaction mixture.
Reaction time 60 h.
Under our current conditions, the tandem reaction is capricious when the sequence is initiated with substituted ynamides. Although we were able to generate a tri-substituted amino cyclopropyl carbinol (entry 6), the isolated yield was low (28%) and reaction time long (60 h). Cyclopropanation of β-alkoxy enamines generated from aromatic aldehydes or aryl-substituted ynamides did not proceed, resulting in isolation of β-hydroxy enamines.
The diastereomeric ratios in Table 4 were determined by 1H NMR of crude reaction products. As mentioned earlier, the absolute and relative configuration of 2f was determined by X-ray diffraction (see SI). The structure revealed that cyclopropanation occurred syn to the carbinol, as observed in the cyclopropanation of allylic alcohols via the Simmons–Smith cyclopropanation.19, 64, 65, 106, 107 A possible transition state for the directed cyclopropanation is illustrated in Figure 1.106, 107
Figure 1.

Proposed transition states for diastereoselective cyclopropanation of β-alkoxy enamines. The interaction of R with the substituents is responsible for the diastereoselectivity.
The amino vinylation/cyclopropanation method introduced above affords amino cyclopropyl carbinols containing trans-disubstituted cyclopropane motifs. The products are formed with good to excellent enantioselectivities, very high diastereoselectivities, and good yields. This one-pot procedure results in generation of three C–C bonds and three continuous stereocenters and is conducted without isolation of any intermediates.
3.3.1. Diastereoselective Hydrogenation of β-Hydroxy Enamines
1,3-Amino alcohols are common structural motifs in many biologically active compounds55–58 and are extensively used in asymmetric synthesis both as chiral ligands and auxiliaries.108 There are many methods available for the synthesis of enantio- and diastereoenriched 1,3-disubstituted-1,3-amino alcohols.109–113 In contrast, the stereoselective synthesis 1,2-disubstituted-1,3-amino alcohols remains a significant challenge.6, 114–122 The best approach to enantioenriched 1,2-disubstituted-1,3-amino alcohols is addition of lithium arylacetonitriles to aldehydes123 leading to β-hydroxy nitriles with moderate enantio- and diastereoselectivities. This method employs greater than stoichiometric amounts of chiral ligand. Efficient methods for the synthesis of enantio- and diastereoenriched 1,2-disubstituted-1,3-amino alcohols would facilitate their use in medicinal chemistry and asymmetric catalysis. We envisioned a catalytic diastereoselective hydrogenation of β-hydroxy enamines as a straightforward route to 1,2-disubstituted-1,3-amino alcohols.
Our initial efforts to reduce β-hydroxy enamines focused on homogenous hydrogenation catalysts and variation of the solvent, hydrogen pressure, and reaction time (Table 5). Both Crabtree's124 and Wilkinson's125 catalysts resulted in loss of the hydroxy group, probably by elimination to form an α,β-unsaturated iminium intermediate. This intermediate could then undergo subsequent hydrogenation resulting in formation of the observed alkyl sulfonamide 4 (entries 1–6). These examples illustrate the sensitivity of β-hydroxy enamines to elimination even under very mild conditions. Next we turned to common heterogeneous catalysts. PtO2 did not catalyze the hydrogenation at room temperature, but caused decomposition of the hydroxy enamine at elevated temperature or pressure (entries 7–9). On the other hand, rhodium on alumina (5 mol %) was completely inactive (entry 10). Palladium on carbon (10 mol %) was the most promising catalyst, providing the desired 1,3-amino alcohol as the major product. Atmospheric pressure at room temperature (entry 11) or above (entry 12) was not sufficient for full conversion, so higher pressure was used (entries 13 and 14). With 10 mol % of Pd/C catalyst in methanol at 8 MPa of hydrogen the reaction provided 90% isolated yield with excellent diastereoselectivity (>20:1, entry 14). Unlike the β-hydroxy enamines with aliphatic carbinols, which undergo highly diastereoselective reductions in methanol, we found that the enamine hydrogenation of substrates with benzylic hydroxyl groups led to elimination/reduction in methanol. Therefore the choice of the solvent was crucial and dependent on the substituent attached to the carbinol. We reasoned that protic solvents would hydrogen bond to the hydroxyl of the β-hydroxy enamine and facilitate the elimination pathway. To reduce elimination, β-hydroxy enamines with aromatic carbinols were hydrogenated in aprotic solvents. After screening several solvents, ethyl acetate was determined to be the most effective. Interestingly, hydrogenation of β-hydroxy enamines with aliphatic carbinols in ethyl acetate resulted in lower conversions (entry 15) than methanol.
Table 5.
Optimization of the Diastereoselective Hydrogenation of β-Hydroxy Enamines
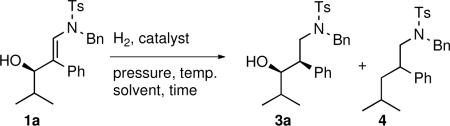
| entry | catalyst (mol %)/solvent | pressure (MPa) | temp. (°C) | time (h) | product | isolated yield (%) | dra |
|---|---|---|---|---|---|---|---|
| 1 | Crabtree's cat. (10)/CH2Cl2 | 0.1 | 20 | 12 | 4 | 40 | - |
| 2 | Crabtree's cat. (10)/MeOH:CH2Cl2, 1:1 | 0.1 | 20 | 12 | 4 | 50 | - |
| 3 | Crabtree's cat. (10)/MeOH:CH2Cl2, 1:1 | 1.0 | 20 | 5 | 4 | 50 | - |
| 4 | Crabtree's cat. (10)/MeOH:CH2Cl2, 1:1 | 8.0 | 20 | 12 | 4 | 60 | - |
| 5 | Wilkinson's cat. (1.0)/benzene | 0.1 | 20 | 12 | NR | 0 | - |
| 6 | Wilkinson's cat. (10)/benzene | 1.0 | 20 | 12 | 4 | 60 | - |
| 7 | PtO2 (10)/MeOH | 0.1 | 20 | 12 | NR | 0 | - |
| 8 | PtO2 (10)/MeOH | 0.1 | 50 | 12 | dec. | 0 | - |
| 9 | PtO2 (10)/MeOH | 8.0 | 20 | 12 | dec. | 0 | - |
| 10 | 5%Rh/Al2O3 (10)/AcOEt | 1.0 | 20 | 12 | NR | 0 | - |
| 11 | 10%Pd/C (10)/MeOH | 0.1 | 20 | 12 | 3a | 10 | >20:1 |
| 12 | 10%Pd/C (10)/MeOH | 0.1 | 50 | 5 | 3a | 14 | >20:1 |
| 13 | 10%Pd/C (10)/MeOH | 1.0 | 20 | 12 | 3a | 55 | >20:1 |
| 14 | 10%Pd/C (10)/MeOH | 8.0 | 20 | 12 | 3a | 90 | >20:1 |
| 15 | 10%Pd/C (10)/AcOEt | 8.0 | 10 | 10 | 3a | 14 : | >20:1 |
NR: only starting material recovered, dec: starting material decomposed to an inseparable mixture.
Diastereoselectivity determined by 1H NMR analysis of crude reaction mixture.
3.3.2. Substrate Scope of the Diastereoselective Hydrogenation of β-Hydroxy Enamines
The results of the diastereoselective hydrogenation of β-hydroxy enamines are shown in Table 6. Catalytic hydrogenation of β-hydroxy enamines with aliphatic (entries 1, 2, 3, 7 and 9) or aromatic (entries 4, 5, 6, 8 and 10) carbinol substituents and aromatic (entries 1–6) or heteroaromatic (entries 7 and 8) substituents in the 2-position led to 1,2-disubstituted-1,3-aminoalcohols with good isolated yields (80–92%) and excellent diastereoselectivities (>20:1). When an alkyl group was in the 2-position of the β-hydroxy enamine, however, the diastereoselectivity was only moderate (entries 9 and 10). The diastereomeric ratios in Table 6 were determined by 1H NMR of the crude reaction products. The ee of the β-hydroxy enamines 1a, 1e and 1j were determined before the hydrogenation to be 92, 91 and 98%. Their hydrogenation products 3a, 3e and 3j were found to have ee's of 91, 90 and 95%, respectively. The absolute and relative stereochemistries in Table 6 were based on the X-ray structural determination of 3a, which revealed that the hydrogenation is syn-selective (see the Supporting Information).
Table 6.
Diastereoselective Hydrogenation of β-Hydroxy Enamines

| entry | product | isolated yield (%) | dra |
|---|---|---|---|
| 1 |
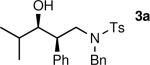
|
90 | >20:1b |
| 2 |
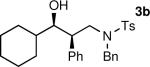
|
80 | >20:1 |
| 3 |
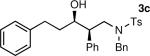
|
80 | >20:1 |
| 4 |
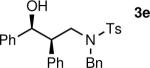
|
92 | >20:1c |
| 5 |
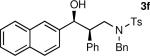
|
86 | >20:1 |
| 6 |
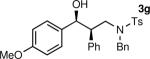
|
90 | >20:1 |
| 7 |
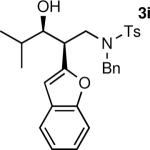
|
88 | >20:1 |
| 8 |
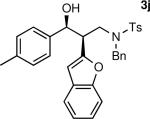
|
82 | >20:1d |
| 9 |
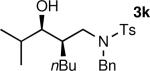
|
85 | 3:1 |
| 10 |
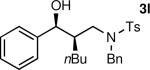
|
90 | 4:1 |
Diastereoselectivity determined by 1H NMR analysis of crude reaction mixture.
91% ee (HPLC).
90% ee (HPLC).
95% ee.
For aliphatic R1: MeOH;for aromatic R1: EtOAc.
The diastereoselective outcome of the hydrogenation can be rationalized by a conformational analysis of the β-hydroxy enamine when associated with the surface of the palladium catalyst (Figure 2, left). The β-hydroxy enamine binds to the surface of the catalyst through coordination of the hydroxy group. The substrate then adopts a conformation that minimizes unfavorable interaction between R1 and the hydroxyl, as would be predicted for the free substrate in solution. The dihydrogen is then delivered to give the syn-product. It is possible to disfavor the association of the hydroxyl group with the metal catalyst by introduction of bulky protecting group. In this case, the two conformers differ in steric interactions between iPr–Ph and OTBS-Ph (Figure 2, right). The difference in energy between the conformers is not sufficient for good diastereoselection, resulting in 0.7:1 syn:anti mixture of diastereomers in case of TBS and 1:1 mixture in case of TIPS. It is noteworthy that the N-benzyl group is not hydrogenolysed under these reaction conditions.
Figure 2.

A: proposed conformation of hydroxy enamines on the surface of catalyst to rationalize the syn selective hydrogenation. B: conformation of TBS protected β-hydroxy enamine slightly favors anti diastereomer.
3.4. Enantio- and Diastereoselective Formal Synthesis of Derivatives of PRC200-SS
The 1,2-disubstituted-1,3-amino alcohol moiety is present in many biologically active compounds such as the antidepressants Venlafaxine (Effexor, Wyeth) and Desvenlafaxine (Pristiq, Wyeth), which are both serotonin-norepinephrine reuptake inhibitors (SNRIs). Most antidepressants prescribed at this time target serotonergic and noradrenergic neurotransmission, resulting in enhanced dopamine neurotransmission. Influencing the combination of serotonergic, noradrenergic and dopaminergic neurotransmission (SNDRIs) can result in significant improvement of depressive symptoms. There are no approved SNDRIs on the market yet, but several are in development. One family of SNDRIs includes PRC200-SS126–129 and its derivatives.130 To demonstrate the utility of our methods, we performed a short formal enantio- and diastereoselective synthesis of derivatives of PRC200-SS.130 Our synthesis (Scheme 7) starts with the copper catalyzed oxidative coupling of commercially available 2-naphtyl acetylene with N-methyl toluene sulfonamide, which proceeded in 87% yield. Hydroboration of the resulting 2-naphtyl ynamide 5 with diethylborane followed by transmetalation to zinc and enantioselective addition to benzaldehyde promoted by the (+)-MIB-based catalyst provided β-hydroxy enamine 6 in high yield (92%) and excellent enantioselectivity (97%). Due to the presence of the N-methyl in place the of N-benzyl in the examples in Tables 1–5, the hydrogenation of β-hydroxy enamine 6 under the original conditions (Table 7, entry 1) or related conditions (entries 2 and 3) provided only mediocre diastereoselectivity of 7 (2:1). Upon screening additional heterogeneous catalysts, however, it was found that 6 mol % of 20% Pd(OH)2/C in methanol under 1 atm of hydrogen provided the syn-product 7 of high diastereoselectivity (dr=10.8:1, 1H NMR) in 65% yield and with 95% ee (entry 4). When higher hydrogen pressure (entry 5) or higher catalyst loading (entry 6) was applied, partial hydrogenation of 2-naphtyl group was observed and 7' was the major product. Structure of 7' was confirmed by X-ray diffraction (see Supporting Information). The diastereomers formed in entry 4 were easily separable by column chromatography, allowing isolation of the pure syn isomer. Detosylation with sodium naphthalenide afforded 8 in 89% yield. Biologically active chloride, fluoride and azide derivatives can be obtained from the common intermediate 8, as shown in Scheme 8.130 Mitsunobu reaction of 7 and subsequent hydrolysis afforded 9 in 79% yield with 95% ee (Scheme 9). After the detosylation PRC200-SS was isolated in 76% yield.
Scheme 7.

Synthesis of the Key Intermediate 8 in the Synthesis of PRC200-SS and Derivatives.
Table 7.
Optimization of Diastereoselective Hydrogenation of β-Hydroxy Enamine 6
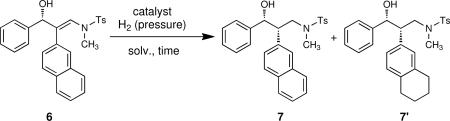
| entry | catalyst (mol %)/solvent | pressure (MPa) | time (h) | major product | isolated yield (%) | dra |
|---|---|---|---|---|---|---|
| 1 | 10%Pd/C (10)/AcOEt | 8.0 | 5 | 7 | 48 | 2:1 |
| 2 | 10%Pd/C (10)/MeOH | 0.1 | 48 | 7 | 64 | 4:1 |
| 3 | 10%Pd/C (10)/MeOH | 8.0 | 5 | 7 | 83 | 2:1 |
| 4 | 10% Pd(OH)2/C (6)/MeOH | 0.1 | 48 | 7 | 65 | 10.8:1 |
| 5 | 10% Pd(OH)2/C (10)/MeOH | 8.0 | 8 | 7' | 78 | 2:1 |
| 6 | 10% Pd(OH)2/C (20)/MeOH | 0.1 | 48 | 7' | 68 | 3:1 |
Diastereoselectivity determined by 1H NMR analysis of the crude reaction mixture.
Scheme 8.

Known Conversion of 8 to Bioactive Derivatives
Scheme 9.

Conversion of 7 to 9 and PCR200-SS
In contrast to the original 9-step synthesis of 8 (Scheme 10), which involved resolution of racemic intermediate 10 and late-stage separation of diastereomers (Scheme 10, step 8), our procedure is highly efficient and both enantio- and diastereoselective, affording 8 in four steps from commercially available materials in 46% overall yield. Although the original anti-selective nitrile aldol method allows synthesis of PRC200-SS, the absolute configuration of carbinol must be inverted to prepare the bioactive chloride, fluoride and azide derivatives. Our syn-selective method opens the door to direct synthesis of PRC200-SS derivatives without the need for a Mitsunobu reaction.
Scheme 10.

Carlier's synthesis of PRC200-SS and Derivatives.
4. Summary and Outlook
Herein is presented the first general catalytic and highly enantioselective method of synthesis of β-hydroxy enamines. The versatility of this method was demonstrated by its wide substrate scope, combining aliphatic, α,β-unsaturated, aromatic and heteroaromatic aldehydes with aliphatic, aromatic or heteroaromatic ynamides. The resulting β-hydroxy enamines were obtained with high yields and good to excellent ee's. We have shown that β-hydroxy enamines are viable substrates for alkoxy and hydroxyl directed reactions. An efficient catalytic enantio- and diastereoselective method for the synthesis of amino cyclopropyl carbinols is outlined. This convenient one-pot method enables the synthesis of amino cyclopropyl carbinols with good yields, good to excellent enantioselectivities and excellent diastereoselectivities. Additionally, an efficient syn diastereoselective hydrogenation of β-hydroxy enamines has been developed that leads to 1,2-disubstituted-1,3-amino alcohols, a common structural motif in many biologically active compounds. This method has made possible the efficient synthesis of the precursor to PRC200-SS derivatives, which are potent serotonin-norepinephrine-dopamine reuptake inhibitors.
Supplementary Material
Acknowledgement
This work was financially supported by the NIH (National Institute of General Medical Sciences GM58101) and the NSF (CHE–0848467). Funds for instrumentation were provided by the NIH for a MS (1S10RR023444) and NSF for an X-ray diffractometer (CHE–0840438). We would like to thank to Dr. Hun Young Kim for preliminary results and Prof. Marisa C. Kozlowski for use of the hydrogenation apparatus. PV thanks the Isabel and Alfred Bader Foundation for a graduate fellowship.
Footnotes
Supporting Information Available. Experimental procedures, synthesis and full characterization of new compounds and conditions for resolution of racemates (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Ogawa Y, Konishi T. Chem. Pharm. Bull. 2009;57:1110. doi: 10.1248/cpb.57.1110. [DOI] [PubMed] [Google Scholar]
- (2).Zhang Y, Cichewicz RH, Nair MG. Life Sci. 2004;75:753. doi: 10.1016/j.lfs.2004.03.002. [DOI] [PubMed] [Google Scholar]
- (3).Munter T, Curieux FL, Sjoholm R, Kronberg L. Chem. Res. Toxicol. 1999;12:1205. doi: 10.1021/tx990116c. [DOI] [PubMed] [Google Scholar]
- (4).Taylor DE, Trieber CA, Trescher G, Bekkering M. Antimicrob. Agents Ch. 1998;42:59. doi: 10.1128/aac.42.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Yoshikawa K, Nagai M, Arihara S. Phytochemistry. 1994;35:1057. doi: 10.1016/s0031-9422(00)90428-x. [DOI] [PubMed] [Google Scholar]
- (6).Hirano S, Fukudome Y, Tanaka R, Sato F, Urabe H. Tetrahedron. 2006;62:3896. [Google Scholar]
- (7).Barbazanges M, Meyer C, Cossy J. Org. Lett. 2007;9:3245. doi: 10.1021/ol0711725. [DOI] [PubMed] [Google Scholar]
- (8).Barbazanges M, Meyer C, Cossy J. Tetrahedron Lett. 2008;49:2902. [Google Scholar]
- (9).Tanaka R, Hirano S, Urabe H, Sato F. Org. Lett. 2003;1:67. doi: 10.1021/ol027209x. [DOI] [PubMed] [Google Scholar]
- (10).Hirano S, Tanaka R, Urabe H, Sato F. Org. Lett. 2004;6:727. doi: 10.1021/ol036396b. [DOI] [PubMed] [Google Scholar]
- (11).Oppolzer W, Kingma AJ, Pillai SK. Tetrahedron Lett. 1991;32:4893. [Google Scholar]
- (12).De Korver KA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y, Hsung RP. Chem. Rev. 2010;110 doi: 10.1021/cr100003s. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Evano G, Coste A, Jouvin K. Angew. Chem. Int. Ed. 2010;49:2840. doi: 10.1002/anie.200905817. [DOI] [PubMed] [Google Scholar]
- (14).Hamada T, Ye X, Stahl SS. J. Am. Chem. Soc. 2008;130:833. doi: 10.1021/ja077406x. [DOI] [PubMed] [Google Scholar]
- (15).Hussain MM, Walsh PJ. Acc. Chem. Res. 2008;41:883. doi: 10.1021/ar800006h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kelly AR, Lurain AE, Walsh PJ. J. Am. Chem. Soc. 2005;127:14668. doi: 10.1021/ja051291k. [DOI] [PubMed] [Google Scholar]
- (17).Lurain AE, Carroll PJ, Walsh PJ. J. Org. Chem. 2005;70:1262. doi: 10.1021/jo048345d. [DOI] [PubMed] [Google Scholar]
- (18).Jeon S-J, Chen YK, Walsh PJ. Org. Lett. 2005;7:1729. doi: 10.1021/ol050255n. [DOI] [PubMed] [Google Scholar]
- (19).Kim HY, Lurain AE, Garcia PG, Carrol PJ, Walsh PJ. J. Am. Chem. Soc. 2005;127:13138. doi: 10.1021/ja0539239. [DOI] [PubMed] [Google Scholar]
- (20).Oppolzer W, Radinov RN. Helv. Chim. Acta. 1992;75:170. [Google Scholar]
- (21).Soai K, Takahashi K. J. Chem. Soc., Perkin Trans. 1. 1994;10:1257. [Google Scholar]
- (22).Wipf P, Ribe S. J. Org. Chem. 1998;63:6454. [Google Scholar]
- (23).Soai K, Niwa S. Chem. Rev. 1992;92:833. [Google Scholar]
- (24).Pu L, Yu H. Chem. Rev. 2001;101:757. doi: 10.1021/cr000411y. [DOI] [PubMed] [Google Scholar]
- (25).Oppolzer W, Radinov R. Tetrahedron Lett. 1988;32:5777. [Google Scholar]
- (26).von dem Bussche-Hunnefeld JL, Seebach D. Tetrahedron. 1992;48:5719. [Google Scholar]
- (27).Shibata T, Nakatsui K, Soai K. Inorg. Chim. Acta. 1999;296:33. [Google Scholar]
- (28).Wipf P, Xu W. Tetrahedron Lett. 1994;35:5197. [Google Scholar]
- (29).Wipf P, Xu W. Org. Synth. 1996;74:205. [Google Scholar]
- (30).Hoveyda AH, Evans DA, Fu GC. Chem. Rev. 1993;93:1307. [Google Scholar]
- (31).Wessjohann LA, Brandt W, Thiemann T. Chem. Rev. 2003;103:1625. doi: 10.1021/cr0100188. [DOI] [PubMed] [Google Scholar]
- (32).Salaun J. Top. Curr. Chem. 2000;207:1. [Google Scholar]
- (33).Salaun J, Baird MS. Curr. Med. Chem. 1995;2:511. [Google Scholar]
- (34).Aggarwal VK, de Vicente J, Bonnert RV. Org. Lett. 2001;3:2785. doi: 10.1021/ol0164177. [DOI] [PubMed] [Google Scholar]
- (35).de Meijere A, Kozhushkov SI, Savchenko AI. J. Organomet. Chem. 2004;689:2033. [Google Scholar]
- (36).Ye A, Komarov IV, Kirby AJ, Jones MJ., Jr. J. Org. Chem. 2002;67:9288. doi: 10.1021/jo0206014. [DOI] [PubMed] [Google Scholar]
- (37).Vanier SF, Larouche G, Wurz RP, Charette AB. Org. Lett. 2010;12:672. doi: 10.1021/ol9026528. [DOI] [PubMed] [Google Scholar]
- (38).Wurz RP, Charette AB. J. Org. Chem. 2004;69:1262. doi: 10.1021/jo035596y. [DOI] [PubMed] [Google Scholar]
- (39).Lebel H, Marcoux J-F, Molinaro C, Charette AB. Chem. Rev. 2003;103:977. doi: 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]
- (40).Lavallo V, Mafhouz J, Canac Y, Donnadieu B, Schoeller WW, Bertrand G. J. Am. Chem. Soc. 2004;126:8670. doi: 10.1021/ja047503f. [DOI] [PubMed] [Google Scholar]
- (41).Ogawa A, Takami N, Nanke T, Ohya S, Hirao T. Tetrahedron. 1997;53:12895. [Google Scholar]
- (42).Bégis G, Sheppard TD, Cladingboel DE, Motherwell WB, Tocher TA. Synthesis. 2005;19:3186. [Google Scholar]
- (43).Vilsmaier E. In: The Chemistry of the Cyclopropyl Group. Rappoport Z, editor. Wiley; New York: 1987. p. 1341. [Google Scholar]
- (44).de Meijere A, editor. Houben-Weyl. Vol. E17a–c. Stuttgart; Thieme: 1997. [Google Scholar]
- (45).Ha JD, Lee J, Blackstock SC, Cha JK. J. Org. Chem. 1998;63:8510. [Google Scholar]
- (46).Williams CM, de Meijere A. J. Chem. Soc., Perkin Trans. 1. 1998;22:3699. [Google Scholar]
- (47).Lee HB, Sung MJ, Blackstock SC, Cha JK. J. Am. Chem. Soc. 2001;123:11322. doi: 10.1021/ja017043f. [DOI] [PubMed] [Google Scholar]
- (48).Campos PJ, Soldevilla A, Sampedro D, Rodriguez MA. Tetrahedron Lett. 2002;43:8811. [Google Scholar]
- (49).Voigt T, Winsel H, de Meijere A. Synlett. 2002;8:1362. [Google Scholar]
- (50).Tsai CC, Hsieh IL, Cheng TT, Tsai PK, Lin KW, Yan TH. Org. Lett. 2006;8:2261. doi: 10.1021/ol0604365. [DOI] [PubMed] [Google Scholar]
- (51).Wiedemann S, Rauch K, Savchenko A, Marek I, de Meijere A. Eur. J. Org. Chem. 2004;3:631. [Google Scholar]
- (52).Jimenez JM, Rife J, Ortuno RM. Tetrahedron: Asymmetry. 1996;7:537. [Google Scholar]
- (53).Declerck D, Josse S, Van Nhien AN, Postel D. Tetrahedron Lett. 2009;50:2171. [Google Scholar]
- (54).Lu T, Hayashi R, Hsung RP, De Korver KA, Lohse AG, Song Z, Tang Y. Org. Biomol. Chem. 2009;7:3331. doi: 10.1039/b908205k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Peter A, Kaman J, Fiilop F, van der Eycken J, Armstrong DW. J. Chromatogr. A. 2001;79:919. doi: 10.1016/s0021-9673(01)00791-9. [DOI] [PubMed] [Google Scholar]
- (56).Rife J, Ortuno RM. Tetrahedron: Asymmetry. 1999;10:4245. [Google Scholar]
- (57).Saotome C, Ono M, Akita H. Tetrahedron: Asymmetry. 2000;11:4137. [Google Scholar]
- (58).Barrett S, O'Brien P, Steffens HC, Towers TD, Voith M. Tetrahedron. 2000;56:9633. [Google Scholar]
- (59).Bruckner D. Tetrahedron. 2006;62:3809. [Google Scholar]
- (60).Srebnik M. Tetrahedron Lett. 1991;32:2449. [Google Scholar]
- (61).Chen YK, Lurain AE, Walsh PJ. J. Am. Chem. Soc. 2002;124:12225. doi: 10.1021/ja027271p. [DOI] [PubMed] [Google Scholar]
- (62).Lurain AE, Walsh PJ. J. Am. Chem. Soc. 2003;125:10677. doi: 10.1021/ja035213d. [DOI] [PubMed] [Google Scholar]
- (63).Lurain AE, Maestri A, Kelly AR, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2004;126:13608. doi: 10.1021/ja046750g. [DOI] [PubMed] [Google Scholar]
- (64).Kim HY, Salvi L, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2009;131:954. doi: 10.1021/ja806989n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Kim HY, Salvi L, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2010;132:402. doi: 10.1021/ja907781t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Valenta P, Drucker NA, Bode JW, Walsh PJ. Org. Lett. 2009;10:2117. doi: 10.1021/ol9005757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Witulski B, Buschmann N, Bergstraser U. Tetrahedron. 2000;56:8473. [Google Scholar]
- (68).Hoffmann RW, Bruckner D. New J. Chem. 2001;25:369. [Google Scholar]
- (69).Nugent WA. J. Chem. Soc., Chem. Commun. 1999;15:1369. [Google Scholar]
- (70).Chen YK, Jeon S-J, Walsh PJ, Nugent WA. Organic Syntheses. 2005;82:87. [Google Scholar]
- (71).Soai K, Hatanaka T, Yamashita T. J. Chem. Soc., Chem. Commun. 1992;13:927. [Google Scholar]
- (72).Watanabe M, Soai K. J. Chem. Soc., Perkin Trans. 1. 1994;21:3125. [Google Scholar]
- (73).Delair P, Einhorn C, Dinhorn J, Luche JL. Tetrahedron. 1995;51:165. [Google Scholar]
- (74).Goralski CT, Chrisman W, Hasha DL, Nicholson LW, Rudolf PR, Zakett D, Singaram B. Tetrahedron: Asymmetry. 1997;8:3863. [Google Scholar]
- (75).Lawrence CF, Nayak SK, Thijs L, Zwanenburg B. Synlett. 1999;10:1571. [Google Scholar]
- (76).Dehli JR, Legros J, Bolm C. Chem. Commun. 2005;8:973. doi: 10.1039/b415954c. [DOI] [PubMed] [Google Scholar]
- (77).Berrisford DJ, Bolm C, Sharpless BK. Angew. Chem. Int. Ed. 1995;34:1059. [Google Scholar]
- (78).Muniz K, Bolm C. Chem. Eur. J. 2000;6:2309. doi: 10.1002/1521-3765(20000703)6:13<2309::aid-chem2309>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- (79).Rodriguez-Escrich S, Reddy KS, Jimeno C, Colet G, Rodriguez-Escrich C, Sola L, Vidal-Ferran A, Pericas MA. J. Org. Chem. 2008;73:5340. doi: 10.1021/jo800615d. [DOI] [PubMed] [Google Scholar]
- (80).Garcia-Delgado N, Fontes M, Pericas MA, Riera A, Verdaguer X. Tetrahedron: Asymmetry. 2004;15:2085. [Google Scholar]
- (81).Jimeno C, Pasto M, Riera A, Pericas MA. J. Org. Chem. 2003;68:3130. doi: 10.1021/jo034007l. [DOI] [PubMed] [Google Scholar]
- (82).Sola L, Reddy KS, Vidal-Ferran A, Moyano A, Pericas MA, Riera A, Alvarez-Larena A, Piniella J-F. J. Org. Chem. 1998;63:7078. doi: 10.1021/jo981336i. [DOI] [PubMed] [Google Scholar]
- (83).Yamakawa M, Noyori R. Organometallics. 1999;18:128. [Google Scholar]
- (84).Yamakawa M, Noyori R. J. Am. Chem. Soc. 1995;117:6327. [Google Scholar]
- (85).Gnad F, Reiser O. Chem. Rev. 2003;103:1603. doi: 10.1021/cr010015v. [DOI] [PubMed] [Google Scholar]
- (86).Burgess K-H, Ho K-K, Moye-Sherman D. Synlett. 1994;8:575. [Google Scholar]
- (87).Moreau B, Charette AB. J. Am. Chem. Soc. 2005;127:18014. doi: 10.1021/ja056192l. [DOI] [PubMed] [Google Scholar]
- (88).Fujita T. J. Med. Chem. 1973;16:923. doi: 10.1021/jm00266a012. [DOI] [PubMed] [Google Scholar]
- (89).Campoli-Richards DM, Monk JP, Price A, Benfield P, Todd PA, Ward A. Drugs. 1988;35:373. doi: 10.2165/00003495-198835040-00003. [DOI] [PubMed] [Google Scholar]
- (90).Wise R, Andrews JM, Edwards LJ. Antimicrob. Chemother. 1983;23:559. doi: 10.1128/aac.23.4.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Brighty KE. WO Patent 91/02526, 1991; EU Patent 413.455, 1991; U. S. Patent 5.164.402. :199.
- (92).Brighty KE, Castaldi MJ. Synlett. 1996;8:1097. [Google Scholar]
- (93).Vilsmaier E, Goerz T. Synthesis. 1998;29:739. [Google Scholar]
- (94).Barrett AGM, Seefeld MA. Tetrahedron. 1993;49:7857. [Google Scholar]
- (95).Tanguy C, Bertus P, Szymoniak J, Larionov OV, de Meijere A. Synlett. 2006;18:3164. [Google Scholar]
- (96).Adams LA, Charmant JPH, Cox RJ, Walter M, Whittingham WG. Org. Biomol. Chem. 2004;2:542. doi: 10.1039/b311322a. [DOI] [PubMed] [Google Scholar]
- (97).Bubert C, Reiser O. Tetrahedron Lett. 1997;38:4985. [Google Scholar]
- (98).Böhm C, Schinnerl M, Bubert C, Zabel M, Labahn T, Parisini E, Reiser O. Eur. J. Org. Chem. 2000;16:2955. [Google Scholar]
- (99).Hubner J, Liebscher J, Patzel M. Tetrahedron. 2002;58:10485. [Google Scholar]
- (100).Zhao Y, Yang T, Lee M, Lee D, Newton MG, Chu CK. J. Org. Chem. 1995;60:5236. [Google Scholar]
- (101).Zhao Z, Liu H. J. Org. Chem. 2002;67:2509. doi: 10.1021/jo010994r. [DOI] [PubMed] [Google Scholar]
- (102).Lee MG, Du JF, Chun MW, Chu CK. J. Org. Chem. 1997;62:1991. doi: 10.1021/jo961523l. [DOI] [PubMed] [Google Scholar]
- (103).Song Z, Lu T, Hsung RP, Al-Rashid ZF, Ko K, Tang Y. Angew. Chem. Int. Ed. 2007;46:4069. doi: 10.1002/anie.200700681. [DOI] [PubMed] [Google Scholar]
- (104).Yang ZQ, Lorenz JC, Shi Y. Tetrahedron Lett. 1998;39:8621. [Google Scholar]
- (105).Lorenz JC, Long J, Yang Z, Xue S, Xie Y, Shi Y. J. Org. Chem. 2004;69:327. doi: 10.1021/jo030312v. [DOI] [PubMed] [Google Scholar]
- (106).Charette AB, Beauchemin A. Org. React. 2001;58:1. [Google Scholar]
- (107).Charette AB. In: The Chemistry of Organozinc Compounds. Rappoport Z, Marek I, editors. John Wiley and Sons, Ltd.; West Sussex: 2006. p. 237. [Google Scholar]
- (108).Lait SM, Rankic DA, Keay BA. Chem. Rev. 2007;107:767. doi: 10.1021/cr050065q. [DOI] [PubMed] [Google Scholar]
- (109).Blaser H-U. Chem. Rev. 1992;92:935. [Google Scholar]
- (110).Ager DJ, Prakash I, Schaad DR. Chem. Rev. 1996;96:835. doi: 10.1021/cr9500038. [DOI] [PubMed] [Google Scholar]
- (111).Cho BT, Kim N. Synth. Commun. 1995;25:167. [Google Scholar]
- (112).Eilers J, Wilken J, Martens J. Tetrahedron: Asymmetry. 1996;7:2343. [Google Scholar]
- (113).Arend M, Westermann B, Risch N. Angew. Chem., Int.Ed. Engl. 1998;37:1044. doi: 10.1002/(SICI)1521-3773(19980504)37:8<1044::AID-ANIE1044>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- (114).Martens J, Kossenjans M. Tetrahedron: Asymmetry. 1999;10:3409. [Google Scholar]
- (115).Laumer JM, Kim DK, Beak P. J. Org. Chem. 2002;67:6797. doi: 10.1021/jo0258251. [DOI] [PubMed] [Google Scholar]
- (116).Liu C, Hashimoto Y, Saigo K. Tetrahedron Lett. 1996;37:6177. [Google Scholar]
- (117).Cherkauskas JP, Klos AM, Borzilleri RM, Sisko J, Weinreb SM, Parvez M. Tetrahedron. 1996;52:3135. [Google Scholar]
- (118).Carlier PR, Moon-Lo K, Lo MMC, Williams ID. J. Org. Chem. 1995;60:7511. [Google Scholar]
- (119).Carlier PR, Lo CWS, Lo MMC, Wan NC, Williams ID. Org. Lett. 2000;2:2443. doi: 10.1021/ol006099w. [DOI] [PubMed] [Google Scholar]
- (120).Gotor V, Dehli JR, Rebolledo F. J. Chem. Soc., Perkin Trans. 1. 2000;3:307. [Google Scholar]
- (121).Nino AD, Dalpozzo R, Cupone G, Maiuolo L, Procopio A, Tagarelli A, Bartoli G. Eur. J. Org. Chem. 2002;17:2924. doi: 10.1021/jo0260387. [DOI] [PubMed] [Google Scholar]
- (122).Bartoli G, Bosco M, Dalpozzo R, Marcantoni E, Massaccesi M, Rinaldia S, Sambria L. Tetrahedron Lett. 2001;42:8811. [Google Scholar]
- (123).Carlier PR, Lam WWF, Wan NC, Williams ID. Angew. Chem. Int. Ed. 1998;37:2252. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2252::AID-ANIE2252>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- (124).Crabtree RH, Davis MW. J. Org. Chem. 1986;51:2655. [Google Scholar]
- (125).Osborn JA, Jardine FH, Young JF, Wilkinson GJ. Chem. Soc. A. 1966;12:1711. [Google Scholar]
- (126).Shaw AM, Boules M, Zhang Y, Williams K, Robinson J, Carlier PR, Richelson E. Eur. J. Pharmacol. 2007;555:30. doi: 10.1016/j.ejphar.2006.10.004. [DOI] [PubMed] [Google Scholar]
- (127).Liang Y, Shaw AM, Boules M, Briody S, Robinson J, Oliveros A, Blazar E, Williams K, Zhang Y, Carlier PR, Richelson E. J. Pharmacol. Exp. Ther. 2008;327:573. doi: 10.1124/jpet.108.143610. [DOI] [PubMed] [Google Scholar]
- (128).Carlier PR, Lo MMC, Lo PCK, Richelson E, Tatsumi M, Reynolds IJ, Sharma TA. Bioorg. Med. Chem. Lett. 1998;8:487. doi: 10.1016/s0960-894x(98)00062-6. [DOI] [PubMed] [Google Scholar]
- (129).Richelson E, Carlier PR. PCT Int. Appl. 2003;45 WO 2003007929. [Google Scholar]
- (130).Richelson E, Fauq AH. PCT Int. Appl. 2009;107 WO 2009089479. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.