

Abstract
The mitogen-activated protein kinase-activated protein kinase MK5 is ubiquitously expressed in vertebrates and is implicated in cell proliferation, cytoskeletal remodeling, and anxiety behavior. This makes MK5 an attractive drug target. We tested several diterpenoid alkaloids for their ability to suppress MK5 kinase activity. We identified noroxoaconitine as an ATP competitor that inhibited the catalytic activity of MK5 in vitro (IC50 = 37.5 μM; K i = 0.675 μM) and prevented PKA-induced nuclear export of MK5, a process that depends on kinase active MK5. MK5 is closely related to MK2 and MK3, and noroxoaconitine inhibited MK3- and MK5- but not MK2-mediated phosphorylation of the common substrate Hsp27. Molecular docking of noroxoaconitine into the ATP binding sites indicated that noroxoaconitine binds more strongly to MK5 than to MK3. Noroxoaconitine and derivatives may help in elucidating the precise biological functions of MK5 and may prove to have therapeutic values.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-010-0452-1) contains supplementary material, which is available to authorized users.
Keywords: Mitogen-activated protein kinase-activated protein kinase MK5, Kinase activity, Diterpenoid alkaloid, Noroxoaconitine, MK5, MK3, ATP binding site, Molecular docking
Introduction
Mitogen-activated protein kinase (MAPK) signal transduction pathways are activated by many different stimuli, and they regulate diverse processes ranging from proliferation and differentiation to apoptosis. To date, seven distinct MAPKs pathways have been discovered in mammalian cells, including: extracellular signal-regulated kinases (ERK1 and ERK2); c-Jun N-terminal kinases (JNK1, JNK2 and JNK3); p38MAPK (p38α, p38β, p38γ and p38δ); ERK3 and ERK4; ERK5; and ERK7 and ERK8 [1–9]. The typical MAPKs family consists of at least three-tiered cascades: upstream MAPK kinase kinase (MAP3K, MEKK or MKKK), a MAPK kinase (MAP2K, MEK or MKK), and a downstream MAPK. Each MAPK can phosphorylate a variety of substrates, including protein kinases referred to as MAPK-activated protein kinases (MAPKAPKs or MKs) [3, 5, 10–14]. The human MK family comprises 11 different members: 4 p90 ribosomal S6 kinases (RSK1-4), 2 mitogen- and stress-activated protein kinases (MSK1 and MSK2), 2 MAPK-interacting kinases (MNK1 and MNK2), and 3 MAPK-activated protein kinases (MK2, MK3 and MK5) [15]. According to their sequence, all MKs can be classified into five subgroups: RSKs, MSKs, MNKs, MK2 and MK3, and MK5 [2, 16–20].
MK5 or the human analogue p38-regulated/activated kinase, PRAK, was originally discovered as a sequence tag by the searching of sequences homologous to MK2 [21, 22]. Their catalytic domain displays 48% amino acid identity and their overall sequence identity is 42%. MK3, which possesses 75% overall identity with MK2, is 41% identical with MK5. All three proteins seem to be ubiquitously expressed [16, 23], but the expression level and activity of MK2 are significantly higher than MK3 in the cells and tissues examined [17]. The expression level and activity of MK2 and MK5 in different cells has not been compared. MK2, MK3, and MK5 exert different biological roles as is reflected by their substrate specificity and differences in phenotypes of knock-out mice [15, 24–26]. Nevertheless, they may participate in common functions such as F-actin remodeling (MK2 and MK5) and stimulation of tumor necrosis factor synthesis and p38MAPK stabilization (MK2 and MK3) [26]. Studies in MK5-deficient mouse models and cells have demonstrated that depletion promoted oncogenic transformation by c-Ha-RAS and facilitated dimethylbenzanthrance-induced skin carcinogenesis [24]. Furthermore, MK5 overexpression inhibited proliferation of NIH3T3 cells [27]. MK5 also seems to be involved in PKA-induced F-actin rearrangement and cell migration through Hsp27 phosphorylation, which is suppressed by 14-3-3ε [28–30]. Moreover, transgenic mice expressing constitutive active MK5 displayed anxiety-related traits and locomotor differences compared to wild-type littermates, suggesting the involvement of MK5 in anxiety behavior [31]. In spite of these described functions, the exact biological role of MK5 is still incompletely understood because MK5 knock-out mice do not display any obvious phenotype under normal conditions [24, 25].
The physiological function of protein kinases can be investigated by multiple approaches using targeted gene knock-out and knock-in animal models, RNA interference, overexpression of constitutive active or dominant negative mutants, and by the use of specific inhibitors (reviewed in [4, 32]). In recent years, there has been a globally increasing interest in herbal compounds because of their beneficial effect on human health, but also as potential specific protein kinase inhibitors [33, 34]. One group of them, the diterpenoid alkaloids (DAs), has been tested, but due to their toxic nature there are very few reports about their pharmacological effects. Tyrosinase inhibition and structure–activity relationship (SAR) of 15 DAs, with lycoctonine skeleton, and semisynthetic derivatives have been reported [35]. Some of the DAs were also reported to be active chemotherapeutic agents by inhibiting American trypanosomiasis [36]. To date, no specific MK5 inhibitors have been reported. This prompted us to test several DAs for their ability to suppress enzymatic activity of MK5. One of them, noroxoaconitine, specifically inhibited MK5 but not MK2 with an IC50 = 37.5 μM. This compound may therefore be a valuable tool in MK5 studies.
Materials and methods
Chemicals
The DAs were generously provided by Prof. M. Sultankhudzaev, Uzbek National Academy of Science, Tashkent, Uzbekistan. The compounds are HPLC grade pure and structures have been confirmed by different NMR techniques. The compounds were earlier cited elsewhere [37], and the structures are shown in Fig. 7 (see below, under “Docking of inhibitors at the ATP binding site of MK5”). Compounds 1–8 and 12–14 are natural alkaloids, while compounds 9–11 are semi-synthetic derivatives [37 and references therein]. Forskolin was purchased from Sigma-Aldrich (St. Louis, MO, USA), 32P γ-ATP was obtained from Perkin Elmer (Waltham, MA, USA), while recombinant MK2, MK3, and MK5 were from Invitrogen (Carlsbad, CA, USA). The expression plasmid encoding EGFP-MK5 fusion protein has been previously described [29]. Recombinant Hsp27 was purchased from Signal Chem (Richmond, BC, Canada). The MK5 substrate peptide PRAKtide (KKLRRTLSVA) was purchased from Millipore (Billerica, MA, USA).
Fig. 7.

2D molecular structures of the DAs screened against MK5
Biological studies
Screening and kinase activity
Screening of the DAs for inhibition of MK5 enzymatic activity was performed as follows. MK5 kinase activity was monitored against 30 μM PRAKtide substrate peptide at 30°C for 20 min in 50 μl of buffer containing 40 mM Tris–Cl, pH 7.5, 0.4 mg/ml BSA, 15 mM MgCl2, 0.1 mM cold ATP, and 1 μCi of [γ-32P]ATP (3,000 Ci/mmol), recombinant MK5 (45 ng per assay) in the presence or absence of DAs. The initial screening was done in the presence of 2 mM DA. Following incubation, 15-μl aliquots were spotted onto phosphocellulose disks (Millipore) and washed extensively with 1% phosphoric acid before measurement of radioactive incorporation by scintillation counting. Activity was determined as proposed by Mett et al. [38] where the average of counts in the presence of compounds were calculated, then negative controls (C−) (reaction in the absent of active MK5) were subtracted from these values and divided by the positive control (C+) (reaction in the present of active MK5, but without compounds) minus the negative control, resulting in kinase activity.
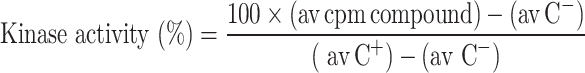 |
1 |
Percentage of inhibition was calculated from kinase activity as follows:
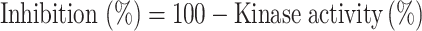 |
2 |
In vitro phosphorylation of Hsp27 by MK2, MK3, and MK5 was performed as previously described [28]. The pSer-78 Hsp27 specific antibodies were purchased from Millipore (cat. no. #04-447).
Kinetic studies
Enzyme inhibition reactions were performed to study the kinetic trends and ATP competitions of the active compounds (11 and 8). The biochemical method was similar to the screening described above. The difference was that multiple inhibitor (0.025, 0.125, 0.25, 1.25, and 2.5 mM) and ATP concentrations (1, 8, 16, and 32 mM) were used.
The kinetic data (V max, K m) were analyzed using Enzyme Kinetic™ module of SigmaPlot™ version 10, integrated with SigmaStat™. The K i values were determined from IC50 of the inhibitors values using the Cheng-Prusoff relationship [39–41].
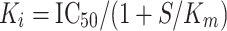 |
3 |
Cell culture and transfection
PC12 cells, a kind gift from Dr. Jaakko Saraste (University of Bergen, Norway), were maintained in F-12K Medium (Kaighn’s Modification of Ham’s F-12 Medium) supplemented with 2 mM l-glutamine, 1.5 g/l sodium bicarbonate, 15% Horse serum (Gibco) and 2.5% fetal calf serum, penicillin (110 U/ml) and streptomycin (100 μg/ml). Cells were transfected using the Nucleofection kit (Amaxa) according to the manufacturer’s instructions. The EGFP-MK5 expression plasmid has been previously described [42]. The plasmid that encodes the EGFP-NES fusion protein with the NES motif of the Rev protein of human immunodeficiency was constructed by cloning the complementary oligonucleotides 5′-CCG GAG ACG CTC TAC CAC CGC TTG AGA GAC TTA CTC TTG ACC GAG CT-3′ and 5′-CGG TCA AGA GTA AGT CTC TCA AGC GGT GGT AGA GCG TCT-3′ into the BspEI/SacI sites of pEGFP-C1 (Clontech, Mountain View, CA, USA).
Confocal microscopy
Cells were fixed for 10 min with 4% formaldehyde. Then cells were washed with PBS and examined using a confocal laser-scanning Zeiss LSM 510 META microscope. Several hundred cells were monitored and representative pictures are presented.
Computational biology
Homology modeling of MK5
The X-ray crystal structure of MK5 is not known. A model of MK5 was therefore constructed by homology with MK2 using the traditional homology modeling approach. The PDB co-ordinates of MK2 (chain A, 2OZA [43]) was obtained from the Protein Databank (PDB, http://www.pdb.org) and used as template for constructing the MK5 model based on the amino acid sequence of MK5 [22]. The model was built using the ICM™ Bioinformatics module. The ICM program constructs the molecular model by homology from core sections defined by the average of Cα atom positions in conserved regions.
Evaluations of model quality
The model quality check was performed using Structural Analysis and Verification Server (SAVS, http://nihserver.mbi.ucla.edu/SAVS/). PROCHECK [44, 45], WHAT_CHECK, ERRAT [46], VERIFY_3D [47, 48], and PROVE, were used to calculate quality factors of both the template (2OZA) and the generated 3D model of MK5. The total, local, burial and contact energies of the predicted models were calculated using predicted structure quality score (PSQS) (http://www1.jcsg.org/psqs) [49–52].
RMSD calculations
The 3D structures of 2OZA and the MK5 model were superimposed and their RMSD (root mean squared deviation) between backbone Cα atoms value was calculated utilizing DaliLite (http://www.ebi.ac.uk/DaliLite) [53] in order to compare their overall 3D structures and active site architecture.
Docking
Compounds 8 and 11 were docked into the MK5, while compound 11 was also docked into the X-ray structures of MK2 and MK3 [54]. The docking studies of the compounds were performed as previously described [55]. The Internal Coordinate Mechanics (ICM) program from Molsoft (http://www.molsoft.com/) was used for the docking studies.
Compounds 8 and 11 were docked into the predicted binding site of MK5, which corresponds to the ATP binding site of MK2 and MK3, while compound 11 was also docked into the ATP binding site of MK2 and MK3. The best docking conformations were selected based on docking energies, RMSD of ligand from initial ligand conformation, and similarities with ATP binding in known X-ray crystal structures (2PHK [56] and 2JBO [57]). The potent MK5 inhibitor 2-(2-quinolin-3-ylpyridin-4-yl)-1,5,6,7-tetrahydro-4H-pyrrolo[3,2-c]pyridine-4-one was also docked into the predicted binding site of the MK5 model and the X-ray structures of MK2 and MK3 using the approach as for compounds 8 and 11.
Results
Biochemical studies of DAs against MK5
Diterpenoid alkaloids were reported as moderate to potent inhibitors of the bifunctional copper containing enzyme tyrosinase [35]. This prompted us to test whether they could inhibit the activity of protein kinases. MK5, which belongs to the group of calcium/calmodulin-dependent protein kinases [58] was selected as a target. To ensure a clear potential inhibition, high concentrations (2 mM) were initially tested. The first screening of different natural and semisynthetic DAs (structures shown in Fig. 7, see below, under “Docking of inhibitors at the ATP binding site of MK5”) displayed inhibitory activity of all examined compounds (Fig. 1). Three of the compounds, aconitine (6), aconine (8) and noroxoaconitine (11) completely blocked MK5 enzyme activity at the tested concentration.
Fig. 1.

Initial screening of some diterpenoid alkaloids (DAs) for their inhibitory activity towards MK5. The percent inhibition of MK5 kinase activity by compounds 1, 3, 4, 6, 8, 11, and 13 at concentration of 2 mM was tested. The activity of MK5 towards the PRAKtide substrate in the absence of inhibitor was arbitrary set as 100% and the residual activity in the presence of inhibitor was related to this. These values then allowed us to calculate the percentage of inhibition
Next, we performed dose–response studies using multiple concentrations of compounds 8 and 11, and determined the IC50 values. While the IC50 value for compound 8 was in the mM range, compound 11 had an IC50 of 37.5 μM (data not shown). These results demonstrate that compound 11 is a more potent MK5 inhibitor than compound. 8.
DA compounds prevent PKA-induced nuclear export of MK5
MK5 contains nuclear export signal (NES) and nuclear localization signal (NLS) motifs, i.e., sequences that mediate subcellular targeting to the cytoplasm and to the nucleus, respectively. Due to these signals, MK5 shuttles between the nucleus and the cytoplasm in a cell. In resting cells, MK5 resides predominantly in the nucleus [59, 60]. Recently, we reported transient nuclear export of MK5 in the presence of active cAMP-dependent protein kinase (PKA) or when overexpressing the catalytic subunit Cα of PKA in PC12 cells. This process requires the kinase activity of both Cα and MK5 as a kinase dead Cα could not trigger nuclear export of MK5, while activation of the cAMP/PKA pathway failed to induce nucleocytoplasmic redistribution of kinase dead MK5 [29]. Hence, we reasoned that inhibition of the MK5 activity by compound 11 would prevent PKA-induced nuclear export of MK5. EGFP-MK5-transfected cells were left untreated or exposed to 20 μM of compound 11. This concentration was not toxic for the cells (results not shown). Cells were subsequently exposed to the cAMP-elevating agent forskolin, and the subcellular distribution of EGFP-MK5 was monitored by confocal microscopy. Thirty minutes after treatment with forskolin, approximately 84% cells that did not receive DA compound 11 displayed both nuclear and cytosolic EGFP-MK5 (Fig. 2b) versus 6% in untreated control cells (Fig. 2a). Pretreatment with compound 11 at a concentration of 20 μM reduced cAMP/PKA-induced nuclear export of MK5 to 20% (Fig. 2c). To exclude that the effect of this compound on the subcellular behavior of MK5 was a result of inhibiting the nuclear export machinery and not the kinase activity, we compared the subcellular localization of the EGFP-NES-MK5 T182A fusion protein in the absence and presence of compound 11. The mutation T182A destroys the kinase activity of MK5 but also makes MK5 unable to shuttle to the cytoplasm. Fusing a functional NES from human immunodeficiency virus Rev protein causes EGFP-NES-MK5 T1882A to reside exclusively in the cytoplasm. So this EGFP-MK5 mutant is a kinase dead MK5 that resides solely in the cytoplasm. Compound 11 did not interfere with the cytoplasmic localization of EGFP-NES-MK5 T182A, indicating that this compound had no effect on the nuclear export machinery (compare panels d and e in Fig. 2). The results suggest that compound 11 does not prevent PKA-induced nuclear export of MK5 by interfering with the nuclear export machinery, but rather by inhibiting the catalytic activity of MK5.
Fig. 2.

Noroxoaconitine (compound 11) prevents PKA-induced nucleocytoplasmic translocation of MK5. a EGFP-MK5 transfected cells in the absence of forskolin and compound 11. b As in (a), but EGFP-MK5 subcellular localization was monitored 30 min after the cells were exposed to 10 μM forskolin. Forskolin treatement resulted in nuclear and cytoplasmic distribution of MK5 in 84% of the examined cells. c As in (b), but cells were pretreated with 20 μM of compound 11. d Cells transfected with EGFP-NES-MK5 T182A. e As in (d), but cells were treated with 20 μM of compound 11. The subcellular location of ectopic expressed EGFP-MK5 and EGFP-NES-MK5 T182A fusion proteins was monitored by EGFP fluorescence (green channel) in a confocal microscope
Compound 8 and 11 are ATP but not substrate competitors
Many of the known protein kinase inhibitors act through competition with ATP for binding to the ATP binding pocket of the enzyme [61]. Enzyme inhibitor kinetic reactions were performed to study the kinetic trends and competitions between the compounds (11 and 8) and ATP. Kinetic studies showed that the K m of MK5 for ATP is 0.146 mM. Enzyme kinetic studies of compounds 8 and 11 against varying ATP concentrations showed that both act on MK5 in an ATP-competitive manner and their K i values were calculated to be 301.0 and 0.675 μM, respectively. These results may indicate that they bind at the ATP binding site of MK5, and confirm that compound 11 is a more potent MK5 inhibitor than compound 8. The Lineweaver–Burk plots of compound 11 (panel a) and compound 8 (panel b) against ATP are shown in Fig. 3. Dose-dependent studies with increasing concentrations of PRAKtide substrate showed that compound 11 did not interfere with the binding of the substrate (results not shown). Taken together, our results indicate that compound 11 acts as a competitor of ATP binding.
Fig. 3.

Compounds 8 and 11 are ATP binding competitors. Enzyme kinetic models of compounds 11 and 8 against increasing concentrations of ATP. The Lineweaver–Burk plots of compound 11 (a) and compound 8 (b) against ATP
Compound 11 abrogates MK3- and MK5-, but not MK2-mediated phosphorylation of Hsp27
Because MK2 and MK3 are most similar to MK5, we wanted to test the effect of compound 11 on the kinase activities of MK2 and MK3. The small heat shock protein Hsp27 has been shown to be an in vitro and in vivo substrate for all three kinases [62], and thus we monitored their ability to phosphorylate recombinant Hsp27 at the relevant serine residue 78 in presence or absence of this compound. These studies showed that compound 11 did not interfere with MK2-mediated phosphorylation of Hsp27 (compare lanes 5 and 6 in Fig. 4), but it almost completely ablated phosphorylation by MK5 and MK3 [compare lanes 1 and 2 (MK5) with lanes 3 and 4 (MK2) in Fig. 4].
Fig. 4.

Compound 11 inhibits Hsp27 phosphorylation by MK3 and MK5, but not by MK2. Recombinant Hsp27 was incubated with purified MK2, MK3, or MK5 in the presence or absence of compound 11 . Hsp27 phosphorylation at serine residue 78 (p-Ser78 Hsp27) was monitored by western blot with phosphoserine-78 specific antibodies. Lanes 1, 3, and 5: Hsp27 incubated with purified MK5, MK3, and MK2, respectively. Lanes 2, 4, and 6: as lanes 1, 3, and 5 but in the presence of 20 μM noroxoaconitine (compound 11)
Homology modeling of MK5
Three-dimensional (3D) X-ray crystal structures of MK2 apoenzyme, truncated and point mutated forms, and of MK2 in complex with p38MAPK, ADP, AMPPNP, or staurosporine have been published [57, 63–65]. Recently, an X-ray structure of MK3 in complex with the pharmaceutical lead compound P4O was also published [54]. In the absence of an available X-ray crystal structure of MK5, we constructed a three-dimensional (3D) model of MK5 by homology with MK2 and MK3 using the homology modeling approach. Multiple sequence alignments using ClustalW [66, 67], showed that MK5 has 43% amino acid sequence identity with MK2 (PDB id: 2OZA) [43], and 41% amino acid sequence identity with MK3 [54]. The structure of MK2 (2OZA) was therefore used as a template for constructing a 3D model of MK5 (Fig. 5).
Fig. 5.

Multiple sequence alignment and the homology-based model of MK5. a The primary sequence alignments of MK5 (accession number NP_003659), MK2 (chain A of 2OZA), and MK3 (accession number NP_849238) are shown and identical residues are indicated with asterisks in the consensus, while analogous residues are shown by colons. The conserved GXGXXG motif is located at the N-terminal part of the proteins. b Ramachandran plot of the MK5 model. c The superimposition of the template (2OZA) and the MK5 model (RMSD value of 0.68 Å)
The structural quality of the model was checked and compared with the template structure (MK2). The Ramachandran plot of the model (panel b of Fig. 5) revealed that 88.7% of the residues were in most favored regions, which is better than the template X-ray structure (86.9%). Verify_3D showed that 71.6% of the residues had an averaged 3D–1D score of more than 0.2, whereas the corresponding value for the template was 82%. During the assessment of PROVE scores for both the model and template, the mean Z score was found to be 0.086 and 0.278, respectively, which also indicates a structurally high quality model.
Finally, the 3D MK5 model was superimposed onto the 3D structure of the template 2OZA (shown in panel c of Fig. 5), giving a RMSD of 0.68 Å between backbone Cα atoms of template and model. The structural deviations between the model and the template were mainly seen in some loop regions, but secondary structure elements (not shown) and the ATP binding site (GXGXXG) were highly conserved (Fig. 5).
Docking of inhibitors at the ATP binding site of MK5
Prior to docking, putative binding pockets at the MK5 model were predicted using the ‘pocket finder’ algorithm of ICM. The best predicted binding pocket is shown in panel a of Fig. 6, and corresponds to the ATP binding pocket of MK2 and MK3. The predicted binding pocket included the following amino acids of MK5: Leu28, Ile32, Gly34, Val36, Ala49, Lys51, Ile32, Gly34, Val36, Ala49, Lys51, Met102, Met105, Glu152, Asn153, Leu155, Cys168 and Asp169. The enzyme kinetic studies indicated that compounds 8 and 11 are ATP competitive, and they were therefore docked into the predicted binding pocket of MK5 that corresponds to the ATP binding pocket of MK2 and MK3. Docking poses were evaluated based on docking energy, ligand conformation and similarities with ATP binding in known X-ray structure complexes. The highest scored docking complex of compound 8 had a docking energy of −72.26 kcal/mol, while the binding energy (ΔG) was calculated to −0.89 kcal/mol. For compound 11, the highest scored docking pose (panels c and d of Fig. 6) showed a binding mode very similar to the ATP binding in the X-ray structure complex of MK2 with ATP (PDB code: 2PHK [56]). The docking energy of compound 11 was −90.4 kcal/mol, while (ΔG) was calculated to −9.0 kcal/mol, indicating that compound 11 binds more strongly to MK5 than compound 8.
Fig. 6.

Docking of compound 11 at the ATP binding site of MK5. a The predicted binding pocket of MK5 which corresponds to the ATP binding site of MK2 and MK3; b,c the best docking pose of compound 11; d superimposition of the compound 11 (in dark-blue stick model) and 8 (in dark-yellow stick model) in their best docking poses
Finally, the docked complexes of both compounds were superimposed and their binding modes compared. The RMSD between identical parts of their structure was 0.2 Å (shown in panel d of Fig. 6). The major difference between the binding modes of the compounds was due to the presence of a phenyl ring in compound 11 lacking in compound 8 (see Fig. 7), which probably plays a major role of the inhibitory activities against MK5. The phenyl ring of compound 11 interacted with MK5 in a quite hydrophobic region of the binding pocket consisting of Leu28, Ala49, Met102, Met105 and Leu155 (Fig. 6).
The docking indicated that compound 8 has several hydrophobic interactions with amino acids in the ATP binding pocket. Leu28, Gly29, Gly31 Leu155 and Met105 of MK5 all interacted with compound 8 after docking. A hydroxyl oxygen of compound 8 formed a hydrogen bond with a side chain oxygen of Asp169. The hydroxyl group of compound 11 (Fig. 7) had a hydrogen bond with the backbone carbonyl oxygen of Glu152. Compound 11 also formed a hydrogen bond with the backbone carbonyl group of Leu28.
Compound 11 was also docked into the ATP binding sites of the X-ray structure of MK2 (PDB id: 2OZA) [43] and MK3 [54]. The docking indicated that compound 11 may bind both MK2 and MK3, but with weaker affinity than for MK5. The docking energy for MK2 was −79.1 kcal/mol, while the calculated binding energy was −5.2 kcal/mol. The corresponding values for MK3 were −72.1 kcal/mol and −4.2 kcal/mol, respectively.
To validate our docking procedure, the potent inhibitor of MK2, MK3 and MK5 2-(2-quinolin-3-ylpyridin-4-yl)-1,5,6,7-tetrahydro-4H-pyrrolo[3,2-c]pyridine-4-one was docked into the ATP binding pockets of MK2, and MK3 and the predicted pocket of the MK5 model. This inhibitor has IC50 values of 0.008.5 μM for MK2, 0.21 μM for MK2 and 0.081 μM for MK5 [68]. The calculated binding energies (ΔG) for MK2, MK3 and MK5 were −10.3 kcal/mol, −3.8 kcal/mol and −7.3 kcal/mol, respectively. The ΔG and the IC50 values were compared using linear regression and found to be highly correlated (R 2 = 0.93).
The calculated binding energy (ΔG) of compound 11 is overestimated compared with the experimental observation. Compound 11 contains functional groups with strong hydrogen bonding capacity, and a reason for the overestimations may be imperfect parameterization of these reactive groups. However, the results obtained for the potent binder 2-(2-quinolin-3-ylpyridin-4-yl)-1,5,6,7-tetrahydro-4H-pyrrolo[3,2-c]pyridine-4-one show the validity of our docking approaches.
Discussion
Protein kinases, including protein kinases of the MAP kinase pathways are attractive therapeutic targets for development of specific inhibitors because they often display perturbed activity in clinical conditions such as cancer, cardiac conditions, neurological and inflammatory diseases [69–72]. The search for highly specific MAP kinase inhibitors is therefore a major challenge for pharmaceutical companies. Unfortunately, many of the protein kinase inhibitors developed today display little specificity and have therefore not entered the clinic [73–75].
MK5 is a compound of the MAP kinase pathways, but studies aimed at elucidating the role of MK5 have been hampered by the lack of a specific inhibitor. DAs are a group of complex plant chemicals with similar structure to that of terpene. A large number of these alkaloids were isolated from Aconitum, Delphinium and Consolida. The DAs demonstrate plethora of pharmacological properties, such as analgesic, anti-arrhythmic, anti-inflammatory, arrhythmogenic, curariform, hypotensive, local anesthetic, neurotropic, psychotropic, and spasmolytic (reviewed in [36, 76, 77]). Therefore, we investigated whether they possess the potential to inhibit MK5. Our screening showed that some of the DAs can suppress the catalytic activity of MK5. Of the tested compounds, noroxoaconitine (compound 11) displayed the strongest MK5 inhibition with an IC50 = 37.5 μM, and a very favorable interaction energy predicted by docking studies. The IC50 value is in the same range as that of the two previously described unspecific MK5 inhibitors: epigallocatechin gallate (EGCG) from green tea (IC50 < 10 μM) and flavokavain A (IC50 = 10 μM) from kava (Piper methysticum) [74, 78]. In accordance with flavokavain A, noroxoaconite inhibited MK5 and MK3 kinase activity, but had no effect on MK2 [78]. The effect of EGCG on the enzymatic activity of MK2 and MK3 was not tested [74]. Both EGCG and flavokavain, however, are rather unspecific. EGCG at a concentration of 10 μM was shown to inhibit the in vitro kinase activity, by ~90%, of ERK2, DYRK1A, ROCK-II, and PDK1 [74]. The same concentration of flavokavain A also inhibited the protein kinases Aurora B, DYRK1A, IKKβ, and MK3 more or to a similar extent, while approximately 40–70% inhibition was measured for ERK8, GSK3β, RSK2, MK2, MSK1, p70S6K, PDK1, PHK, PKA, PKB2, PLK1, p38δ, and smMLCK [78]. While our studies suggest that 20 μM noroxoaconitine can inhibit the kinase activity of MK5 in cells, the effect of EGCG and flavokavain on MK5 in vivo has not been investigated.
The theoretical docking studies indicated that noroxoaconite fitted nicely into the ATP binding pocket of MK3 and MK5 in agreement with the experimental studies. The docking indicated that noroxoaconite binds a bit differently in MK3 and MK5. In MK3, the phenyl ring of noroxoaconite binds in between Leu52 in a β-sheet region, and Glu172 located four amino acids C-terminal of the catalytic RD region. In MK5, the phenyl ring of noroxoaconite interacted in a quite hydrophobic region of the binding pocket consisting of Leu28, Ala49, Met102, Met105 and Leu155 (Fig. 6). In MK5, a more bulky region of the inhibitor interacts inbetween Ala30 and Glu152 that corresponds to Leu52 and Glu172 in MK3. The main reason for the differences in orientation seems to be that the cleft between Leu52 and Glu172 in MK3 is much narrower than the corresponding cleft between Ala30 and Glu152 in MK5. The atomic distance between the Cα-atoms of Leu52 and Glu172 in MK3 is 8.6 Å, while the corresponding distance between Ala30 and Glu152 in MK5 is 12.1 Å, which might explain our docking predictions indicating that noroxoaconite interacts with MK3 with the phenylring inbetween Leu52 and Glu172, while it interacts with MK5 with a more bulky region between Ala30 and Glu152.
The experimental studies showed that noroxoaconite did not prevent MK2-mediated phosphorylation of recombinant Hsp27 at serine residue 78. However, this was not predicted by the docking studies since noroxoaconite bound MK2 similar to the binding mode in MK5. At present, we cannot explain the specificity of compound 11 for MK3 and MK5, but flavokavain A also inhibited MK3 and MK5, but not MK2, despite the higher degree of overall identity between MK2 and MK3 than between MK3 and MK5 [55]. Similarly, the MK2 inhibitor 2-(2-quinolin-3-ylpyridin-4-yl)-1,5,6,7-tetrahydro-4H-pyrrolo[3,2-c]pyridin-4-one (IC50 = 8.5 nM) is a more potent inhibitor for MK5 (IC50 = 81 nM) than for MK3 (IC50 = 210 nM), even though the ATP binding pockets of MK2 and MK3 show most identity [68].
In an effort to determine the specificity of noroxoaconite, a screen was performed by the company ProQinase (Freiburg, Germany) with 25 μM of this compound against 252 different protein kinases. While significant inhibition of only two other protein kinases was detected (GRK2 and S6Kβ), no inhibition was found for MK3 and MK5 (see Electronic supplementary material, Table S1). At present, we cannot explain why this company failed to detect inhibition of MK5, while we clearly observed an inhibition using different experimental approaches. Differences in assay, source of MK5, conditions, and substrate may account for this discrepancy. While we used Praktide or Hsp27 as specific substrates for MK5, ProQinase used the peptide RBER-CHKtide, which was also used for 56 other protein kinases in their screen.
In conclusion, we have identified a potent novel MK5 inhibitor, noroxoaconitine (compound 11), which most probably acts by competing with ATP for the ATP binding pocket in the enzyme. Noroxoaconitine also reduced MK3 but not MK2 kinase activity towards Hsp27 in vitro. As MK3 is less expressed and less active than MK2 [22], the use of this inhibitor may allow testing the individual involvement of MK2 and MK5 in cellular responses to stress which are know to activate both enzymes [15]. Rationally designed derivatives of noroxoaconitine may then be synthesized and experimentally tested for their potency and specificity to inhibit MK5, and may enable us to elucidate the biological roles of MK5. In addition, such inhibitors may prove therapeutic values in diseases with aberrant MK5 functions.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We are grateful to Prof. M. Sultankhudzaev, Uzbek National Academy of Science, Tashkent, Uzbekistan, for kindly supplying us the compounds reported here. We are also grateful to Dr. Thomas Hesterkamp Evotec AG, Germany, and Dr. John Barker Evotec Ltd., UK, for providing the X-ray structure coordinates of MK3. We are also grateful to Dr. Mona Johannessen, University of Tromsø. S.K and M.T.H.K. were fellows of the PhD School for Molecular and Structural Biology, University of Tromsø, Norway. This work was supported by the Norwegian Cancer Society (A01037) and the Mohn Foundation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- DA
Diterpenoid alkaloid
- DMBA
Dimethylbenzanthrance
- ERK
Extracellular signal-regulated kinase
- ICM
Internal coordinate mechanics
- JNK
c-Jun N-terminal kinase
- MAP2K, MEK or MKK
MAP kinase kinase
- MAP3K, MEKK or MKKK
Mitogen-activated protein kinase kinase kinase
- MAPK
Mitogen-activated protein kinase
- MK
MAPK-activated protein kinase
- MNK
MAPK-interacting kinase
- MSK
Mitogen- and stress-activated protein kinases
- PDB
Protein Databank
- PRAK
p38-regulated/activated kinase
- PSQS
Predicted structure quality score
- RMSD
Root mean squared deviation
- RSK
p90 ribosomal S6 kinase
- SAR
Structure–activity relationship
- SAVS
Structural Analysis and Verification Server
- 3D
Three-dimensional
Footnotes
S. Kostenko and M. T. H. Khan contributed equally to this work.
References
- 1.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/er.22.2.153. [DOI] [PubMed] [Google Scholar]
- 2.Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 4.Dhanasekaran DN, Johnson GL. MAPKs: function, regulation, role in cancer and therapeutic targeting. Oncogene. 2007;26:3097–3099. doi: 10.1038/sj.onc.1210395. [DOI] [PubMed] [Google Scholar]
- 5.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/S0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 6.Imajo M, Tsuchiya Y, Nishida E. Regulatory mechanisms and functions of MAP kinase signaling pathways. IUBMB Life. 2006;58:312–317. doi: 10.1080/15216540600746393. [DOI] [PubMed] [Google Scholar]
- 7.Klevernic IV, Stafford MJ, Morrice N, Peggie M, Morton S, Cohen P. Characterization of the reversible phosphorylation and activation of ERK8. Biochem J. 2006;394:365–373. doi: 10.1042/BJ20051288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. MAP kinases. Chem Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 9.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/S0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 10.Dhanasekaran N, Premkumar RE. Signaling by dual specificity kinases. Oncogene. 1998;17:1447–1455. doi: 10.1038/sj.onc.1202251. [DOI] [PubMed] [Google Scholar]
- 11.Fanger GR, Johnson NL, Johnson GL. MEK kinases are regulated by EGF and selectively interact with Rac/Cdc42. EMBO J. 1997;16:4961–4972. doi: 10.1093/emboj/16.16.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/S0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 13.Gutkind JS. Cell growth control by G protein-coupled receptors: from signal transduction to signal integration. Oncogene. 1998;17:1331–1342. doi: 10.1038/sj.onc.1202186. [DOI] [PubMed] [Google Scholar]
- 14.Minden A, Karin M. Regulation and function of the JNK subgroup of MAP kinases. Biochim Biophys Acta. 1997;1333:F85–F104. doi: 10.1016/s0304-419x(97)00018-8. [DOI] [PubMed] [Google Scholar]
- 15.Gaestel M. MAPKAP kinases—MKs—two’s company, three’s a crowd. Nat Rev Mol Cell Biol. 2006;7:120–130. doi: 10.1038/nrm1834. [DOI] [PubMed] [Google Scholar]
- 16.Perander M, Keyse SM, Seternes OM. Does MK5 reconcile classical and atypical MAP kinases? Front Biosci. 2008;13:4617–4624. doi: 10.2741/3027. [DOI] [PubMed] [Google Scholar]
- 17.Ronkina N, Kotlyarov A, Gaestel M. MK2 and MK3—a pair of isoenzymes? Front Biosci. 2008;13:5511–5521. doi: 10.2741/3095. [DOI] [PubMed] [Google Scholar]
- 18.Buxade M, Parra-Palau JL, Proud CG. The Mnks: MAP kinase-interacting kinases (MAP kinase signal-integrating kinases) Front Biosci. 2008;13:5359–5373. doi: 10.2741/3086. [DOI] [PubMed] [Google Scholar]
- 19.Arthur JS. MSK activation and physiological roles. Front Biosci. 2008;13:5866–5879. doi: 10.2741/3122. [DOI] [PubMed] [Google Scholar]
- 20.Carriere A, Ray H, Blenis J, Roux PP. The RSK factors of activating the Ras/MAPK signaling cascade. Front Biosci. 2008;13:4258–4275. doi: 10.2741/3003. [DOI] [PubMed] [Google Scholar]
- 21.New L, Jiang Y, Zhao M, Liu K, Zhu W, Flood LJ, Kato Y, Parry GC, Han J. PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J. 1998;17:3372–3384. doi: 10.1093/emboj/17.12.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ni H, Wang XS, Diener K, Yao Z. MAPKAPK5, a novel mitogen-activated protein kinase (MAPK)-activated protein kinase, is a substrate of the extracellular-regulated kinase (ERK) and p38 kinase. Biochem Biophys Res Commun. 1998;243:492–496. doi: 10.1006/bbrc.1998.8135. [DOI] [PubMed] [Google Scholar]
- 23.Gerits N, Shiryaev A, Kostenko S, Klenow H, Shiryaeva O, Johannessen M, Moens U. The transcriptional regulation and cell-specific expression of the MAPK-activated protein kinase MK5. Cell Mol Biol Lett. 2009;14:548–574. doi: 10.2478/s11658-009-0020-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun P, Yoshizuka N, New L, Moser BA, Li Y, Liao R, Xie C, Chen J, Deng Q, Yamout M, Dong MQ, Frangou CG, Yates JR, III, Wright PE, Han J. PRAK is essential for ras-induced senescence and tumor suppression. Cell. 2007;128:295–308. doi: 10.1016/j.cell.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 25.Shi Y, Kotlyarov A, Laabeta K, Gruber AD, Butt E, Marcus K, Meyer HE, Friedrich A, Volk HD, Gaestel M. Elimination of protein kinase MK5/PRAK activity by targeted homologous recombination. Mol Cell Biol. 2003;23:7732–7741. doi: 10.1128/MCB.23.21.7732-7741.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ronkina N, Kotlyarov A, Dittrich-Breiholz O, Kracht M, Hitti E, Milarski K, Askew R, Marusic S, Lin LL, Gaestel M, Telliez JB. The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol Cell Biol. 2007;27:170–181. doi: 10.1128/MCB.01456-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Q, Zhang N, Zhang D, Wang Y, Lin T, Wang Y, Zhou H, Ye Z, Zhang F, Lin SC, Han J. Determinants that control the distinct subcellular localization of p38alpha-PRAK and p38beta-PRAK complexes. J Biol Chem. 2008;283:11014–11023. doi: 10.1074/jbc.M709682200. [DOI] [PubMed] [Google Scholar]
- 28.Tak H, Jang E, Kim SB, Park J, Suk J, Yoon YS, Ahn JK, Lee JH, Joe CO. 14-3-3epsilon inhibits MK5-mediated cell migration by disrupting F-actin polymerization. Cell Signal. 2007;19:2379–2387. doi: 10.1016/j.cellsig.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 29.Gerits N, Mikalsen T, Kostenko S, Shiryaev A, Johannessen M, Moens U. Modulation of F-actin rearrangement by the cyclic AMP/cAMP-dependent protein kinase (PKA) pathway is mediated by MAPK-activated protein kinase 5 and requires PKA-induced nuclear export of MK5. J Biol Chem. 2007;282:37232–37243. doi: 10.1074/jbc.M704873200. [DOI] [PubMed] [Google Scholar]
- 30.Kostenko S, Johannessen M, Moens U. PKA-induced F-actin rearrangement requires phosphorylation of Hsp27 by the MAPKAP kinase MK5. Cell Signal. 2009;21:712–718. doi: 10.1016/j.cellsig.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 31.Gerits N, Van BW, Moens U. Transgenic mice expressing constitutive active MAPKAPK5 display gender-dependent differences in exploration and activity. Behav Brain Funct. 2007;3:58. doi: 10.1186/1744-9081-3-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stockwell BR. Exploring biology with small organic molecules. Nature. 2004;432:846–854. doi: 10.1038/nature03196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kazi A, Smith DM, Daniel K, Zhong S, Gupta P, Bosley ME, Dou QP. Potential molecular targets of tea polyphenols in human tumor cells: significance in cancer prevention. In Vivo. 2002;16:397–403. [PubMed] [Google Scholar]
- 34.Brisdelli F, D’Andrea G, Bozzi A. Resveratrol: a natural polyphenol with multiple chemopreventive properties. Curr Drug Metab. 2009;10:530–546. doi: 10.2174/138920009789375423. [DOI] [PubMed] [Google Scholar]
- 35.Sultankhodzhaev MN, Khan MTH, Choudhary MI, Atta-ur-Rahman S. Tyrosinase inhibition studies of diterpenoid alkaloids and their derivatives: structure–activity relationships. Nat Prod Res. 2005;19:517–522. doi: 10.1080/14786410512331330585. [DOI] [PubMed] [Google Scholar]
- 36.Gonzalez P, Marin C, Rodriguez-Gonzalez I, Illana A, Mateo H, Longoni SS, Rosales MJ, Gonzalez-Coloma A, Reina M, Sanchez-Moreno M. Diterpenoid alkaloid derivatives as potential chemotherapeutic agents in American trypanosomiasis. Pharmacology. 2006;76:123–128. doi: 10.1159/000090600. [DOI] [PubMed] [Google Scholar]
- 37.Sultankhodzhaev MN, Khan MT, Moin M, Choudhary MI, >Atta-ur-Rahman S. Tyrosinase inhibition studies of diterpenoid alkaloids and their derivatives: structure-activity relationships. Nat Prod Res. 2005;19(5):517–522. doi: 10.1080/14786410512331330585. [DOI] [PubMed] [Google Scholar]
- 38.Mett H, Holscher K, Degen H, Esdar C, De Neumann BF, Flicke B, Freudenreich T, Holzer G, Schinzel S, Stamminger T, Stein-Gerlach M, Marschall M, Herget T. Identification of inhibitors for a virally encoded protein kinase by 2 different screening systems: in vitro kinase assay and in-cell activity assay. J Biomol Screen. 2005;10:36–45. doi: 10.1177/1087057104270269. [DOI] [PubMed] [Google Scholar]
- 39.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 40.Gaucher JF, Selkti M, Tiraboschi G, Prange T, Roques BP, Tomas A, Fournie-Zaluski MC. Crystal structures of alpha-mercaptoacyldipeptides in the thermolysin active site: structural parameters for a Zn monodentation or bidentation in metalloendopeptidases. Biochemistry. 1999;38(39):12569–12576. doi: 10.1021/bi991043z. [DOI] [PubMed] [Google Scholar]
- 41.Selkti M, Tomas A, Gaucher JF, Prange T, Fournie-Zaluski MC, Chen H, Roques BP. Interactions of a new alpha-aminophosphinic derivative inside the active site of TLN (thermolysin): a model for zinc-metalloendopeptidase inhibition. Acta Crystallogr D Biol Crystallogr. 2003;59(Pt 7):1200–1205. doi: 10.1107/S0907444903010060. [DOI] [PubMed] [Google Scholar]
- 42.Gerits N, Van Belle W, Moens U. Transgenic mice expressing constitutive active MAPKAPK5 display gender-dependent differences in exploration and activity. Behav Brain Funct. 2007;3:58. doi: 10.1186/1744-9081-3-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White A, Pargellis CA, Studts JM, Werneburg BG, Farmer BT., 2nd Molecular basis of MAPK-activated protein kinase 2:p38 assembly. Proc Natl Acad Sci USA. 2007;104(15):6353–6358. doi: 10.1073/pnas.0701679104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 45.Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Stereochemical quality of protein structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 46.Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bowie JU, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 48.Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 49.Godzik A, Kolinski A, Skolnick J. Are proteins ideal mixtures of amino acids? Analysis of energy parameter sets. Protein Sci. 1995;4:2107–2117. doi: 10.1002/pro.5560041016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Godzik A. Knowledge-based potentials for protein folding: what can we learn from known protein structures? Structure. 1996;4:363–366. doi: 10.1016/S0969-2126(96)00041-X. [DOI] [PubMed] [Google Scholar]
- 51.Pawlowski K, Jaroszewski L, Bierzynski A, Godzik A (1997) Multiple model approach—dealing with alignment ambiguities in protein modeling. Pac Symp Biocomput 328–339 [PubMed]
- 52.Jaroszewski L, Pawlowski K, Godzik A. Multiple model approach: exploring the limits of comparative modeling. J Mol Model. 1998;4:294–309. doi: 10.1007/s008940050087. [DOI] [Google Scholar]
- 53.Holm L, Park J. DaliLite workbench for protein structure comparison. Bioinformatics. 2000;16:566–567. doi: 10.1093/bioinformatics/16.6.566. [DOI] [PubMed] [Google Scholar]
- 54.Cheng R, Felicetti B, Palan S, Toogood-Johnson I, Scheich C, Barker J, Whittaker M, Hesterkamp T. High-resolution crystal structure of human Mapkap kinase 3 in complex with a high affinity ligand. Protein Sci. 2010;19:168–173. doi: 10.1002/pro.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khan MT, Fuskevag OM, Sylte I. Discovery of potent thermolysin inhibitors using structure based virtual screening and binding assays. J Med Chem. 2009;52:48–61. doi: 10.1021/jm8008019. [DOI] [PubMed] [Google Scholar]
- 56.Lowe ED, Noble ME, Skamnaki VT, Oikonomakos NG, Owen DJ, Johnson LN. The crystal structure of a phosphorylase kinase peptide substrate complex: kinase substrate recognition. EMBO J. 1997;16:6646–6658. doi: 10.1093/emboj/16.22.6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hillig RC, Eberspaecher U, Monteclaro F, Huber M, Nguyen D, Mengel A, Muller-Tiemann B, Egner U. Structural basis for a high affinity inhibitor bound to protein kinase MK2. J Mol Biol. 2007;369:735–745. doi: 10.1016/j.jmb.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 58.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 59.New L, Jiang Y, Han J. Regulation of PRAK subcellular location by p38 MAP kinases. Mol Biol Cell. 2003;14:2603–2616. doi: 10.1091/mbc.E02-08-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seternes OM, Mikalsen T, Johansen B, Michaelsen E, Armstrong CG, Morrice NA, Turgeon B, Meloche S, Moens U, Keyse SM. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO J. 2004;23:4780–4791. doi: 10.1038/sj.emboj.7600489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gerits N, Moens U. Protein kinase inhibitors. In: Offermanns S, Rosenthal W, editors. Encyclopedia of molecular pharmacology. Berlin: Springer; 2008. pp. 1008–1012. [Google Scholar]
- 62.Kostenko S, Moens U. Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell Mol Life Sci. 2009;66:3289–3307. doi: 10.1007/s00018-009-0086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Underwood KW, Parris KD, Federico E, Mosyak L, Czerwinski RM, Shane T, Taylor M, Svenson K, Liu Y, Hsiao CL, Wolfrom S, Maguire M, Malakian K, Telliez JB, Lin LL, Kriz RW, Seehra J, Somers WS, Stahl ML. Catalytically active MAP KAP kinase 2 structures in complex with staurosporine and ADP reveal differences with the autoinhibited enzyme. Structure. 2003;11:627–636. doi: 10.1016/S0969-2126(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 64.ter Haar E, Prabhakar P, Liu X, Lepre C. Crystal structure of the p38 alpha-MAPKAP kinase 2 heterodimer. J Biol Chem. 2007;282:9733–9739. doi: 10.1074/jbc.M611165200. [DOI] [PubMed] [Google Scholar]
- 65.Argiriadi MA, Sousa S, Banach D, Marcotte D, Xiang T, Tomlinson MJ, Demers M, Harris C, Kwak S, Hardman J, Pietras M, Quinn L, DiMauro J, Ni B, Mankovich J, Borhani DW, Talanian RV, Sadhukhan R. Rational mutagenesis to support structure-based drug design: MAPKAP kinase 2 as a case study. BMC Struct Biol. 2009;9:16. doi: 10.1186/1472-6807-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thompson JD, Gibson TJ, Higgins DG (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinforma Chapter 2:Unit 2.3 [DOI] [PubMed]
- 67.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed]
- 68.Anderson DR, Meyers MJ, Vernier WF, Mahoney MW, Kurumbail RG, Caspers N, Poda GI, Schindler JF, Reitz DB, Mourey RJ. Pyrrolopyridine inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2) J Med Chem. 2007;50:2647–2654. doi: 10.1021/jm0611004. [DOI] [PubMed] [Google Scholar]
- 69.Gaestel M, Kotlyarov A, Kracht M. Targeting innate immunity protein kinase signalling in inflammation. Nat Rev Drug Discov. 2009;8:480–499. doi: 10.1038/nrd2829. [DOI] [PubMed] [Google Scholar]
- 70.Pandya N, Santani D, Jain S. Role of mitogen-activated protein (MAP) kinases in cardiovascular diseases. Cardiovasc Drug Rev. 2005;23:247–254. doi: 10.1111/j.1527-3466.2005.tb00169.x. [DOI] [PubMed] [Google Scholar]
- 71.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 72.Samuels IS, Saitta SC, Landreth GE. MAP’ing CNS development and cognition: an ERKsome process. Neuron. 2009;61:160–167. doi: 10.1016/j.neuron.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen FZ, Chen DL, Chen QH, Wang FP. Diterpenoid alkaloids from Delphinium majus. J Nat Prod. 2009;72:18–23. doi: 10.1021/np800439a. [DOI] [PubMed] [Google Scholar]
- 77.Nisar M, Ahmad M, Wadood N, Lodhi MA, Shaheen F, Choudhary MI. New diterpenoid alkaloids from Aconitum heterophyllum Wall:selective butyrylcholinestrase inhibitors. J Enzym Inhib Med Chem. 2009;24:47–51. doi: 10.1080/14756360801906202. [DOI] [PubMed] [Google Scholar]
- 78.Folmer F, Blasius R, Morceau F, Tabudravu J, Dicato M, Jaspars M, Diederich M. Inhibition of TNFalpha-induced activation of nuclear factor kappaB by kava (Piper methysticum) derivatives. Biochem Pharmacol. 2006;71:1206–1218. doi: 10.1016/j.bcp.2005.12.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.