

Abstract
An in vitro assay for nucleocytoplasmic transport was established in which signal-dependent protein import is reproduced faithfully by isolated purified nuclei. The assay permits the precise quantification of import kinetics and the discrimination between translocation through the nuclear envelope and intranuclear transport. Nuclei were manually isolated from Xenopus oocytes and after manual purification incubated with a medium containing a green fluorescent transport substrate, karyopherins α2 and β1, a red fluorescent control substrate, an energy mix and, for keeping an osmotic balance, 20% (wt/vol) BSA. Import of transport substrates into the nucleus and exclusion of the control substrate were monitored simultaneously by two-color confocal microscopy. Two widely differing import substrates were used: the recombinant protein P4K [480 kDa, four nuclear localization sequences (NLSs) per P4K tetramer], and NLS-BSA (90 kDa, 15 NLSs). The measurements suggested that import, at the specific conditions used in this study, consisted of two consecutive processes: (i) the rapid equilibration of the concentration difference across the nuclear envelope, a process involving binding and translocation of substrate by the nuclear pore complex , and (ii) the dissipation of the intranuclear concentration difference by diffusion.
Both the strict separation of the nuclear contents from the cytoplasm by the nuclear envelope and the tightly regulated exchange of selected molecules between nucleus and cytoplasm via the nuclear pore complex (NPC) are of vital importance for the eukaryotic cell. It is crucial, for instance, that only fully matured ribonucleoproteins are released from the nucleus into the cytoplasm. Likewise, the selective import of proteins into the nucleus is essential for the regulation of transcription, processing, replication, and DNA repair. At the center of nucleocytoplasmic transport is the NPC, a large molecular machine spanning the nuclear envelope. It has become apparent in recent years that the NPC is a transporter with very special properties (for review, see refs. 1–6). On one hand, it has a passive permeability permitting molecules up to approximately 10 nm in diameter to pass between cytoplasm and nuclear contents by diffusion (7, 8). On the other hand, the NPC translocates molecules larger than approximately 10 nm only if the molecules contain either a nuclear localization signal (NLS; ref. 9) or a nuclear export signal (NES; refs. 10, 11). It appears that each NPC can operate in all three modes. However, whether passive transport, signal-dependent import, and signal-dependent export can proceed simultaneously, whether they are sequential processes, and how translocation occurs on a molecular basis have remained obscure so far.
Independent of its profound physiological and pathophysiological implications, the field of nucleocytoplasmic transport provides fascinating examples for the impact of methodological developments on scientific progress. Thus, methods for the mass isolation of highly purified cell nuclei by cell homogenization and density centrifugation were developed (12) when the field was in an early stage. Such nuclei made possible many biochemical and physical studies but have not yet been used successfully in transport studies. In fact, up to the present day, application of any biochemical preparation of isolated nuclei in transport studies has remained elusive, although many attempts have been made. Therefore, microinjection of intact living cells was the method of choice for a long time. In microinjection studies, very large cells such as amphibian oocytes played an important role (13–18), because microinjection could be relatively well quantified and combined with microdissection methods, biochemical analysis, and electron microscopy. In contrast, manually isolated nuclei of Xenopus oocytes were considered to be unsuited for transport studies. Paine et al. (19) found that nuclei isolated in buffer rapidly lost the large majority of nuclear proteins. As a remedy, Paine et al. (20) designed a method for isolating Xenopus oocyte nuclei under oil. Transport studies were performed by pairing an isolated nucleus with an aqueous agarose bead or a bolus of oocyte cytoplasm under oil (21). Otherwise, isolated Xenopus oocyte nuclei have, to the best of our knowledge, been used only by Clapham and coworkers (22) to show on a purely qualitative basis that a 10-kDa dextran, which normally enters the nucleus by passive diffusion through the NPC, is excluded from the nucleus when the perinuclear space is depleted of calcium ions. However, the introduction of semiintact cells in which the plasma membrane, but not the nuclear envelope, was made permeable for macromolecules by treatment with the detergent digitonin was a methodological breakthrough (23). It became immediately apparent (23) that soluble factors are required for nucleocytoplasmic transport, in addition to NLS-containing substrates and NPC. This observation has led to the discovery and characterization of the large family of transport factors, referred to as karyopherins or importins/exportins/transportins (24–26), as well as to the role of the small GTPase Ras-related nuclear protein (Ran) and its regulatory proteins (27, 28). Although digitonin permeabilized cells are currently used most frequently in nucleocytoplasmic transport studies, a need exists for methods giving better control over the medium bathing the nuclear faces of the NPC and providing more quantitative data. A decisive step in this direction was the development of optical single-transporter recording (29, 30) which has been used recently to measure transport through single NPCs (31, 32).
In the present study, an in vitro system for nucleocytoplasmic transport was established in which signal-dependent protein import is reproduced faithfully by isolated purified nuclei. This method, which is convenient, fast, and accurate, opens possibilities for the quantitative analysis of nucleocytoplasmic transport and closes the gap between methods using semiintact cells (23) or isolated nuclear envelopes (32). The method was used here to analyze basic kinetic parameters of signal-dependent protein import.
Materials and Methods
Materials.
Texas red-labeled 70-kDa dextran (TRD70) was obtained from Molecular Probes. NLS–BSA was a gift of Achim Dickmanns, Max-Planck-Institut für Biophysikalische Chemie, Göttingen, Germany and was generated by chemically coupling the peptide CGGGPKKKRKVED to BSA according to standard procedures (33). NLS–BSA had a mean molecular mass of approximately 90 kDa, thus containing approximately 15 signal peptides per BSA molecule on average. It was labeled with fluorescein isothiocyanate. The recombinant protein P4K was prepared as described (34) and conjugated with the dye Alexa-488 (Molecular Probes), according to the procedure recommended by the manufacturer. Karyopherins α2 and β1 were prepared as described (35). All manipulations of the nucleus and the transport measurements were performed in a mock intracellular medium containing 90 mM KCl, 10 mM NaCl, 2 mM MgCl2, 0.1 mM CaCl2, 1.0 mM of the calcium chelator N-(2-hydroxyethyl)ethylene-diaminetriacetic acid and 10 mM Hepes (pH 7.3). Because this mock intracellular medium was designed to have a concentration of free Ca2+ of 3.0 μM, it is referred to as mock 3 in this paper. The transport media were made up of mock 3 containing either 0.5 μM of NLS-BSA or 0.5 μM of Alexa-488-P4K and always 2.0 μM TRD70, 0.5 μM karyopherin α2, 0.5 μM karyopherin β1, an energy mix (2mM ATP/25 mM phosphocreatine/30 units/ml creatine phosphokinase/200 μM GTP), and 20% (wt/vol) BSA. The BSA added to the transport media had been dialyzed overnight against mock 3. The ATPase apyrase was obtained from Sigma and used at a final concentration of 0.4 units/ml.
Isolation and Purification of Oocyte Nuclei.
Stage VI oocytes were prepared from Xenopus laevis as described (31) and kept for further use in amphibian Ringer's solution containing 88 mM NaCl, 1 mM KCl, 0.8 mM MgSO4, 1.4 mM CaCl2, and 5 mM Hepes (pH 7.4), as well as 10.000 units/ml penicillin and 100 mg/liter streptomycin. For the preparation of a nucleus, an oocyte was transferred from amphibian Ringer's solution into mock 3. Under a stereomicroscope, the oocyte was gripped at the boundary between animal and vegetal pole by two pairs of sharp forceps. By slight tension, the oocyte was opened and the nucleus was set free. For further purification, the isolated nucleus was transferred into a dish containing 4 ml of fresh mock 3 and gently touched repeatedly with the side of the tip of a microcapillary. Thus, residual yolk and other adhering material were removed without damaging the nuclear envelope. All manipulations, as well as microscopic measurements, were performed at a room temperature of approximately 22°C.
Transport Measurements and Data Evaluation.
A microchamber was prepared by drilling a hole of 2.5 mm in diameter into the bottom of a cell-culture dish and by gluing a coverslip to the bottom of the dish. The microchamber had a depth of 1.0 mm and a volume of 5 μl. It was filled with mock 3. By using a microliter pipette, a purified nucleus was deposited at the bottom of the microchamber, carefully avoiding contact with the air–water interface. The medium in the microchamber was exchanged with transport medium by using a micropipette equipped with a very fine plastic tip (geloader tips, Eppendorf). By bringing the pipette tip close to the nucleus in the microchamber, 15 μl of transport medium was deposited on and around the nucleus so that the mock 3 was completely displaced and, together with the surplus of transport medium, spilled over the edge of the microchamber. The surplus of mock 3 and transport medium was removed and either the dish was closed with its lid (when using an inverted microscope) or the microchamber was closed with a coverslip (when using an upright microscope).
A microchamber containing a nucleus and loaded with transport medium was placed on the stage of a confocal laser scanning microscope. Either an inverted or an upright microscopic setup was used, both of the TCS line of Leica (Heidelberg, Germany). Alternating measurements of the same sample with both microscopes yielded identical results. The nucleus was imaged with a low-power objective (HC PL Fluotar, ×10, 0.30 NA) yielding a scanning field of 1,000 μm × 1,000 μm with the confocal aperture set to 1.5 optical units. By determination of the point-spread function and by three-dimensional reconstruction, it was found that the thickness of the optical section amounted to 3.0 μm, and that contribution of object regions, other than the optical section, were negligible. Fluorescence was excited by the 488- and 568-nm lines of argon/krypton lasers. The fluorescence of the Alexa-488-labeled transport substrate was monitored in channel 1 by using a 515- to 540-nm bandpass filter, and the fluorescence of the Texas red-labeled control substrate was monitored in channel 2 by using a 590-nm bandpass filter. An overspill of fluorescence from channels 1 to 2 was kept negligibly small by using relatively high concentrations of the control substrate and a small voltage of the photomultiplier of channel 2. At these conditions, the largest perimeter of the nucleus was brought into focus and the first scan obtained 1 or 2 min after addition of the transport medium to the nucleus. Further scans were acquired at 5, 10, 15, 20, and 30 min after transport substrate addition.
The software scion image (Scion, Frederick, MD) was used to derive six parameters as a function of time from the confocal scans: Fsi, the mean intranuclear fluorescence intensity of the transport substrate; Fci, the mean intranuclear fluorescence intensity of the control substrate; F, the mean fluorescence intensity of the substrate in the nuclear rim; Fse, the mean extranuclear fluorescence intensity of the transport substrate; and Fce, the mean extranuclear concentration of the control substrate, and the radius rn of the nucleus. From these parameters, the number Ni(t) of imported substrate molecules was calculated according to:
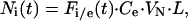 |
1 |
where Fi/e is
Fi/Fe;
Ce is the molar concentration of
substrate in the extranuclear space, i.e., in the transport medium;
VN is the volume of the nucleus equal
to 4/3πr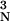 where L is
Avogadro's number. Ni(t)
was fitted by the equation for the diffusion into a sphere in which the
initial concentration is zero and the external concentration is
constant (equation 8 of chapter 9.3 in ref. 36):
where L is
Avogadro's number. Ni(t)
was fitted by the equation for the diffusion into a sphere in which the
initial concentration is zero and the external concentration is
constant (equation 8 of chapter 9.3 in ref. 36):
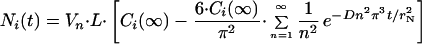 |
2 |
Ci(∞) is the intranuclear substrate concentration at long time amounting to approximately 0.7 Ce for P4K and NLS–BSA. D is the translational diffusion coefficient of the substrate inside the nucleus.
Results and Discussion
Preparation of Xenopus Oocyte Nuclei for Transport Measurements.
The steps of the specimen preparation are illustrated in Fig. 1. In the mock 3 buffer, a stage VI Xenopus oocyte (Fig. 1a) was opened, and the nucleus was freed gently from most of the yolk (Fig. 1b). By repeatedly touching the nucleus with the side of a fine-glass needle, residual yolk and other adhering material were removed. The purified nucleus was transferred into a 5-μl microchamber (Fig. 1c), and the mock 3 buffer was exchanged with a transport medium containing one of the transport substrates together with the control substrate Texas red-labeled dextran of 70 kDa, transport factors, an energy mix and, for osmotic balancing, BSA, as specified in Materials and Methods. The procedure was completed within 5 to 10 min, a relatively short time, thus reducing a loss of intranuclear components required for signal-dependent transport. An abundance of transport factors in the nuclear contents may explain why the exogenous addition of Ran to the import mix was not required in these experiments, although the process is Ran-dependent (27). Western blot analysis of isolated Xenopus oocyte nuclei demonstrated an abundance of Ran (data not shown).
Figure 1.

Preparation of an isolated Xenopus oocyte nucleus for transport measurement. The nucleus of a stage VI Xenopus oocyte (a) was isolated manually (b) and, after manual purification with a fine-glass needle, deposited in a microchamber (c). The microchamber (d) had a volume of 5 μl. Transport was monitored by confocal scans by using either an inverted or an upright setup.
Measurement of Import Kinetics.
A microchamber containing a nucleus and loaded with transport medium was placed on the stage of a confocal microscope. At low magnification (×10, NA 0.3 objective lens), the focus was adjusted to the largest perimeter of the nucleus, and a first two-color scan was made to simultaneously monitor the green fluorescence of the transport substrate (channel 1) and the red fluorescence of the control substrate (channel 2). Further scans were acquired at time points suiting the transport kinetics, i.e., at 2, 5, 10, 15, 20, and 30 min after addition of the transport medium. The thickness of the optical section was determined to be approximately 3.0 μm. Thus, the scans represented a thin slice through the center of the nuclei and were essentially free of fluorescence contributions from out-of-focus regions of the object.
A typical import experiment involving the recombinant NLS protein P4K is shown in Fig. 2. Already in the first scan, which was acquired 2 min after exchanging mock 3 buffer by transport medium, an accumulation of P4K at the nuclear periphery was observed. By this rim staining, the nuclear periphery could be recognized clearly. With time, the rim staining increased while P4K appeared simultaneously inside the nucleus. However, in early phases, a steep concentration gradient of P4K existed between the periphery and the center of the nucleus. The concentration gradient leveled out with time, so that after an import time of 30 min, the intranuclear P4K concentration became relatively homogeneous. Notably, the P4K concentration always remained smaller inside the nucleus than in the external medium. The control substrate (Fig. 2 Right) was excluded from the nucleus, indicating that the NPC and nuclear membranes were intact.
Figure 2.

Example of a typical import measurement. At zero time, a microchamber containing an isolated and purified nucleus was loaded with a transport medium containing the green fluorescent NLS protein P4K (480 kDa) as transport substrate, the red fluorescent control substrate Texas red-labeled dextran of 70 kDa (TRD70) as well as karyopherins α2 and β1, an energy mix, and, for osmotic balancing, BSA. It can be seen that the transport substrate rapidly accumulated at the nuclear periphery. At initial stages of import, a concentration gradient between nuclear periphery and center existed that dissipated within approximately 30 min. The control substrate was excluded from the nucleus, indicating that NPC and nuclear membranes had remained intact.
Data Evaluation.
Confocal scans such as those shown in Fig. 2 were evaluated by image analysis. Thus, from scans by which the import of P4K was monitored (Fig. 2 Left), intensity profiles were obtained along a straight line through the center of the nucleus. The profiles (Fig. 3a) substantiate the qualitative conclusions made by inspection of the scans. Furthermore, they suggest that the fluorescence of P4K increased very rapidly at the inner side of the nuclear envelope. It appears that even before acquisition of the first scan (transport time 2 min), the fluorescence intensity at this location reached a level corresponding to approximately 70% of the extranuclear P4K fluorescence. With increasing transport time, the fluorescence intensity seemed to remain approximately constant at the inner side of the nuclear envelope but increased continuously in the nuclear interior, approaching a constant level.
Figure 3.

Quantitative evaluation of an import experiment. (a) From the confocal scans monitoring the import of P4K (Fig. 2 Left), intensity profiles were obtained along a straight line through the center of the nucleus. P4K fluorescence in the extranuclear space (e.s.), the rim, and the intranuclear space can be discriminated clearly. It appears that at the intranuclear side of the nuclear rim, the concentration of P4K rapidly rose to reach, perhaps before the first scan, a constant value amounting to approximately 70% of the extranuclear fluorescence. With time, P4K seemed to fill the intranuclear space from the rim. The intranuclear concentration changes can be described by diffusion into a sphere with zero initial concentration and constant external concentration. (b) The confocal scans monitoring the import of P4K (Fig. 2 Left) were further evaluated for five parameters: (Top to Bottom) the mean fluorescence intensities of the control substrate in the extranuclear medium (open triangles), the mean concentration of the transport substrate in the nuclear rim (filled squares), the mean concentration of the transport substrate in the extranuclear medium (open squares), the mean concentration of the transport substrate in the intranuclear spaces (open circles), and the mean fluorescence intensity of the control substrate in the intranuclear space (open diamonds). In addition, the nuclear radius was measured (not shown) and was found to remain virtually constant over the 30-min time period.
The confocal scans of Fig. 2 were further evaluated for the time dependence of six parameters (Fig. 3b): the mean fluorescence intensities of the transport substrate and the control substrate in both the intranuclear and the extranuclear space, the mean fluorescence intensity of the substrate in the nuclear rim, and the radius of the nucleus. As expected, four of the parameters, including the nuclear radius (not shown in Fig. 3b), remained fairly constant over the measuring time. Only the mean fluorescence of the transport substrate increased continuously in both the nuclear rim and intranuclear space.
The data of Fig. 3, which are typical for all of the data obtained with P4K, suggest that the import of P4K involves two steps: (i) At the nuclear rim, P4K accumulated rapidly by a process that may involve binding and translocation of P4K by the NPC. The effect of the binding/translocation processes seems to be that the concentration difference, which initially existed between the extra- and intranuclear side of the nuclear envelope, is equilibrated almost instantaneously. That the intranuclear concentration of P4K reached only 70% of its extranuclear concentration may simply be because the proportion of solvent water available for P4K is smaller in the intra- than in the extranuclear space, i.e., the space taken by chromatin and other nuclear constituents may be only partially accessible to molecules as large as P4K. (ii) The concentration difference between the inside of the nuclear envelope and the center of the nucleus seems to be equilibrated by simple diffusion. The diffusion coefficient of P4K in water is 33 μm2/s (34), whereas in the Xenopus oocyte nucleus, the diffusion coefficients of macromolecules are reduced by a factor of approximately 4 (7). Thus, we estimate the diffusion coefficient of P4K in the intranuclear space to be approximately 8 μm2/s. This figure agrees with the measured kinetics, as discussed in the next paragraph.
Import Kinetics of Two NLS-Containing Proteins.
By using the methods described above, we have measured the import kinetics of two proteins. One was P4K (34), a recombinant protein we have used previously. P4K is a fusion of a segment (amino acids 111–135) of the simian virus 40 large T antigen containing the NLS with Escherichia coli β-galactosidase. The protein forms stable tetramers and thus has four NLSs and a mass of 480 kDa. The other was NLS–BSA, a frequently used import substrate generated by chemically coupling an NLS-containing peptide to BSA. The conjugate had a mean molecular mass of 90 kDa and contained 15 NLSs on average.
Results pertaining to P4K are collected in Fig. 4. Fi/e values, i.e., the ratio of the intra- and extranuclear fluorescence intensities, are given in Fig. 4a for several experiments. In the presence of karyopherins α2 and β1 (Fig. 4a, open circles), Fi/e increased steadily with time, approaching a plateau of approximately 0.6 after 30 min. In the absence of karyopherins α2 and β1 but in the presence of the energy mix (Fig. 4a, open squares), there was only a small increase of Fi/e within 30 min, amounting to approximately 10% of the increase in the presence of karyopherins α2 and β1. When the energy mix was omitted and replaced by the potent ATPase apyrase, import was also very weak, even though it was in the presence of karyopherins α2 and β1 (individual measurements are not shown, but mean values of imported molecules are given in Fig. 4b).
Figure 4.

Import kinetics of P4K. (a) For a number of individual measurements, the ratio Fi/e of intra- over extranuclear fluorescence was plotted vs. time. In the presence of karyopherins α2 and β1 and an energy mix (open circles), P4K was imported, approaching a plateau at 30 min that corresponded to approximately 60–70% of the extranuclear fluorescence intensity. In absence of karyopherins α2 and β1 but presence of the energy mix (open squares), import was negligibly small. It may be noted that part of the scattering of the data is caused by variation in nuclear volume. (b) From the data shown in a, the number of imported molecules was derived by taking the nuclear volume into account according to Eq. 1. The experimental data (symbols and bars) represent mean ± SD. The data for the import of P4K in the presence of karyopherins α2 and β1 (open circles) were fitted by Eq. 2 (full line), which holds for diffusion into a sphere with zero initial concentration and fixed external concentration. The fit yielded a diffusion coefficient of approximately 9 μm2/s. In the absence of karyopherins α2 and β1 (but in the presence of the energy mix, open squares), import was strongly reduced. Depletion of nuclei from metabolic energy by addition of the ATPase apyrase also strongly reduced import (open triangles), even in the presence of karyopherins α2 and β1.
The mean number of P4K molecules imported per nucleus was calculated according to Eq. 1 (Fig. 4b, symbols and bars representing mean ± SD). The data for the import of P4K in the presence of karyopherins α2 and β1 (Fig. 4b, open circles) were fitted (Fig. 4b, full line) by Eq. 2, which holds for diffusion into a sphere with zero initial concentration and constant external concentration. The fit yielded a diffusion coefficient of 9 μm2/s, which agrees with the expectation as stated above. When karyopherins α2 and β1 were omitted (Fig. 4b, open squares), import was strongly reduced, even though it was in the presence of the energy mix. Addition of apyrase and the omission of the energy mix (Fig. 4b, open triangles) also strongly reduced import, even in the presence of karyopherins α2 and β1.
The data for NLS–BSA (not shown) were remarkably similar to the data for P4K. It seemed that the concentration of NLS–BSA at the internal side of the nuclear envelope almost instantaneously reached a value of approximately 70% of the extranuclear concentration, that this value remained constant, and that the intranuclear concentration equilibrated by diffusion with time. Effective import of NLS–BSA required karyopherins α2 and β1 and metabolic energy.
Paine and coworkers (21), by using Xenopus oocyte nuclei isolated under oil, characterized the import of the karyophilic protein nucleoplasmin as facilitated transport through the nuclear envelope followed by intranuclear binding. Our results are consistent with the first mechanism, facilitated transport. However, we do not see intranuclear binding. Future studies may address the questions of whether and how facilitated transport can be reconciled with what is already known about the components and mechanisms of signal-dependent protein import. It may be particularly instructive to explore whether an accumulation of the transport substrate in the nucleus over its extranuclear concentration can be induced, e.g., by generating high intranuclear Ran–GTP concentrations.
Distinctive Properties of the Method.
In recent years, semiintact, digitonin-permeabilized cultured mammalian cells (23) have been used in nucleocytoplasmic transport studies. In comparison, the method described here uses isolated and purified nuclei of primary, perfectly synchronized cells. No detergents are involved. A comparison of nucleocytoplasmic transport in semiintact cells and isolated nuclei may provide important clues for the involvement of cytoplasmic structures in nucleocytoplasmic transport.
The large size of the oocyte nuclei has many favorable consequences for transport studies: (i) The preparation of nuclei is convenient and fast. (ii) Microscopic measurements can be performed at low magnification, thereby greatly enhancing mechanical stability. Thus, during a measuring time of 30 min, refocusing was usually not necessary, which suggests that the measuring process can be automated easily and even extended to parallel measurements on several nuclei to speed up data collection. (iii) The large working distance of low-power objective lenses permits the use of either inverted or upright microscopic setups. (iv) Transient intranuclear concentration gradients of an imported substrate can be discerned easily, which makes it possible to differentiate between processes occurring at the NPC and in the nuclear interior. (v) The method can be combined with biochemical methods such as gel electrophoresis and biophysical methods such as fluorescence microphotolysis, fluorescence correlation spectroscopy, or single-particle tracking methods. (vi) The method can be extended to export measurements by microinjection. An additional benefit of the method is that the Xenopus oocyte NPC has been the favorite object of structural and functional studies in the past and is therefore better characterized than the NPC of any other cell type.
Acknowledgments
We thank Dr. Achim Dickmanns, Max-Planck-Institut für Biophysikalische Chemie, Göttingen, Germany, for providing us with NLS–BSA. Financial support by Deutsche Forschungsgemeinschaft Grant Pe138/17–1 is acknowledged gratefully.
Abbreviations
- NES
nuclear export signal
- NLS
nuclear localization signal
- NPC
nuclear pore complex
- Ran
Ras-related nuclear protein
References
- 1.Mattaj I J, Englmeier L. Annu Rev Biochem. 1998;67:265–306. doi: 10.1146/annurev.biochem.67.1.265. [DOI] [PubMed] [Google Scholar]
- 2.Doye V, Hurt E. Curr Opin Cell Biol. 1997;9:401–411. doi: 10.1016/s0955-0674(97)80014-2. [DOI] [PubMed] [Google Scholar]
- 3.Daneholt B. Cell. 1997;88:585–588. doi: 10.1016/s0092-8674(00)81900-5. [DOI] [PubMed] [Google Scholar]
- 4.Pemberton L F, Blobel G, Rosenblum J S. Curr Opin Cell Biol. 1998;10:392–399. doi: 10.1016/s0955-0674(98)80016-1. [DOI] [PubMed] [Google Scholar]
- 5.Görlich D, Kutay U. Annu Rev Cell Dev Biol. 1999;15:607–660. doi: 10.1146/annurev.cellbio.15.1.607. [DOI] [PubMed] [Google Scholar]
- 6.Blobel G, Wozniak R W. Nature (London) 2000;403:835–836. doi: 10.1038/35002687. [DOI] [PubMed] [Google Scholar]
- 7.Paine P L, Moore L C, Horowitz S B. Nature (London) 1975;254:109–114. doi: 10.1038/254109a0. [DOI] [PubMed] [Google Scholar]
- 8.Peters R. EMBO J. 1984;3:1831–1836. doi: 10.1002/j.1460-2075.1984.tb02055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalderon D, Richardson W D, Markham A F, Smith A E. Nature (London) 1984;311:33–38. doi: 10.1038/311033a0. [DOI] [PubMed] [Google Scholar]
- 10.Wen W, Meinkoth J L, Tsien R Y, Taylor S S. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- 11.Fischer U, Huber J, Boelens W C, Mattaj I W, Lührmann R. Cell. 1995;82:475–483. doi: 10.1016/0092-8674(95)90436-0. [DOI] [PubMed] [Google Scholar]
- 12.Blobel G, Potter V R. Science. 1966;154:1662–1665. doi: 10.1126/science.154.3757.1662. [DOI] [PubMed] [Google Scholar]
- 13.Feldherr C M. J Cell Biol. 1962;14:65–72. doi: 10.1083/jcb.14.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gurdon J B. Proc R Soc London Ser B. 1970;176:303–314. doi: 10.1098/rspb.1970.0050. [DOI] [PubMed] [Google Scholar]
- 15.Bonner W M. J Cell Biol. 1975;64:431–437. doi: 10.1083/jcb.64.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dingwall C, Sharnik S V, Laskey R A. Cell. 1982;30:449–458. doi: 10.1016/0092-8674(82)90242-2. [DOI] [PubMed] [Google Scholar]
- 17.Zasloff M, Rosenberg M, Santos T. Nature (London) 1982;300:81–84. doi: 10.1038/300081a0. [DOI] [PubMed] [Google Scholar]
- 18.Feldherr C M, Kallenbach E, Schultz N. J Cell Biol. 1984;99:2216–2222. doi: 10.1083/jcb.99.6.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paine P L, Austerberry C F, Desjarlais L J, Horowitz S B. J Cell Biol. 1983;97:1240–1242. doi: 10.1083/jcb.97.4.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paine P L, Johnson M E, Lau Y-T, Tluczek J M, Miller D. BioTechniques. 1992;13:238–246. [PubMed] [Google Scholar]
- 21.Vancurowa I, Lou W, Paine T M, Paine P L. Eur J Cell Biol. 1993;62:22–33. [PubMed] [Google Scholar]
- 22.Stehno-Bittel L, Perez-Terciz C, Clapham D E. Science. 1995;270:1835–1838. doi: 10.1126/science.270.5243.1835. [DOI] [PubMed] [Google Scholar]
- 23.Adam S, Stern-Marr G, Gerace L. J Cell Biol. 1990;111:807–816. doi: 10.1083/jcb.111.3.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adam S A, Gerace L. Cell. 1991;66:837–847. doi: 10.1016/0092-8674(91)90431-w. [DOI] [PubMed] [Google Scholar]
- 25.Adam E J H, Adam S A. J Cell Biol. 1994;125:547–555. doi: 10.1083/jcb.125.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moroianu J, Blobel G, Radu A. Proc Natl Acad Sci USA. 1995;92:2008–2011. doi: 10.1073/pnas.92.6.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore M S, Blobel G. Nature (London) 1993;365:661–663. doi: 10.1038/365661a0. [DOI] [PubMed] [Google Scholar]
- 28.Avis J M, Clarke P R. J Cell Sci. 1996;109:2423–2427. doi: 10.1242/jcs.109.10.2423. [DOI] [PubMed] [Google Scholar]
- 29.Peters R, Sauer H, Tschopp J, Fritzsch G. EMBO J. 1990;9:2447–2451. doi: 10.1002/j.1460-2075.1990.tb07421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tschödrich-Rotter M, Peters R. J Microsc (Oxford) 1998;192:114–125. doi: 10.1046/j.1365-2818.1998.00411.x. [DOI] [PubMed] [Google Scholar]
- 31.Keminer O, Peters R. Biophys J. 1999;77:217–228. doi: 10.1016/S0006-3495(99)76883-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keminer O, Siebrasse J-P, Zerf K, Peters R. Proc Natl Acad USA. 1999;96:11842–11847. doi: 10.1073/pnas.96.21.11842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marshallsay C. Mol Biol Rep. 1995;20:163–171. doi: 10.1007/BF00990549. [DOI] [PubMed] [Google Scholar]
- 34.Rihs H-P, Peters R. EMBO J. 1989;8:1479–1484. doi: 10.1002/j.1460-2075.1989.tb03531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moroianu J, Blobel G, Radu A. Proc Natl Acad Sci USA. 1995;92:2008–2011. doi: 10.1073/pnas.92.6.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carslaw H S, Jaeger J C. Conduction of Heat in Solids. Oxford, U.K.: Oxford Univ. Press; 1959. [Google Scholar]
- 37.Maul G G. Int Rev Cytol. 1977;Suppl. 6:S75–S186. [PubMed] [Google Scholar]