

Abstract
The histidine-rich calcium binding protein (HRC) is a novel regulator of sarcoplasmic reticulum (SR) Ca2+-uptake, storage and release. Residing in the SR lumen, HRC binds Ca2+ with high capacity but low affinity. In vitro phosphorylation of HRC affects ryanodine affinity of the ryanodine receptor (RyR), suggesting a functional role of HRC on SR Ca2+-release. Indeed, acute HRC overexpression in isolated rodent cardiomyocytes decreases Ca2+-induced Ca2+-release, increases SR Ca2+-load, and impairs contractility. The HRC effects on RyR may be regulated by the Ca2+-sensitivity of its interaction with triadin. However, HRC also affects the SR Ca2+-ATPase, as shown by HRC overexpression in transgenic mouse hearts, which resulted in reduced SR Ca2+-uptake rates, cardiac remodeling and hypertrophy. In fact, in vitro generated evidence suggests that HRC directly interacts with SR Ca2+-ATPase2, supporting a dual role of HRC in Ca2+-homeostasis: regulation of both SR Ca2+-uptake and Ca2+-release. Furthermore, HRC plays an important role in myocyte differentiation and in antiapoptotic cardioprotection against ischemia/reperfusion induced cardiac injury. Interestingly, HRC has been linked with familiar cardiac conduction disease and an HRC polymorphism was shown to associate with malignant ventricular arrhythmias in the background of idiopathic dilated cardiomyopathy. This review summarizes studies, which have established the critical role of HRC in Ca2+-homeostasis, suggesting its importance in cardiac physiology and pathophysiology.
Keywords: HRC, Calcium-homeostasis, Contractility, Heart failure, Arrhythmia
1. Introduction
A universal characteristic of the failing cardiomyocytes is aberrant intracellular Ca2+-handling, manifested by prolonged intracellular Ca2+-transients and changes in peak systolic and diastolic Ca2+ levels [1]. Rapid and effective regulation of the cardiomyocyte Ca2+ steady state is orchestrated by the coordinated function of the sarcoplasmic reticulum (SR) proteins. Therefore, targeting these molecules has been suggested to constitute promising therapy for the treatment of heart failure and malignant arrhythmias [2,3]. The histidine-rich calcium-binding protein (HRC), expressed predominantly in striated muscle [4], is a good candidate for such an approach because it is a SR component implicated in Ca2+-uptake [5,6 ], storage [7] and release [5,8,9]. Early studies by Hofmann et al. [4], showed that HRC is expressed in the SR of rabbit skeletal and cardiac muscles. The 160 kD HRC protein stains metachromatically blue with Stainsall, a cationic dye staining positive for Ca2+ binding proteins and it binds 45Ca2+ on nitrocellulose membranes with high capacity and low affinity [4,10]. Although it was found that this molecule could interact with low density lipoproteins, such a biological function for HRC was rejected because of its localization within the SR lumen, where it has no access to plasma lipoproteins. Instead, it was proposed that HRC may have a role in Ca2+-homeostasis [4]. This notion was later supported by further experimental evidence that placed HRC at the junctional SR, which contains the ryanodine receptor channels (RyR), recognizing its involvement in excitation-contraction coupling [11–15].
The localization of HRC has been the subject of extensive debates. It was initially described as a SR lumen component [4], although Saccheto et al. [16] proposed that HRC is a cytosolic protein which interacts with the triadin cytoplasmic domain and it is phosphorylated by the SR membrane anchored calmodulin kinase II (CaMKII) on the cytoplasmic side. However, HRC is not likely to be a cytoplasmic protein, based on several lines of evidence: a) electron microscopy studies indicate a diffuse localization pattern of HRC within the terminal cisternae and the longitudinal tubules of SR [4]; b) HRC can only be released by sodium carbonate treatment of the SR, consistent with its luminal location [4]; c) HRC is present in microsomal fractions following subcellular fractionation [18]; and d) Lee et al. [8] demonstrated that HRC binds to the carboxyl-terminal luminal KEKE motif of triadin, providing further support for its SR luminal localization. In addition, several studies indicate that HRC is phosphorylated by the lumenally located casein kinase 2 (CKII) [9,15] and phosphoproteomic analysis of human skeletal muscle also confirmed that CKII is involved in HRC phosphorylation [17].
Functionally, HRC has an important role in SR Ca2+-handling and in the overall cardiac physiology as it has been shown to improve the SR Ca2+-storage capacity and enhance cardiomyocyte survival after ischemia [19], to alter Ca2+-cycling after acute [20] and chronic overexpression [6], and to be implicated in human arrhythmia as a modifying factor [21,22].
2. Histidine-rich calcium-binding protein (HRC)
2.1. HRC gene structure and transcription
The HRC gene, located at 19q13.3 in human, and 7 B4 in mouse, consists of 6 exons, with almost 90% of the entire coding region being in its first exon [23,24]. The transcription of this gene is tissue specific, with high levels of expression in cardiac, skeletal, and to a lesser extend in arteriolar smooth muscles [23,25,26]. Three potential CpG islands could be identified at 2.1 kb, 2.5 kb, and 3.2 kb upstream of the transcriptional initiation site of HRC, but there is no identifiable TATA box. At -224 there is a GGCTGGGG sequence which is also present upstream of the sarco(endo)plasmic reticulum Ca2+-ATPase genes 1 and 2 (ATP2A1 and ATP2A2, that encode the SERCA1 and SERCA2 proteins, respectively), the fast-twitch skeletal muscle calsequestrin, dihydropyridine receptor and ryanodine receptor, suggesting that it may serve as a SR signal [27–31]. Importantly, HRC transcription during embryonic development in mouse heart is regulated by a 2.6 kb genomic region upstream of the transcriptional initiation site, that contains a small, highly conserved sequence with a consensus MEF2 binding site [32]. In contrast, the minimal genomic region, which facilitates gene expression in skeletal muscle, is 770 bp upstream of the transcriptional initiation site [32]. Since additional upstream cis-acting enhancer sequences upstream (−770) are necessary for HRC gene expression in developing cardiomyocytes, it appears that its fine regulation in human heart is of critical importance. Notably, the transcription initiation site of HRC is located within 2.3 kb from the transcription initiation site of the transient receptor potential cation channel (subfamily M, member 4, TRPM4) [33]. This Ca2+-activated non-selective cation channel has been associated with human progressive familial heart block type I [34]. It appears that the HRC promoter is bidirectional regulating the transcription of both HRC and TRPM4.
The HRC protein is not detectable in undifferentiated, proliferating rat thigh muscle isolated myoblast cultures. However, HRC accumulates rapidly, when differentiation is induced [4]. This may be attributed to the SOX15-driven regulation of the HRC transcription. SOX15 is a member of the SOX (sex determining region Y-related, high mobility group-box) family of transcriptional factors, which are involved in the regulation of embryonic development, determination of cell fate and cell commitment. SOX15 antagonizes muscle differentiation in myogenic precursor cells, and regulates early myogenesis or fusion of myoblasts to myotubes [35]. SOX15 ablation in mouse embryonic stem cells results in selective overexpression of HRC mRNA, suggesting that SOX15 is a suppressor of the HRC locus [36]. Interestingly, SOX15 null mice exhibit delayed skeletal muscle regeneration after injury, although their myofiber ultrastructure is normal [37]. Similar results are obtained from cultured Sox15-deficient mouse myoblasts, which display a marked delay in differentiation in vitro [37]. The significance of HRC repression by SOX15 in the early stages of myocyte differentiation, especially during myofiber trauma repair, remains to be elucidated.
2.2 HRC structure and physicochemical properties
The HRC protein is a highly charged molecule with 31% acidic and 8% basic amino acid residues. There is a signal peptide at the amino-terminus of HRC (1-28 AA in human), which targets the molecule to the SR. This is followed by domains rich in: a) histidine (59-187 AA); b) glutamic acid and histidine (188-245 AA); c) aspartic acid, glutamic acid and histidine (246-309 AA); d) glutamic acid and histidine (310-468 AA); e) glutamic acid (469-608 AA); and f) cysteine (627-673 AA). An interesting feature of HRC is that it contains a highly repetitive region in the middle of the protein, between AA 106 and 365 in human, composed of ten histidine-rich acidic tandem repeats. These repeats are characterized by two types of motifs. The type A motif, which is repeated four times, starts with an almost invariant hexapeptide (HRH-R/Q-GH), followed by a stretch of acidic residues (8-18 AA in length), and ends with a conservative peptide of 9 AA (STESDRHQA). The type B motif, which is repeated six times, starts with an heptapeptide (HRHQXHG), followed by 1 to 4 amino acids, and ends with an EEDEDVSXEHHHHXPS sequence, as identified in humanHRC [23,38].
HRC lacks a typical Ca2+ binding motif and it is likely that the highly acidic central cores of each repeat may constitute the Ca2+ binding sites, in a manner similar to calsequestrin. This implies that HRC binds Ca2+ through electrostatic interactions with pairs or triplets of acidic amino acids, which form Ca2+ binding surfaces [39]. These surfaces of the ten histidine-rich acidic tandem repeats of motives A and B may be closely packed in the presence of Ca2+. In support of this notion, Ca2+-binding induces changes in the electrophoretic mobility of HRC and suggests Ca2+-induced conformational changes [11,15]. This is highly similar to calsequestrin and the condensed formation of its three thireodoxin-like domains in the presence of high Ca2+ [39,40 ]. Importantly, the HRC protein remains tightly attached to the SR membrane after treatment with 500 mM of potassium chloride, which removes calsequestrin. However, HRC can be readily extracted with 1 mM EDTA, suggesting that the interaction of HRC with the SR membrane components requires Ca2+ [14]. Picello et al. [14] reported that HRC binds 200 nmoles of Ca2+/mg protein with low affinity (KD=1.9 mM). It also binds zinc ions (Zn2+) at different binding sites than Ca2+ while calsequestrin has no affinity for Zn2+ [14]. Zinc has been shown to have negative inotropic effects in the heart, causing prolonged action potentials [41]. The HRC acidic surfaces along with the C-terminal cysteine-rich domain, which is highly conserved among species, provide the means for protein-protein interactions through positively and negatively charged amino acid residues, Ca2+ or cysteine bridges. The lack of three-dimensional structural information on HRC limits our knowledge regarding the domains that bind Ca2+ and Zn2+, as well as the folding changes which may be propagated by different ion saturation states.
2.3 HRC binding partners
The presence of repetitive elements, which are rich in histidine and acidic amino acids, raises the question of whether HRC could be multimeric. Indeed, Suk et al. [42] showed that purified HRC was eluted in increasing molecular size fractions under ascending Ca2+ concentrations. Under physiological Ca2+-levels, HRC appears to be present as a multimer. However, at higher Ca2+-levels and when HRC is saturated with Ca2+, the protein is present as a monomer [42]. Thus, local and rapid changes of Ca2+-levels within the SR lumen [43], may lead to different multimerization states of HRC and affect its interactions with other SR components.
The first SR protein identified to interact directly with HRC was triadin [8,9,16]. Triadin is embedded in the SR membrane, as part of the quaternary Ca2+-release complex [40,44]. Lee et al. [8] demonstrated that HRC binds triadin’s luminal KEKE motif, which is capable of interacting with calsequestrin and the ryanodine receptor [45]. Specifically, the human triadin domain, which contains a EQKKAKTAEKSEEKTKKE sequence and is similar to the EEKKARTKEKIEEKTKKE motif (KEKE-motif) of rabbit triadin, was found to interact with the human HRC [5]. It seems that multiple domains of HRC could interact with triadin, since both the C-terminal cysteine-rich domain [5,9,16] and the histidine-rich and acidic repeats [8] have been identified to target the triadin KEKE motif. Importantly, the HRC/triadin interaction is Ca2+ sensitive and the highest binding occurs at moderate Ca2+ concentrations, similar to the physiological [Ca2+]SR [5,8,9]. As the Ca2+ concentration increases or decreases below that level, the HRC/triadin interaction is disrupted. Therefore, it appears that the basis of HRC/triadin interaction is electrostatic, and conformational changes of HRC due to Ca2+ binding, may disturb it. Binding of HRC to triadin may affect RyR function, through the quaternary (junctin, triadin, calsequestrin, and RyR) Ca2+-release complex [40,44 ], which in turn could lead to SR Ca2+-release perturbations, with detrimental effects on myocyte Ca2+ cycling and contractility.
Indeed, overexpression of HRC in mouse hearts decreased SR Ca2+-uptake rates, without significant change of the SR Ca2+-load, suggesting a suppressive role for HRC on SERCA pumping [6]. HRC was shown to bind to SERCA2a in mouse and human hearts, implicating HRC as a regulator of SR Ca2+-uptake [5]. The domain of human HRC that binds to SERCA2 encompasses the second glutamic acid- and histidine-rich amino acid region. The minimal domain of SERCA required for binding to HRC contains part of the N-terminal region and is in close proximity to the cation transporter domain [46,47], which is identical in all SERCA1 and SERCA2 isoforms [48–51]. The HRC/SERCA interaction is also Ca2+ sensitive, with maximal binding occurring at very low Ca2+ concentrations [5]. Thus, it may be hypothesized that localized reductions of SR luminal Ca2+ [43], promote HRC binding to SERCA. As the concentration of SR Ca2+ is increased, the HRC/SERCA interaction may get attenuated, resulting in dissociation of SERCA from HRC. The released HRC can then bind to triadin, and regulate Ca2+-release through the quaternary RyR complex. Therefore, HRC may function as a sensor of SR Ca2+ changes, and may regulate both SR Ca2+-release and Ca2+-uptake (Fig. 1). However, future studies are needed to further investigate the reversibility of HRC/SERCA and HRC/triadin interactions in vivo and their functional implications on Ca2+-uptake and Ca2+-release, respectively.
Fig. 1.

Schematic representation of HRC interactions within the SR. The interactions are indicated with a star. HRC could directly bind either the RyR quaternary complex partner triadin or SERCA (upper panel). These interactions are Ca2+-sensitive (lower panel) and HRC may function as a sensor of intra-SR Ca2+-changes, regulating SR Ca2+-release and/or uptake.
Another important finding towards our understanding of the role of HRC in cardiac function is the identification of its interaction with a mutant variant of the junction plakoglobin, namely S39_K40insS, which has been shown to cause Naxos disease [22]. Naxos disease is an inherited disorder of arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C), associated with sudden cardiac death [52]. Plakoglobin is a major component of cell-cell adhesion complexes [53], the desmosomes, abundant in tissues exposed to high levels of mechanical stress, such as epidermis and cardiac muscle [54]. Although wild type plakoglobin does not bind HRC, the S39 K40insS mutant does [22]. Furthermore, bioinformatical analysis of the first 45 amino acid residues of the wild type and mutant plakoglobin, using the SignalP v. 3.0 (http://www.cbs.dtu.dk/services/SignalP/) [55], indicate that addition of an extra serine raises the probability for cleavage at position 40. Thus, mutant plakoglobin may potentially move to the SR and interact with HRC affecting Ca2+-homeostasis and thereby promoting myocyte injury and/or contributing to electrical instability [22]. The role of HRC in malignant ventricular arrhythmias may be similar to calsequestrin [56–62] and RyR [63–65] human mutations, which have been suggested as causative factors of catecholaminergic or polymorphic ventricular tachycardia, and arrhythmogenetic right ventricular cardiomyopathy. The mechanism underlying these malignant arrhythmias includes impaired Ca2+ handling, which predisposes to delayed after depolarizations and triggered activity of arrhythmogenesis.
In conclusion, HRC binds directly to SERCA and triadin through different domains and it may regulate both SR Ca2+-sequestration and Ca2+-release. Thus, HRC may serve as a nodal point bridging these two key Ca2+-processes in the SR. Future studies may be designed to elucidate the functional significance of these interactions under physiological and pathophysiological conditions.
2.4 Phosphorylation of HRC
In vivo phosphoproteomic studies of healthy human skeletal muscle [17] showed that HRC is phosphorylated at six serine amino acid residues, namely Ser119, Ser431, Ser563, Ser567, Ser157/Ser159 and Ser170/Ser171. In addition, the authors predicted that the majority of these sites, (Ser119, Ser170 or Ser171, Ser431 and Ser567), may be phosphorylated by the lumenally located CKII, while Ser157 or Ser159 may be phosphorylated by protein kinase B (PKB), and Ser563 by the mitogen-activated protein kinase 8 (MAPK8) or the glycogen synthase kinase 3 beta (GSK3B). This study clearly demonstrated that HRC is phosphorylated in vivo in healthy human skeletal muscle, but its functional significance, as well as the question for potential phosphorylation aberrations under physiological and pathophysiological conditions, remains to be elucidated. Furthermore, endogenous phosphorylation of HRC by CKII in SR membranes isolated from rabbit fast-twitch skeletal muscle has been described and associated with a decreased affinity of RyR for Ca2+ [13,15]. However, this observed effect on RyR activity may not be the direct outcome of HRC phosphorylation, but the result of other SR protein mediators. Given these limited data, the specific contribution of HRC phosphorylation is not clear and further studies are needed to determine its biological significance and its potential implications on SR Ca2+-uptake or release.
2.5 HRC in regulation of Ca2+-homeostasis and contractility
To elucidate the functional significance of HRC, adenoviral gene transfer was used to overexpress HRC in adult rat cardiomyocytes [20]. Acute overexpression was associated with an increase of SR Ca2+ content, but decreased SR Ca2+-release, resulting in depressed cardiomyocyte contractility. HRC overexpression was accompanied by increased levels of triadin and junctin, whereas RyR, calsequestrin, PLN and SERCA remained unaltered [20]. Importantly, a modest increase of HRC expression had a more pronounced effect on Ca2+ transients and cardiomyocyte contractility than a 20-fold overexpression of calsequestrin [67].
Furthermore, Gregory et al. [6] generated a transgenic mouse model with cardiac specific HRC overexpression (3-fold) to investigate and characterize the role of HRC in vivo. In this model, SR Ca2+-uptake rates were impaired and cardiomyocyte Ca2+ decay was attenuated, without significant alteration in peak Ca2+ transient or SR Ca2+-load. This phenotype was accompanied by increased triadin and NCX protein levels, as well as increased Ca2+ extrusion through the NCX, compared to wild types. Although the total L-type Ca2+-channel (LTCC) levels were unaltered, their current density was increased, constituting a potential compensatory mechanism along with NCX upregulation to maintain SR Ca2+-load in the face of depressed SR Ca2+ transport activity. The chronic overexpression mouse model of HRC exhibited compromised heart response to stress factors, and eventually progressed to hypertrophy and heart failure upon aging. Interestingly, the SR Ca2+-uptake rates were attenuated in HRC overexpressing hearts to a similar extend as in SERCA heterozygous deficient hearts [68,69], indicating a major inhibitory effect of HRC on SERCA activity. This may be due to the binding of HRC with the SERCA domain, which is in close proximity to the cation transporter site [46,47], regulating SR Ca2+-sequestration [70]. Thus, increases in the apparent stoichiometry of HRC/SERCA2 may impair SR Ca2+-cycling. Indeed, the depressed Ca2+-handling in human failing hearts may reflect at least partly, a relative increase in the HRC/SERCA2 levels [20,71], due to a lower reduction in HRC (17%), compared to SERCA2 (40%) levels. Such an apparent increase (1.4 fold) in the relative HRC/SERCA2 protein ratio would contribute to depressed SR Ca2+-uptake and impaired Ca2+-cycling in heart failure.
Interestingly, HRC overexpression was associated with significantly improved recovery of post-ischemic contractile function compared to wild type mouse hearts, while the myocardial infract size was smaller, both ex vivo and in vivo [19]. In addition, the HRC transgenic animals exhibited attenuated ischemia/reperfusion-induced apoptosis, compared to wild types. Since no significant changes were observed in SERCA, RyR, triadin, junctin and calsequestrin, before and after ischemia, a novel role of HRC in the apoptotic machinery was suggested. Indeed, the Bcl-2 protein was significantly increased following reperfusion, whereas the levels of Bax were not altered in HRC overexpressing hearts, which may favour the integrity of mitochondria, repressing mitochondrial-mediated apoptotic and necrotic death pathways. This increase of the Bcl-2/Bax ratio may be the result of the decreased free [Ca2+]SR due to HRC overexpression, which leads to reduced intramitochondrial Ca2+-accumulation. Furthermore, the active caspase-3, caspase-9 and caspase-12 were markedly decreased in transgenic hearts after ischemia/reperfusion, possibly due to increased cytosolic Ca2+-levels [19]. These data suggest that inhibition of apoptotic cell death is the mechanism of protection against cardiac ischemia/reperfusion injury in HRC overexpressing animals.
Recently, an HRC knock-out mouse model was generated [72]. The HRC knock-out mice exhibited impaired weight gain, with significant reduction in muscle mass, and triadin protein overexpression. Furthermore, this model was sensitive to isoproterenol-induced cardiac hypertrophy. Thus, alterations in HRC levels, combined with other genetic or environmental factors, may contribute to pathological hypertrophy and heart failure.
It should be noted that the majority of HRC studies in animal models predominantly focused on the role of HRC in cardiac contractility and its potential involvement in heart failure. However, since recent evidence has linked HRC to fatal arrhythmias in human heart failure patients, it becomes important to design future studies addressing the functional significance of HRC in arrhythmogenesis (see below).
2.6 Genetic variants of HRC and heart disease
Several signalling pathways are involved in the induction of pathologic remodelling and heart failure, and many of these pathways are linked to cardiac SR Ca2+-cycling. The failing heart is characterized by impaired removal of cytosolic Ca2+, reduced loading of the SR, and defective SR Ca2+-release, culminating in impaired cardiac diastolic and systolic function [73]. Given the properties of HRC, alterations in its expression levels, function, localization, and/or regulation may disturb intracellular Ca2+-homeostasis, leading to the development or altered progression of heart failure. Indeed, Fan et al. [20] reported decreased HRC protein levels in both human and experimental heart failure in mice. Decreased HRC levels were also found in a canine model of chronic heart failure while calsequestrin levels remained unaltered, compared to healthy controls [74].
In inbred mouse strains, the HRC gene has been proposed to be a candidate responsible for dystrophic cardiac calcification [75]. The working hypothesis is that aberrant expression or mutation of HRC may affect Ca2+-homeostasis, resulting in increased fluctuation or average concentration of Ca2+ within the cell, and thus contribute to the pathology of dystrophic cardiac calcification. In humans, the HRC gene was mapped within the myotonic dystrophy associated locus, and naturally occurring genetic variants have been described [23]. Since an impairment of Ca2+-handling and excitation-contraction coupling may lead to myotonia, a critical role for HRC in human cardiac disease was also suggested. Furthermore, two independent linkage analysis studies for isolated cardiac conduction and progressive familial heart block type 1 diseases, mapped the candidate gene close to the HRC locus [76,77]. Both diseases are characterized by right or left bundle branch block and QRS widening, that may progress to complete atrioventricular block with syncope and sudden cardiac death. Although the connection of isolated cardiac conduction disease with HRC is well established [76], it is not currently clear whether isolated cardiac conduction and progressive familial heart block type 1 diseases may map to the same locus [77].
Recently, a genetic variant of HRC, namely Ser96Ala, showed a statistically significant association with malignant ventricular arrhythmias and sudden cardiac death in a well-characterized cohort of patients with idiopathic dilated cardiomyopathy [21]. Kaplan-Meier survival curves during follow up of these patients indicated a markedly increased number of malignant arrhythmia episodes for the Ala/Ala homozygous patients, when compared with the Ser/Ala heterozygotes and Ser/Ser homozygotes during the first two years of the follow up study (Fig. 2). This finding was independent of medication and the patients’ clinical characteristics, such as atrial fibrillation or left bundle branch block. The mechanism may be associated with impaired phosphorylation at Ser96 by casein kinase II. The abolishment of the phosphorylation site could affect HRC interactions with SERCA2 and/or triadin [5], resulting in RyR destabilization and exacerbation of spontaneous Ca2+-release and delayed after depolarizations, leading to lethal arrhythmias in idiopathic dilated cardiomyopathy. Future studies in isolated cardiomyocytes and transgenic mouse models will further explore the underlying mechanism of ventricular arrhythmia susceptibility in Ala/Ala homozygous dilated cardiomyopathy patients.
Fig. 2.
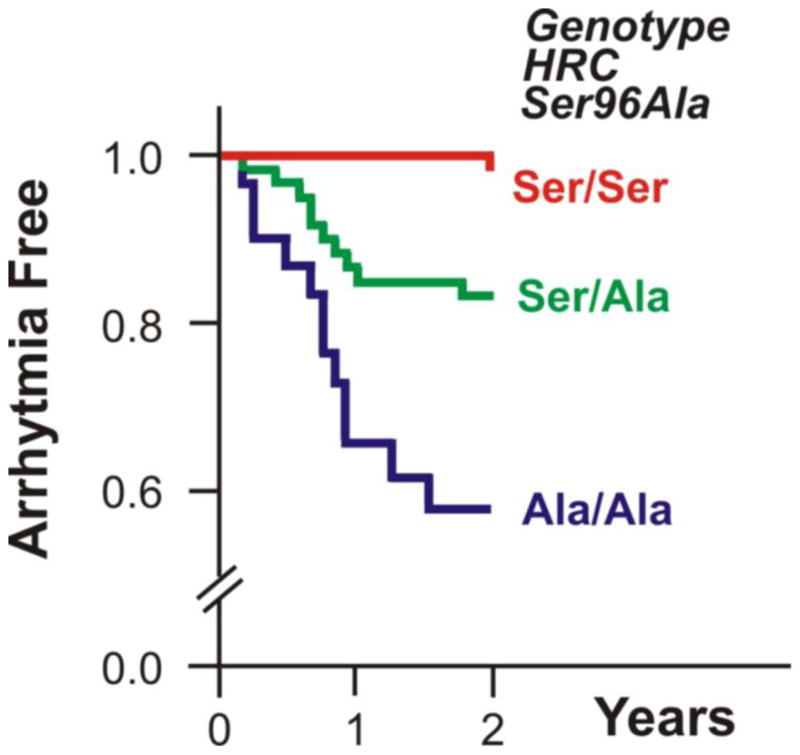
Two year Kaplan-Meier plots for the probability of survival of idiopathic dilated cardiomyopathy (DCM) patients from life-threatening ventricular arrhythmic events, defined as sudden cardiac death, episodes of unstable ventricular tachycardia or ventricular fibrillation. The Ala/Ala-HRC homozygous dilated cardiomyopathy patients were more susceptible to ventricular arrhythmic events, compared with the Ser/Ala-HRC heterozygotes and Ser/Ser-HRC homozygotes.
3. Conclusions and future perspective
Increasing evidence suggests that the impaired function of the SR, consisting of Ca2+-uptake, storage, and release, is a critical characteristic of failing hearts. HRC holds an important role in cardiomyocyte physiology, mediating a fine cross-talk between SR Ca2+-uptake and release, through its direct interactions with SERCA and triadin. Thus, HRC may act as an intra-SR Ca2+-sensor that mediates different responses upon luminal SR Ca2+-fluctuations. Although HRC overexpression is associated with cardiac dysfunction, the protein appears to have a cardioprotective effect after ischemic episodes. The recent identification of the Ser96Ala HRC genetic variant as an independent predictor of susceptibility to arrhythmogenesis in the setting of dilated cardiomyopathy, points to the importance of HRC in cardiac function that merits careful consideration and further analysis under physiological and pathophysiological conditions.
Future studies need to explore the HRC role in myocytes at multiple levels. Biophysical analysis of the three-dimensional structure of HRC protein under different Ca2+ and phosphorylation conditions will allow the identification of altered folding patterns and interactions with protein partners. Furthermore, it would be interesting to explore whether the HRC/SERCA and HRC/triadin interactions are reversible in vivo, under different Ca-luminal conditions. In addition, investigation of the HRC transcriptional regulation during myocyte differentiation and heart disease may reveal important insights in its functional significance. Finally, exploring the mechanisms underlying the Ser96Ala HRC genetic variants and the susceptibility to malignant arrhythmias may provide new insights to pathways of excitation-contraction coupling and action potential control, leading to the development of novel clinical interventions.
Acknowledgments
This work was supported by the European Community’s Seventh Framework Programme FP7/2007–2013 under grant agreement No. HEALTH-F2-2009-241526, EUTrigTreat; the Biomedical Research Foundation, Academy of Athens, the John F. Kostopoulos Foundation, the Hellenic Cardiological Society, the National Institutes of Health, (HL26057, HL64018 and HL77101), the Leducq Foundation Trans-Atlantic alliance and the European Community’s Sixth Framework Programme FP6 under grant agreement #LSHG-CT-2006-037277, VALAPODYN.
Footnotes
DISCLOSURES
No Disclosures
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hasenfuss G. Alterations of calcium-regulatory proteins in heart failure. Cardiovasc Res. 1998;37:279–89. doi: 10.1016/s0008-6363(97)00277-0. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt AG, Edes I, Kranias EG. Phospholamban: a promising therapeutic target in heart failure? Cardiovasc Drugs Ther. 2001;15:387–96. doi: 10.1023/a:1013381204658. [DOI] [PubMed] [Google Scholar]
- 3.Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15:380–3. doi: 10.1038/nm.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hofmann SL, Brown MS, Lee E, Pathak RK, Anderson RG, Goldstein JL. Purification of a sarcoplasmic reticulum protein that binds Ca2+ and plasma lipoproteins. J Biol Chem. 1989;264:8260–70. [PubMed] [Google Scholar]
- 5.Arvanitis DA, Vafiadaki E, Fan GC, Mitton BA, Gregory KN, Del Monte F, et al. Histidine-rich Ca-binding protein interacts with sarcoplasmic reticulum Ca-ATPase. Am J Physiol Heart Circ Physiol. 2007;293:H1581–9. doi: 10.1152/ajpheart.00278.2007. [DOI] [PubMed] [Google Scholar]
- 6.Gregory KN, Ginsburg KS, Bodi I, Hahn H, Marreez YM, Song Q, et al. Histidine-rich Ca binding protein: a regulator of sarcoplasmic reticulum calcium sequestration and cardiac function. J Mol Cell Cardiol. 2006;40:653–65. doi: 10.1016/j.yjmcc.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Kim E, Shin DW, Hong CS, Jeong D, Kim DH, Park WJ. Increased Ca2+ storage capacity in the sarcoplasmic reticulum by overexpression of HRC (histidine-rich Ca2+ binding protein) Biochem Biophys Res Commun. 2003;300:192–6. doi: 10.1016/s0006-291x(02)02829-2. [DOI] [PubMed] [Google Scholar]
- 8.Lee HG, Kang H, Kim DH, Park WJ. Interaction of HRC (histidine-rich Ca(2+)-binding protein) and triadin in the lumen of sarcoplasmic reticulum. J Biol Chem. 2001;276:39533–8. doi: 10.1074/jbc.M010664200. [DOI] [PubMed] [Google Scholar]
- 9.Sacchetto R, Damiani E, Turcato F, Nori A, Margreth A. Ca(2+)-dependent interaction of triadin with histidine-rich Ca(2+)-binding protein carboxyl-terminal region. Biochem Biophys Res Commun. 2001;289:1125–34. doi: 10.1006/bbrc.2001.6126. [DOI] [PubMed] [Google Scholar]
- 10.Campbell KP, MacLennan DH, Jorgensen AO. Staining of the Ca2+-binding proteins, calsequestrin, calmodulin, troponin C, and S-100, with the cationic carbocyanine dye “Stains-all”. J Biol Chem. 1983;258:11267–73. [PubMed] [Google Scholar]
- 11.Damiani E, Margreth A. Subcellular fractionation to junctional sarcoplasmic reticulum and biochemical characterization of 170 kDa Ca(2+)- and low-density-lipoprotein-binding protein in rabbit skeletal muscle. Biochem J. 1991;277 (Pt 3):825–32. doi: 10.1042/bj2770825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damiani E, Tarugi P, Calandra S, Margreth A. Sequential expression during postnatal development of specific markers of junctional and free sarcoplasmic reticulum in chicken pectoralis muscle. Dev Biol. 1992;153:102–14. doi: 10.1016/0012-1606(92)90095-x. [DOI] [PubMed] [Google Scholar]
- 13.Orr I, Shoshan-Barmatz V. Modulation of the skeletal muscle ryanodine receptor by endogenous phosphorylation of 160/150-kDa proteins of the sarcoplasmic reticulum. Biochim Biophys Acta. 1996;1283:80–8. doi: 10.1016/0005-2736(96)00078-8. [DOI] [PubMed] [Google Scholar]
- 14.Picello E, Damiani E, Margreth A. Low-affinity Ca(2+)-binding sites versus Zn(2+)-binding sites in histidine-rich Ca(2+)-binding protein of skeletal muscle sarcoplasmic reticulum. Biochem Biophys Res Commun. 1992;186:659–67. doi: 10.1016/0006-291x(92)90797-o. [DOI] [PubMed] [Google Scholar]
- 15.Shoshan-Barmatz V, Orr I, Weil S, Meyer H, Varsanyi M, Heilmeyer LM. The identification of the phosphorylated 150/160-kDa proteins of sarcoplasmic reticulum, their kinase and their association with the ryanodine receptor. Biochim Biophys Acta. 1996;1283:89–100. doi: 10.1016/0005-2736(96)00079-x. [DOI] [PubMed] [Google Scholar]
- 16.Sacchetto R, Turcato F, Damiani E, Margreth A. Interaction of triadin with histidine-rich Ca(2+)-binding protein at the triadic junction in skeletal muscle fibers. J Muscle Res Cell Motil. 1999;20:403–15. doi: 10.1023/a:1005580609414. [DOI] [PubMed] [Google Scholar]
- 17.Hojlund K, Bowen BP, Hwang H, Flynn CR, Madireddy L, Geetha T, et al. In vivo phosphoproteome of human skeletal muscle revealed by phosphopeptide enrichment and HPLC-ESI-MS/MS. J Proteome Res. 2009;8:4954–65. doi: 10.1021/pr9007267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bousette N, Kislinger T, Fong V, Isserlin R, Hewel JA, Emil A, et al. Large-scale characterization and analysis of the murine cardiac proteome. J Proteome Res. 2009;8:1887–901. doi: 10.1021/pr800845a. [DOI] [PubMed] [Google Scholar]
- 19.Zhou X, Fan GC, Ren X, Waggoner JR, Gregory KN, Chen G, et al. Overexpression of histidine-rich Ca-binding protein protects against ischemia/reperfusion-induced cardiac injury. Cardiovasc Res. 2007;75:487–97. doi: 10.1016/j.cardiores.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 20.Fan GC, Gregory KN, Zhao W, Park WJ, Kranias EG. Regulation of myocardial function by histidine-rich, calcium-binding protein. Am J Physiol Heart Circ Physiol. 2004;287:H1705–11. doi: 10.1152/ajpheart.01211.2003. [DOI] [PubMed] [Google Scholar]
- 21.Arvanitis DA, Sanoudou D, Kolokathis F, Vafiadaki E, Papalouka V, Kontrogianni-Konstantopoulos A, et al. The Ser96Ala variant in histidine-rich calcium-binding protein is associated with life-threatening ventricular arrhythmias in idiopathic dilated cardiomyopathy. Eur Heart J. 2008;29:2514–25. doi: 10.1093/eurheartj/ehn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, McKenna WJ. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2007;81:964–73. doi: 10.1086/521633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hofmann SL, Topham M, Hsieh CL, Francke U. cDNA and genomic cloning of HRC, a human sarcoplasmic reticulum protein, and localization of the gene to human chromosome 19 and mouse chromosome 7. Genomics. 1991;9:656–69. doi: 10.1016/0888-7543(91)90359-m. [DOI] [PubMed] [Google Scholar]
- 24.Brown SD, Chartier F, Johnson K, Cavanna JS. Mapping the Hrc gene to proximal mouse chromosome 7: delineation of a conserved linkage group with human 19q. Genomics. 1993;18:459–61. doi: 10.1006/geno.1993.1502. [DOI] [PubMed] [Google Scholar]
- 25.Raeymaekers L, Verbist J, Wuytack F, Plessers L, Casteels R. Expression of Ca2+ binding proteins of the sarcoplasmic reticulum of striated muscle in the endoplasmic reticulum of pig smooth muscles. Cell Calcium. 1993;14:581–9. doi: 10.1016/0143-4160(93)90058-e. [DOI] [PubMed] [Google Scholar]
- 26.Ridgeway AG, Petropoulos H, Siu A, Ball JK, Skerjanc IS. Cloning, tissue distribution, subcellular localization and overexpression of murine histidine-rich Ca2+ binding protein. FEBS Lett. 1999;456:399–402. doi: 10.1016/s0014-5793(99)00993-x. [DOI] [PubMed] [Google Scholar]
- 27.Zarain-Herzberg A, MacLennan DH, Periasamy M. Characterization of rabbit cardiac sarco(endo)plasmic reticulum Ca2(+)-ATPase gene. J Biol Chem. 1990;265:4670–7. [PubMed] [Google Scholar]
- 28.Takeshima H, Nishimura S, Matsumoto T, Ishida H, Kangawa K, Minamino N, et al. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature. 1989;339:439–45. doi: 10.1038/339439a0. [DOI] [PubMed] [Google Scholar]
- 29.Ellis SB, Williams ME, Ways NR, Brenner R, Sharp AH, Leung AT, et al. Sequence and expression of mRNAs encoding the alpha 1 and alpha 2 subunits of a DHP-sensitive calcium channel. Science. 1988;241:1661–4. doi: 10.1126/science.2458626. [DOI] [PubMed] [Google Scholar]
- 30.Korczak B, Zarain-Herzberg A, Brandl CJ, Ingles CJ, Green NM, MacLennan DH. Structure of the rabbit fast-twitch skeletal muscle Ca2+-ATPase gene. J Biol Chem. 1988;263:4813–9. [PubMed] [Google Scholar]
- 31.Zarain-Herzberg A, Fliegel L, MacLennan DH. Structure of the rabbit fast-twitch skeletal muscle calsequestrin gene. J Biol Chem. 1988;263:4807–12. [PubMed] [Google Scholar]
- 32.Anderson JP, Dodou E, Heidt AB, De Val SJ, Jaehnig EJ, Greene SB, et al. HRC is a direct transcriptional target of MEF2 during cardiac, skeletal, and arterial smooth muscle development in vivo. Mol Cell Biol. 2004;24:3757–68. doi: 10.1128/MCB.24.9.3757-3768.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guinamard R, Chatelier A, Demion M, Potreau D, Patri S, Rahmati M, et al. Functional characterization of a Ca(2+)-activated non-selective cation channel in human atrial cardiomyocytes. J Physiol. 2004;558:75–83. doi: 10.1113/jphysiol.2004.063974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kruse M, Schulze-Bahr E, Corfield V, Beckmann A, Stallmeyer B, Kurtbay G, et al. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J Clin Invest. 2009;119:2737–44. doi: 10.1172/JCI38292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beranger F, Mejean C, Moniot B, Berta P, Vandromme M. Muscle differentiation is antagonized by SOX15, a new member of the SOX protein family. J Biol Chem. 2000;275:16103–9. doi: 10.1074/jbc.275.21.16103. [DOI] [PubMed] [Google Scholar]
- 36.Maruyama M, Ichisaka T, Nakagawa M, Yamanaka S. Differential roles for Sox15 and Sox2 in transcriptional control in mouse embryonic stem cells. J Biol Chem. 2005;280:24371–9. doi: 10.1074/jbc.M501423200. [DOI] [PubMed] [Google Scholar]
- 37.Lee HJ, Goring W, Ochs M, Muhlfeld C, Steding G, Paprotta I, et al. Sox15 is required for skeletal muscle regeneration. Mol Cell Biol. 2004;24:8428–36. doi: 10.1128/MCB.24.19.8428-8436.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong S, Kim TW, Choi I, Woo JM, Oh J, Park WJ, et al. Complementary DNA cloning, genomic characterization and expression analysis of a mammalian gene encoding histidine-rich calcium binding protein. Biochim Biophys Acta. 2005;1727:188–96. doi: 10.1016/j.bbaexp.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 39.Wang S, Trumble WR, Liao H, Wesson CR, Dunker AK, Kang CH. Crystal structure of calsequestrin from rabbit skeletal muscle sarcoplasmic reticulum. Nat Struct Biol. 1998;5:476–83. doi: 10.1038/nsb0698-476. [DOI] [PubMed] [Google Scholar]
- 40.Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem. 1997;272:23389–97. doi: 10.1074/jbc.272.37.23389. [DOI] [PubMed] [Google Scholar]
- 41.Nayler WG, Anderson JE. Effects of Zinc on Cardiac Muscle Contraction. Am J Physiol. 1965;209:17–21. doi: 10.1152/ajplegacy.1965.209.1.17. [DOI] [PubMed] [Google Scholar]
- 42.Suk JY, Kim YS, Park WJ. HRC (histidine-rich Ca2+ binding protein) resides in the lumen of sarcoplasmic reticulum as a multimer. Biochem Biophys Res Commun. 1999;263:667–71. doi: 10.1006/bbrc.1999.1432. [DOI] [PubMed] [Google Scholar]
- 43.Shannon TR, Guo T, Bers DM. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ Res. 2003;93:40–5. doi: 10.1161/01.RES.0000079967.11815.19. [DOI] [PubMed] [Google Scholar]
- 44.Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–8. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo W, Campbell K. Association of Triadin with the Ryanodine Receptor and Calsequestrin in the Lumen of the Sarcoplasmic Reticulum. J Biological Chemistry. 1995;270:9027–9030. doi: 10.1074/jbc.270.16.9027. [DOI] [PubMed] [Google Scholar]
- 46.Toyoshima C, Mizutani T. Crystal structure of the calcium pump with a bound ATP analogue. Nature. 2004;430:529–35. doi: 10.1038/nature02680. [DOI] [PubMed] [Google Scholar]
- 47.Toyoshima C, Nomura H, Tsuda T. Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature. 2004;432:361–8. doi: 10.1038/nature02981. [DOI] [PubMed] [Google Scholar]
- 48.Dally S, Bredoux R, Corvazier E, Andersen JP, Clausen JD, Dode L, et al. Ca2+-ATPases in non-failing and failing heart: evidence for a novel cardiac sarco/endoplasmic reticulum Ca2+-ATPase 2 isoform (SERCA2c) Biochem J. 2006;395:249–58. doi: 10.1042/BJ20051427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kimura T, Nakamori M, Lueck JD, Pouliquin P, Aoike F, Fujimura H, et al. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum Mol Genet. 2005;14:2189–200. doi: 10.1093/hmg/ddi223. [DOI] [PubMed] [Google Scholar]
- 50.Lytton J, MacLennan DH. Molecular cloning of cDNAs from human kidney coding for two alternatively spliced products of the cardiac Ca2+-ATPase gene. J Biol Chem. 1988;263:15024–31. [PubMed] [Google Scholar]
- 51.Zarain-Herzberg A, Alvarez-Fernandez G. Sarco(endo)plasmic reticulum Ca2+-ATPase-2 gene: structure and transcriptional regulation of the human gene. Scientific World Journal. 2002;2:1469–83. doi: 10.1100/tsw.2002.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Protonotarios N, Tsatsopoulou A, Patsourakos P, Alexopoulos D, Gezerlis P, Simitsis S, et al. Cardiac abnormalities in familial palmoplantar keratosis. Br Heart J. 1986;56:321–6. doi: 10.1136/hrt.56.4.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rubenstein A, Merriam J, Klymkowsky MW. Localizing the adhesive and signaling functions of plakoglobin. Dev Genet. 1997;20:91–102. doi: 10.1002/(SICI)1520-6408(1997)20:2<91::AID-DVG2>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 54.Green KJ, Gaudry CA. Are desmosomes more than tethers for intermediate filaments? Nat Rev Mol Cell Biol. 2000;1:208–16. doi: 10.1038/35043032. [DOI] [PubMed] [Google Scholar]
- 55.Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2:953–71. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 56.Terentyev D, Viatchenko-Karpinski S, Gyorke I, Volpe P, Williams SC, Gyorke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: Mechanism for hereditary arrhythmia. Proc Natl Acad Sci U S A. 2003;100:11759–64. doi: 10.1073/pnas.1932318100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Viatchenko-Karpinski S, Terentyev D, Gyorke I, Terentyeva R, Volpe P, Priori SG, et al. Abnormal calcium signaling and sudden cardiac death associated with mutation of calsequestrin. Circ Res. 2004;94:471–7. doi: 10.1161/01.RES.0000115944.10681.EB. [DOI] [PubMed] [Google Scholar]
- 58.Lahat H, Pras E, Eldar M. A missense mutation in CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Ann Med. 2004;36 (Suppl 1):87–91. doi: 10.1080/17431380410032517. [DOI] [PubMed] [Google Scholar]
- 59.Eldar M, Pras E, Lahat H. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Cold Spring Harb Symp Quant Biol. 2002;67:333–7. doi: 10.1101/sqb.2002.67.333. [DOI] [PubMed] [Google Scholar]
- 60.Eldar M, Pras E, Lahat H. A missense mutation in the CASQ2 gene is associated with autosomal-recessive catecholamine-induced polymorphic ventricular tachycardia. Trends Cardiovasc Med. 2003;13:148–51. doi: 10.1016/s1050-1738(03)00025-2. [DOI] [PubMed] [Google Scholar]
- 61.Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378–84. doi: 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song L, Alcalai R, Arad M, Wolf CM, Toka O, Conner DA, et al. Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2007;117:1814–23. doi: 10.1172/JCI31080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 64.Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:485–90. doi: 10.1161/01.cir.103.4.485. [DOI] [PubMed] [Google Scholar]
- 65.Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2) Hum Mol Genet. 2001;10:189–94. doi: 10.1093/hmg/10.3.189. [DOI] [PubMed] [Google Scholar]
- 66.Damiani E, Picello E, Saggin L, Margreth A. Identification of triadin and of histidine-rich Ca(2+)-binding protein as substrates of 60 kDa calmodulin-dependent protein kinase in junctional terminal cisternae of sarcoplasmic reticulum of rabbit fast muscle. Biochem Biophys Res Commun. 1995;209:457–65. doi: 10.1006/bbrc.1995.1524. [DOI] [PubMed] [Google Scholar]
- 67.Sato Y, Ferguson DG, Sako H, Dorn GW, 2nd, Kadambi VJ, Yatani A, et al. Cardiac-specific overexpression of mouse cardiac calsequestrin is associated with depressed cardiovascular function and hypertrophy in transgenic mice. J Biol Chem. 1998;273:28470–7. doi: 10.1074/jbc.273.43.28470. [DOI] [PubMed] [Google Scholar]
- 68.Ji Y, Lalli MJ, Babu GJ, Xu Y, Kirkpatrick DL, Liu LH, et al. Disruption of a single copy of the SERCA2 gene results in altered Ca2+ homeostasis and cardiomyocyte function. J Biol Chem. 2000;275:38073–80. doi: 10.1074/jbc.M004804200. [DOI] [PubMed] [Google Scholar]
- 69.Periasamy M, Reed TD, Liu LH, Ji Y, Loukianov E, Paul RJ, et al. Impaired cardiac performance in heterozygous mice with a null mutation in the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 (SERCA2) gene. J Biol Chem. 1999;274:2556–62. doi: 10.1074/jbc.274.4.2556. [DOI] [PubMed] [Google Scholar]
- 70.Gregory KN, Ginsburg KS, Bodi I, Hahn H, Marreez YM, Song Q, et al. Histidine-rich Ca binding protein: a regulator of sarcoplasmic reticulum calcium sequestration and cardiac function. J Mol Cell Cardiol. 2006;40:653–65. doi: 10.1016/j.yjmcc.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 71.Dash R, Frank KF, Carr AN, Moravec CS, Kranias EG. Gender influences on sarcoplasmic reticulum Ca2+-handling in failing human myocardium. J Mol Cell Cardiol. 2001;33:1345–53. doi: 10.1006/jmcc.2001.1394. [DOI] [PubMed] [Google Scholar]
- 72.Jaehnig EJ, Heidt AB, Greene SB, Cornelissen I, Black BL. Increased susceptibility to isoproterenol-induced cardiac hypertrophy and impaired weight gain in mice lacking the histidine-rich calcium-binding protein. Mol Cell Biol. 2006;26:9315–26. doi: 10.1128/MCB.00482-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morgan JP, Erny RE, Allen PD, Grossman W, Gwathmey JK. Abnormal intracellular calcium handling, a major cause of systolic and diastolic dysfunction in ventricular myocardium from patients with heart failure. Circulation. 1990;81:III21–32. [PubMed] [Google Scholar]
- 74.Gupta RC, Mishra S, Rastogi S, Wang M, Rousso B, Mika Y, et al. Ca(2+)-binding proteins in dogs with heart failure: effects of cardiac contractility modulation electrical signals. Clin Transl Sci. 2009;2:211–5. doi: 10.1111/j.1752-8062.2009.00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van den Broek FA, Bakker R, den Bieman M, Fielmich-Bouwman AX, Lemmens AG, van Lith HA, et al. Genetic analysis of dystrophic cardiac calcification in DBA/2 mice. Biochem Biophys Res Commun. 1998;253:204–8. doi: 10.1006/bbrc.1998.9776. [DOI] [PubMed] [Google Scholar]
- 76.de Meeus A, Stephan E, Debrus S, Jean MK, Loiselet J, Weissenbach J, et al. An isolated cardiac conduction disease maps to chromosome 19q. Circ Res. 1995;77:735–40. doi: 10.1161/01.res.77.4.735. [DOI] [PubMed] [Google Scholar]
- 77.Brink PA, Ferreira A, Moolman JC, Weymar HW, van der Merwe PL, Corfield VA. Gene for progressive familial heart block type I maps to chromosome 19q13. Circulation. 1995;91:1633–40. doi: 10.1161/01.cir.91.6.1633. [DOI] [PubMed] [Google Scholar]