

Abstract
Otitis media (OM) is the most prevalent childhood disease in developed countries. Involvement of innate immunity mediated by Toll-like receptors (TLRs) in OM has been implicated primarily in cell lines and by association studies of innate immune gene polymorphisms with OM prevalence. However, the precise role of innate immunity in OM is incompletely understood. We review recent research that has advanced our understanding of how innate immunity in the middle ear is mediated by the interaction of pathogen molecules with receptors such as the TLRs, leading to the activation of adaptor molecules and production of proinflammatory cytokines. TLR genes and signaling molecules are upregulated in OM in a murine model. Deletion of several key innate immune genes results in persistent OM in mice, coupled with an inability to clear bacterial infection from the middle ear. It is concluded that an intact innate immune signaling system is critical to recovery from bacterial OM.
Keywords: Pathogenesis, Innate immunity, Otitis media, TLR, TNF
Introduction
Otitis media (OM) is one of the most prevalent diseases of childhood and a major public health problem contributing to childhood mortality in developing countries [1]. In more developed societies, OM accounts for more office visits and drug purchases than any other childhood disease and has an estimated annual cost of more than 5 billion dollars [2, 3].
Acute OM typically resolves swiftly, but patients suffering from chronic, recurrent OM may suffer long-term conductive hearing loss and permanent hearing loss due to scarring of the middle ear conductive apparatus and sensorineural damage to the inner ear. The reasons for the chronicity of middle ear disease in 10% to 20% of children are not clear. However, although many factors can contribute to OM [4, 5], bacterial infection is considered to be a dominant etiology, producing inflammation characterized by mucosal hyperplasia, effusion, and leukocytic infiltration of the middle ear [6, 7].
The most commonly isolated bacteria in OM are nontypeable Haemophilus influenzae, Streptococcus pneumoniae, and Moraxella catarrhalis. Since the advent of pneumococcal vaccines, nontypeable H. influenzae has become the most prevalent of these [8]. Bacteria, including nontypeable H. influenzae, activate innate immune receptors that are considered a key component of innate immunity, providing the first line of host defense against infection [9, 10]. Whereas middle ear responses to innate immune ligands such as lipopolysaccharide (LPS) and bacterial lipoproteins are well-documented, the role played by innate immune receptors and their downstream signaling molecules in OM is not yet fully understood. The present review focuses on the role of innate immunity in OM pathogenesis and recovery against the background that Toll-like receptor (TLR) deficiencies have been shown to induce significant abnormalities in the recovery from bacterial infection at other locations [11, 12].
Otitis Media Treatment
As noted above, OM is a very common condition, but optimal treatment is currently under considerable discussion. In uncomplicated, acute OM, antibiotic therapy is no longer recommended [13] because despite short-term benefit, evidence-based studies show only minimal long-term improvement in the recovery rate [14]. Only for more serious, exacerbated OM is antibiotic therapy currently prescribed. Persistent, recurrent, and chronic forms of OM affect about 15% of children and represent a more difficult problem. Prophylactic antibiotic treatment may fail to resolve chronic OM and increases the burden of antibiotic resistance to therapy [15]. Ventilation tubes can be effective in the treatment of chronic/recurrent OM, but they are not always effective and there is controversy regarding long-term benefit and the potential for late adverse outcomes such as tympanic membrane abnormalities.
Etiology and Pathogenesis of Otitis Media
The development of more effective treatments is complicated by the fact that OM seems to be a multifactorial disease. Obstruction of the eustachian tube with resultant negative pressure and exudation of serum seems to be a major factor [16]. Bacterial infection via the eustachian tube, perhaps facilitated by the presence of serum as a growth medium, is also well-recognized as a component in this complex etiology. OM is strongly associated with prior upper respiratory viral infection [15], which increases the likelihood of subsequent bacterial middle ear infection for reasons that are unclear but could include increased bacterial adherence in the nasopharynx and reduced ciliary activity in the eustachian tube. Inflammatory reactions associated with immune response to bacterial antigens, bacterial polysaccharides, and disorders of inflammatory pathways, such as the cytokine network and the complement cascade, may also contribute to OM pathogenesis, especially in its chronic forms. The complex etiology of OM may explain why susceptibility and treatment outcomes can be so variable, and why OM is self-limiting in most children but becomes persistent in others. However, the central role of infection in OM suggests that immunity may be a key to OM resistance and resolution.
Immunity can involve the sensitization of lymphocytes to recognize particular molecular patterns, or epitopes, a process known as cognate immunity. After sensitization, these cells can mount an efficient adaptive immune response whenever the epitope is subsequently encountered. However, another form of immunity, termed innate immunity, does not require prior experience. A significant portion of such innate immunity is mediated by cellular receptors that are genetically encoded to respond to specific types of molecules produced by pathogens. Because they operate without the need for prior sensitization, these receptors form a critical first line of defense against infection.
Immune Response in the Middle Ear
Resistance to infection of the middle ear is the basis for avoidance of OM and, if infection ensues, recovery from middle ear disease. As at other locations, such resistance entails a cooperative response involving innate and adaptive immunity. However, as very few lymphocytes are found in the healthy middle ear, innate immunity presumably mediates the initial host response to and defense against infection [16]. This allows the middle ear to clear many infections rapidly, before significant cognate immune response has the opportunity to develop. This is especially important in the middle ear, as adaptive immunity appears to be poorly sensitized by antigen that is first presented at this site [16]. In contrast, existing systemic immunity is very strongly expressed in the middle ear. In animals that have been systemically immunized, antigenic challenge of the middle ear triggers an immediate and massive middle ear response of IgG. This is associated with a transudation of serum and infiltration by lymphocytes from peripheral blood [16]. However, in a naïve host, until an adaptive immune response is finally generated, innate immune responses bear the burden of protecting the middle ear against bacterial invasion. Moreover, it also has been shown at other locations that signaling through innate immune receptors is required for the effective sensitization needed to generate adaptive immunity [17•]. It can be presumed that this is also true in the middle ear, and deficiencies in innate immunity might be expected to lead to increased incidence, severity, and duration of OM.
Innate Immune Signaling
Multiple cell types, particularly those that interact with the external environment, express innate immune receptors, including the TLRs. TLRs are found on or within surface epithelial cells, mast cells, and dendritic cells, which are prominent at mucosal surfaces such as that lining the middle ear. TLRs are members of the pattern-recognition receptors family that detect specific molecules associated with microbial pathogens. Their activation leads to the mobilization of other innate immune molecules such as cytokines, chemokines, and interferons (IFNs), as well as proteases, defensins, collectins, lysozyme, lactoferrin, and other antimicrobial intermediates.
TLRs are evolutionally conserved and act as the first line of host defense against infection. In general, TLRs belong to the family of type I transmembrane receptors, which are characterized by an N-terminal extracellular leucine-rich repeat domain and a cytoplasmic Toll/interleukin (IL)-1 receptor (TIR) domain [18]. To date, 13 TLRs have been identified in mammalian organisms, each of which seems to mediate distinct functions in pathogen recognition. TLR activation results in the recruitment and activation of adaptor molecules, which include myeloid differentiation factor-88 (MyD88) and TIR domain containing adaptor inducing IFN-β (TRIF). All TLRs except TLR3 can signal via the MydD88-dependent pathway, leading, via nuclear factor-κB (NF-κB) activation, to the production of proinflammatory cytokines such as tumor necrosis factor (TNF) and the ILs. The MydD88-independent TRIF pathway is dominated by the production of type I IFNs via IFN regulatory factor-3, although it also induces delayed cytokine gene expression [19]. These mediators are key components of the innate immune response and prepare to initiate the subsequent adaptive immune responses via leukocyte chemotaxis and activation of lymphocytes.
Toll-Like Receptors in Otitis Media
Nontypeable H. influenzae, one of the most common isolated pathogens of human OM, activates TLR signaling pathways. Nontypeable H. influenzae contains molecular patterns that serve as ligands for several TLRs. Peptidoglycans and peptidoglycan-associated proteins, such as outer membrane protein P6, are ligands for TLR2 [20], which frequently forms heterodimers with TLR1 or TLR6. Lipooligosaccharide, closely related to LPS, activates cellular signals via TLR2 and TLR4 [21]. Injection of peptidoglycan or LPS into the middle ear can mimic pathological changes of OM: mucosal inflammation, leukocytosis, edema, middle ear pressure abnormalities, and an infiltrate of macrophages into the subepithelial space [22, 23]. Whereas middle ear responses to these TLR2 and TLR4 ligands have long been recognized, the role played by TLR receptors in infection-mediated OM pathogenesis has only recently been addressed.
For example, an association between polymorphisms in TLRs and their co-receptor, CD14, with OM proneness has been described in children [24, 25]. Polymorphisms in the anti-inflammatory cytokine IL-10, which often opposes the proinflammatory results of TLR signaling, are also associated with increased incidence of OM [24].
In middle ear epithelial cell lines, nontypeable H. influenzae induces TLR2 expression [20], and the activation of TLR2 leads to the expression of proinflammatory cytokines and mucin genes [26, 27].
Experimentally, reduced short-term responses to nontypeable H. influenzae from 6 h to 3 days were reported in the middle ears of C3H/HeJ mice, which express a nonfunctional TLR4 [28]. However, because TLR deficiencies have been shown to induce significant abnormalities in the recovery from bacterial infection at other sites [11, 12], longer-term studies of nontypeable H. influenzae-induced OM were undertaken. They demonstrated that TLR signaling is critical for efficient host defense of the middle ear.
Genes involved in innate immune responses were identified that, when absent, result in transition from the self-limiting characteristic of acute OM to persistent middle ear disease in mice [29, 30]. These genes include those encoding members of the TLR family itself, such as TLR2 and TLR4. In addition, defects in genes for the TLR signaling molecules MyD88 and TRIF, as well as the important downstream target molecule TNF, result in abnormal OM recovery. The effects of these gene defects underscore the importance of innate immune responses to the normal processes of OM resolution.
Altered Otitis Media in Toll-Like Receptor-Deficient Mice
Recent data illustrate a critical role for TLR2 and TLR4 signaling via a MyD88-dependent pathway in the middle ear inflammatory responses to bacteria, and they are particularly important during the recovery process from OM. However, the individual TLRs appear to play quite different roles.
Mucosal Hyperplasia
A hallmark of OM is hyperplasia of the middle ear mucosa, which transitions from a single layer of simple squamous epithelial cells and a rudimentary stroma to a pseudostratified respiratory epithelium with extensive stroma during OM [29]. After inoculation of the middle ear with nontypeable H. influenzae, wild-type mice display significant thickening of the middle ear mucosa at day 2 (Fig. 1). Mucosal thickness recovers and the middle ear is histologically normal by day 10. In contrast, while mice with deficiencies in TLR2 or MyD88 initially show similar initial increases in mucosal thickness, the mucosa in such mice continues to increase in thickness and fails to return to normal for at least 3 weeks after inoculation with nontypeable H. influenzae (Fig. 1). Mice deficient in TLR4 or TRIF exhibit more modest delays in mucosal recovery.
Fig. 1.
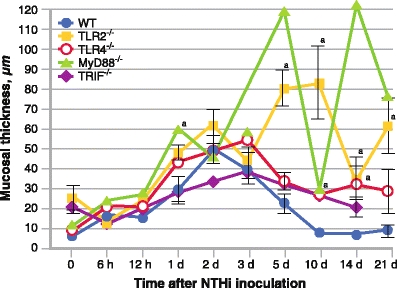
A quantitative evaluation of mucosal thickness in the middle ear cavity throughout the course of otitis media. Mucosal thickness is greater in mice lacking key Toll-like receptor (TLR) signaling genes than in wild-type (WT) mice (n = 6–8 middle ears per time point). Bars represent the mean ± SEM. aSignificantly different from WT (P < 0.05). MyD88—myeloid differentiation factor-88; NTHi—nontypeable Haemophilus influenzae; TRIF—Toll/interleukin-1 receptor domain containing adaptor inducing interferon-β
Leukocyte Recruitment and Function
Although there are few leukocytes in the resting middle ear, these cells enter the tympanic cavity in large numbers during OM. Inflammatory infiltrates during bacterial OM consist primarily of neutrophils and macrophages, with neutrophils dominating the first 24 h after inoculation, and macrophages predominant from 48 to 72 h. Most cells leave the normal middle ear by 5 days after initial infection. In the absence of TLR2 or MyD88, initial neutrophil recruitment is significantly delayed; however, neutrophils that do enter the ME persist in TLR2 or MyD88-deficient mice longer than in wild-type mice. Macrophage recruitment is also delayed in TLR2- and MyD88-deficient middle ears. However, in response to bacterial infection, these cells eventually enter the middle ear in far greater numbers and persist far longer than in wild types. TLR4- and TRIF-deficient mice exhibit modest delays in middle ear recruitment or clearance of neutrophils and macrophages.
Bacterial Clearance and Toll-Like Receptor Signaling
Mice deficient in TLR signaling display distinct deficits in bacterial clearance after infection with nontypeable H. influenzae (Table 1). In wild-type mice, culture-positive middle ears increased from four of six on day 1 after inoculation to six of six on day 2 and dropped to three of six on day 3. Thereafter, bacteria have been cleared from the middle ear, and nontypeable H. influenzae can no longer be cultured from them. TLR2–/– middle ears were culture positive throughout the 21-day observation period, with two to three of six positive middle ears from 1 to 21 days after inoculation. All MyD88–/– middle ears were culture positive from 1 to 14 days after inoculation. At days 21 and 42, two of six middle ears were positive. In contrast, TLR4–/– and TRIF–/– middle ears display culture positivity similar to that of wild types.
Table 1.
Impaired bacterial clearance of middle ears in mice deficient in Toll-like receptor signalinga
| Day after NTHi | C57 culture + MEs | C57, mean CS | TLR2–/– culture + MEs | TLR2–/–, mean CS | TLR4–/– culture + MEs | TLR4–/–, mean CS | MyD–/– culture + MEs | MyD–/–, mean CS | TRIF–/– culture + MEs | TRIF–/–, mean CS |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 4/6 | 4 | 3/6 | 4 | 4/6 | 2.3 | 6/6 | 4 | 4/6 | 4 |
| 2 | 6/6 | 3 | 3/6 | 1 | 4/6 | 1.3 | 6/6 | 4 | 5/6 | 3 |
| 3 | 3/6 | 1 | 3/6 | 2 | 3/6 | 3.3 | 6/6 | 4 | 3/6 | 2 |
| 5 | 0/6 | 0 | 2/6 | 1 | 4/6 | 3.5 | 6/6 | 4 | 1/6 | 1 |
| 10 | 0/6 | 0 | 3/6 | 1.3 | 3/6 | 2 | 6/6 | 4 | 1/6 | 1 |
| 14 | 0/6 | 0 | 3/6 | 2 | 1/6 | 1 | 6/6 | 2.7 | 0/6 | 0 |
| 21 | 0/6 | 0 | 3/6 | 1 | 0/6 | 0 | 2/6 | 2 | 0/6 | 0 |
aOne culture plate was generated for each of 6 middle ears (MEs)/strains, and the number of plates that were positive for nontypeable Haemophilus influenzae (NTHi) were evaluated. Mean bacterial colonization of the positive-culture plates was evaluated using semiquantitative analysis of bacterial colonization (CS): 0 indicates no CFUs, 1 indicates 1 quadrant with CFUs, 2 indicates 2 quadrants with CFUs, 3 indicates 3 quadrants with CFUs, and 4 indicates 4 quadrants with CFUs. Data represent positive-culture plates out of 6
MyD—myeloid differentiation factor; TLR—Toll-like receptor; TRIF—Toll/interleukin-1 receptor domain containing adaptor inducing interferon-β
The middle ears of TLR2- and MyD88-deficient mice thus fail to clear nontypeable H. influenzae despite the presence of large numbers of macrophages, which might be expected to phagocytose and kill bacteria. However, when bacterial phagocytosis and intracellular killing of nontypeable H. influenzae were evaluated in macrophages isolated from MyD88–/– and TNF–/– mice, these processes were found to be severely deficient in both genetic-deficient mice [29, 30].
Toll-Like Receptors and Otitis Media Pathogenesis
TLRs, MyD88, and TRIF are known to play crucial roles in inflammatory responses to nontypeable H. influenzae [20, 26]. However, neither TLRs nor their signaling molecules singly appear to be essential for the initial expression of nontypeable H. influenzae-mediated inflammation in the middle ear. Both mucosal hyperplasia and leukocyte infiltration are similar to controls in all the knockouts studied (days 1–3). This presumably reflects the presence of multiple innate immune receptors, including alternative TLRs such as NOD-like receptors, as well as bacterial DNA receptors, in the middle ear.
Although the initial phases of OM do not depend on TLRs, the striking increase in duration of OM shows that MyD88 and TLR2 are particularly critical to the process of middle ear recovery from infection. This is closely related to failure of bacterial clearance, especially in MyD88-null animals. In the absence of clearance, prolonged inflammation mediated by bacteria presumably leads to prolonged and increased hyperplasia and leukocyte recruitment.
Toll-Like Receptors and Inflammatory Cytokines and Chemokines in the Middle Ear
The activation of TLRs in turn leads to the activation of various transcription factors, such as NF-κB, that mediate the expression of genes encoding inflammatory cytokines, including TNF, IL-1β, IL-6, IL-10, and the chemokines CXCL2 (macrophage inflammatory protein-2) and CCL3 (macrophage inflammatory protein-1a).
TNF is one of the major proinflammatory cytokines with a plausible pathophysiologic role for this mediator in OM. For example, in rats, transtympanic injection of TNF into the middle ears induces a subsequent inflammation [31], while administration of a TNF antagonist, such as soluble TNF receptor type I, decreases the severity of LPS-, TNF- and immune-mediated OM in these animals [32–35]. In children, polymorphisms in TNF have been associated with an increased incidence of OM [36]. Also, in mice, during experimental OM, persistent inflammatory responses of TLR2–/– and TLR4–/– mice were associated with an initial lack of induction of TNF after infection [30].
Importantly, TNF is also known to regulate the expression of TLRs and of molecules essential for TLR signaling, including NF-κB, via positive [9] and negative [37] feedback mechanisms. TNF thus modulates innate immunity and the complex networks of downstream cytokines and chemokines, and participates in the orchestration of the inflammatory response by controlling the migration, activation, and biological properties of various effector cells, particularly mononuclear phagocytes, and neutrophilic polymorphonuclear leukocytes [38, 39]. TNF also directly stimulates production of CCL3, a member of the CC chemokine family known to be an important chemotactic and regulatory agent for macrophages [40, 41].
TNF-deficient mice display abnormal OM that is similar in many respects to that seen in TLR2 and MyD88 deficiency. Similarly to those gene deletions, TNF-null macrophages exhibit reduced nontypeable H. influenzae phagocytosis and killing [42•]. Because lack of TLR2 or MyD88 results in reduced TNF induction in the middle ear, this suggests that TNF is a major effector of TLR signaling during experimental, nontypeable H. influenzae-induced OM. However, application of recombinant TNF to TNF-deficient macrophages does not result in restoration of normal levels of phagocytosis and killing by these cells. Interestingly, recombinant CCL3 not only restores MyD88-, TLR2-, and TNF-deficient macrophage function to normal, but it also reduces the deficit in OM recovery observed in TNF-null mice [42•]. This identifies CCL3 as another key TLR effector molecule in the middle ear, operating downstream from TNF.
Toll-Like Receptor Interactions in the Middle Ear
Whereas mutation of the TLR renders Drosophila spp highly susceptible to fungal and other infections [43], mutation of individual TLRs in mammals has not been found to be as deleterious, suggesting that there is a degree of functional redundancy among mammalian TLRs [44]. Studies have demonstrated an interaction between TLRs such that induction of TLR2 or TLR4 may enhance expression of TLR4 or TLR2, respectively [45], and the activation of TLR2 by the skin commensal bacteria byproduct lipoteichoic acid leads to the expression of negative regulator, TNF receptor-associated factor-1, which in turn suppresses TLR3-dependent inflammation after skin injury [46]. In the middle ear, genetic loss of TLR2 or TLR4 is associated with prolonged and persistent OM. Evaluation of middle ear gene expression in mutant mice revealed that early TLR4 signaling is required for the nontypeable H. influenzae-mediated upregulated expression of TLR2 and is necessary to influence downstream molecular targets TNF and IL-10. These findings demonstrate a predominant role for TLR2 (as opposed to TLR4) in nontypeable H. influenzae-induced OM, with TLR2 induction primed by nontypeable H. influenzae interaction with TLR4 [30]. TLR4 signaling is required for the initial expression of TLR2 in response to nontypeable H. influenzae in wild-type mice, but other signaling mechanisms eventually upregulate TLR2, even in the absence of TLR4.
Conclusions
In summary, TLR signaling is critical for the timely resolution of bacterial OM. Expression of proinflammatory cytokines and the activation of macrophages to phagocytose and kill bacteria are severely disabled in mice lacking key TLR signaling molecules. This leads to failure of bacterial clearance and to persistent OM. OM recovery deficits are most prominent when certain key elements of TLR signaling are absent, particularly TLR2, MyD88, and TNF. This suggests a dominant axis of TLR signaling in OM. However, it is noteworthy that significant deficits in recovery from nontypeable H. influenzae-induced OM were observed in mice lacking any of the elements of TLR signaling that have been tested. This suggests that the entire TLR signaling network must work in concert to ensure timely recovery from OM. Outcome, coupled with the existence of polymorphisms in TLR signaling genes that have been associated with OM proneness in patients may explain why so many children have difficulty with recurrent, persistent, or chronic middle ear disease.
Acknowledgments
This work was supported by National Institutes of Health/National Institute on Deafness and Other Communication Disorders grants no. DC006279 (to Dr. Wasserman) and no. DC000129 (to Dr. Ryan), the VA Research Service (Dr. Ryan), and grant no. E37-2010 (to Dr. Leichtle) from the University of Lübeck, Germany.
Disclosure
Dr. Wasserman has served as a consultant for Genentech and Merck & Co., has been employed by the American Board of Allergy and Immunology, has received grant support from Merck & Co. and Schering-Plough, and has been paid for providing expert testimony by various law firms. Drs. Leichtle, Lai, Wollenberg, and Ryan reported no potential conflicts of interest relevant to this article.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Contributor Information
Anke Leichtle, Email: anke.leichtle@uk-sh.de.
Yuping Lai, Email: laiyuping@126.com.
Barbara Wollenberg, Email: barbara.wollenberg@uk-sh.de.
Stephen I. Wasserman, Email: swasserman@ucsd.edu
Allen F. Ryan, Email: afryan@ucsd.edu
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
- 1.World Health Organization . World Bank. 1993. World Development Report 1993: Investing in Health. New York: Oxford University Press; 1993. [Google Scholar]
- 2.Infante-Rivard C, Fernandez A. Otitis media in children: frequency, risk factors, and research avenues. Epidemiol Rev. 1993;15:444–465. doi: 10.1093/oxfordjournals.epirev.a036129. [DOI] [PubMed] [Google Scholar]
- 3.Klein JO. The burden of otitis media. Vaccine. 2000;1:S2–S8. doi: 10.1016/S0264-410X(00)00271-1. [DOI] [PubMed] [Google Scholar]
- 4.Seibert JW, Danner CJ. Eustachian tube function and the middle ear. Otolaryngol Clin North Am. 2006;39:1221–1235. doi: 10.1016/j.otc.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Mandel EM, Doyle WJ, Winther B, et al. The incidence, prevalence and burden of OM in unselected children aged 1–8 years followed by weekly otoscopy through the “common cold” season. Int J Pediatr Otorhinolaryngol. 2008;72:491–499. doi: 10.1016/j.ijporl.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leibovitz E. Acute otitis media in children aged less than 2 years: drug treatment issues. Paediatr Drugs. 2006;8:337–346. doi: 10.2165/00148581-200608060-00002. [DOI] [PubMed] [Google Scholar]
- 7.Leibovitz E. The challenge of recalcitrant acute otitis media: pathogens, resistance, and treatment strategy. Pediatr Infect Dis J. 2007;26:S8–S11. doi: 10.1097/INF.0b013e318154b24a. [DOI] [PubMed] [Google Scholar]
- 8.Leibovitz E, Jacobs MR, Dagan R. Haemophilus influenzae: a significant pathogen in acute otitis media. Pediatr Infect Dis J. 2004;23:1142–1152. [PubMed] [Google Scholar]
- 9.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 10.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 11.Wieland CW, Florquin S, Maris NA, et al. The MyD88-dependent, but not the MyD88-independent, pathway of TLR4 signalling is important in clearing nontypeable Haemophilus influenzae from the mouse lung. J Immunol. 2005;175:6042–6049. doi: 10.4049/jimmunol.175.9.6042. [DOI] [PubMed] [Google Scholar]
- 12.Leendertse M, Willems RJ, Giebelen IA, et al. TLR2-dependent MyD88 signalling contributes to early host defense in murine Enterococcus faecium peritonitis. J Immunol. 2008;180:4865–4874. doi: 10.4049/jimmunol.180.7.4865. [DOI] [PubMed] [Google Scholar]
- 13.Mandel E, Casselbrant ML. Antibiotics for otitis media with effusion. Minerva Pediatr. 2004;56:481–495. [PubMed] [Google Scholar]
- 14.Johnston LC, Feldman HM, Paradise JL, et al. Tympanic membrane abnormalities and hearing levels at the ages of 5 and 6 years in relation to persistent otitis media and tympanostomy tube insertion in the first 3 years of life: a prospective study incorporating a randomized clinical trial. Pediatrics. 2004;114:e58–e67. doi: 10.1542/peds.114.1.e58. [DOI] [PubMed] [Google Scholar]
- 15.Alper CM, Winther B, Mandel EM, et al. Temporal relationships for cold-like illnesses and otitis media in sibling pairs. Pediatr Infect Dis J. 2007;26:778–781. doi: 10.1097/INF.0b013e318124aa31. [DOI] [PubMed] [Google Scholar]
- 16.Ryan AF, Catanzaro A, Wasserman SI, et al. Complement depletion reduces immunologically mediated middle ear effusion and inflammation. Clin Immunol Immunopathol. 1986;40:410–421. doi: 10.1016/0090-1229(86)90185-6. [DOI] [PubMed] [Google Scholar]
- 17.•.Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev. 2009;227:221–233. doi: 10.1111/j.1600-065X.2008.00731.x. [DOI] [PubMed] [Google Scholar]
- 18.Medzithov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 19.Beutler B. Inferences, questions and possibilities in TLR signaling. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- 20.Shuto T, Xu H, Wang B, et al. Activation of NF-kappa B by nontypeable Haemophilus influenzae is mediated by toll-like receptor 2-TAK1-dependent NIK-IKK alpha /beta-I kappa B alpha and MKK3/6-p38 MAP kinase signaling pathways in epithelial cells. Proc Natl Acad Sci U S A. 2001;98:8774–8779. doi: 10.1073/pnas.151236098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 22.Leake ER, Holmes K, Lim DJ, et al. Peptidoglycan isolated from nontypeable Haemophilus influenzae induces experimental otitis media in the chinchilla. J Infect Dis. 1994;170:1532–1538. doi: 10.1093/infdis/170.6.1532. [DOI] [PubMed] [Google Scholar]
- 23.DeMaria TF, Apicella MA, Nichols WA, et al. Evaluation of the virulence of nontypeable Haemophilus influenzae lipooligosaccharide htrB and rfaD mutants in the chinchilla model of otitis media. Infect Immun. 1997;65:4431–4435. doi: 10.1128/iai.65.11.4431-4435.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emonts M, Veenhoven RH, Wiertsema SP, et al. Genetic polymorphisms in immunoresponse genes TNFA, IL6, IL10, and TLR4 are associated with recurrent acute otitis media. Pediatrics. 2007;120:814–823. doi: 10.1542/peds.2007-0524. [DOI] [PubMed] [Google Scholar]
- 25.Wiertsema SP, Khoo SK, Baynam G, et al. Association of CD14 promoter polymorphism with otitis media and pneumococcal vaccine responses. Clin Vaccine Immunol. 2006;13:892–897. doi: 10.1128/CVI.00100-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe T, Jono H, Han J, et al. Synergistic activation of NF-kappaB by nontypeable Haemophilus influenzae and tumor necrosis factor alpha. Proc Natl Acad Sci U S A. 2004;101:3563–3568. doi: 10.1073/pnas.0400557101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen R, Lim JH, Jono H, et al. Nontypeable Haemophilus influenzae lipoprotein P6 induces MUC5AC mucin transcription via TLR2-TAK1-dependent p38 MAPK-AP1 and IKKbeta-IkappaBalpha-NF-kappaB signaling pathways. Biochem Biophys Res Commun. 2004;324:1087–1094. doi: 10.1016/j.bbrc.2004.09.157. [DOI] [PubMed] [Google Scholar]
- 28.Hirano T, Kodama S, Fujita K, et al. Role of Toll-like receptor 4 in innate immune responses in a mouse model of acute otitis media. FEMS Immunol Med Microbiol. 2007;49:75–83. doi: 10.1111/j.1574-695X.2006.00186.x. [DOI] [PubMed] [Google Scholar]
- 29.Hernandez M, Leichtle A, Pak K, et al. Myeloid differentiation primary response gene 88 is required for the resolution of otitis media. J Infect Dis. 2008;198:1862–1869. doi: 10.1086/593213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leichtle A, Hernandez M, Pak K, et al. TLR4-mediated induction of TLR2 signaling is critical in the pathogenesis and resolution of otitis media. Innate Immun. 2009;15:205–215. doi: 10.1177/1753425909103170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Catanzaro A, Ryan A, Batcher S, Wasserman SI. The response to human rIL-1, rIL-2 and rTNF in the middle ear of guinea pigs. Laryngoscope. 1991;101:271–275. doi: 10.1288/00005537-199103000-00008. [DOI] [PubMed] [Google Scholar]
- 32.Jeon EJ, Park YS, Choi YC, et al. Effect of inhibitor of tumor necrosis factor-alpha on experimental otitis media with effusion. Ann Otol Rhinol Laryngol. 2001;110:917–921. doi: 10.1177/000348940111001005. [DOI] [PubMed] [Google Scholar]
- 33.Lee DH, Park YS, Jung TT, et al. Effect of tumor necrosis factor-alpha on experimental otitis media with effusion. Laryngoscope. 2001;111:728–733. doi: 10.1097/00005537-200104000-00030. [DOI] [PubMed] [Google Scholar]
- 34.Kim KH, Park YS, Jeon EJ, et al. Effects of tumor necrosis factor alpha antagonist, platelet activating factor antagonist, and nitric oxide synthase inhibitor on experimental otitis media with effusion. Ann Otol Rhinol Laryngol. 2006;115:617–623. doi: 10.1177/000348940611500808. [DOI] [PubMed] [Google Scholar]
- 35.Park YS, Auo HJ, Jeon EJ, et al. Effect of tumor necrosis factor-alpha and oxatomide on immune mediated otitis media. Laryngoscope. 2006;116:1642–1646. doi: 10.1097/01.mlg.0000232545.08028.b3. [DOI] [PubMed] [Google Scholar]
- 36.Patel JA, Nair S, Revai K, et al. Association of proinflammatory cytokine gene polymorphisms with susceptibility to otitis media. Pediatrics. 2006;118:2273–2279. doi: 10.1542/peds.2006-0764. [DOI] [PubMed] [Google Scholar]
- 37.Jono H, Lim JH, Chen LF, et al. NF-kappaB is essential for induction of CYLD, the negative regulator of NF-kappaB: evidence for a novel inducible autoregulatory feedback pathway. J Biol Chem. 2004;279:36171–36174. doi: 10.1074/jbc.M406638200. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi H, Tashiro T, Miyazaki M, et al. An essential role of macrophage protein 1 alpha/CCL3 on the expression of host’s innate immunities against infectious complications. J Leukoc Biol. 2002;72:1190–1197. [PubMed] [Google Scholar]
- 39.Montecucco F, Steffens S, Burger F, et al. Tumor necrosis factor-alpha induces integrin CD11b/CD18 (Mac-1) up- regulation and migration to the CC chemokine CCL3 (MIP-1 alpha) on human neutrophils through defined signaling pathways. Cell Signal. 2008;20:557–568. doi: 10.1016/j.cellsig.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 40.Otonello L, Montecucco F, Bertolotto M, et al. CCL3 (MIP-1 alpha) induces in vitro migration of GM-CSF primed human neutrophils via CCR5-dependent activation of ERK 1-2. Cell Signal. 2005;17:355–363. doi: 10.1016/j.cellsig.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 41.Broxmeyer HD, Sheery B, Lu L, et al. Myelopoeitic enhancing effects of murine macrophage inflammatory proteins 1 and 2 on colony formation in vitro by murine and human bone marrow granulocyte/ macrophage progenitor cells. J Exp Med. 1989;170:1583–1594. doi: 10.1084/jem.170.5.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.•.Leichtle A, Hernandez M, Ebmeyer J, et al. CC chemokine ligand 3 overcomes the bacteriocidal and phagocytic defect of macrophages and hastens recovery from experimental otitis media in TNF-/- Mice. J Immunol. 2010;184:3087–3097. doi: 10.4049/jimmunol.0901167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lemaitre B, Nicolas E, Michaut L, et al. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/S0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 44.Fritz JH, Girardin SE. How Toll-like receptors and Nod-like receptors contribute to innate immunity in mammals. J Endotoxin Res. 2005;11:390–394. doi: 10.1179/096805105X76850. [DOI] [PubMed] [Google Scholar]
- 45.Fan J, Frey RS, Malik AB. TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J Clin Invest. 2003;112:1234–1243. doi: 10.1172/JCI18696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lai Y, Di Nardo A, Nakatsuji T, et al. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat Med. 2009;15:1377–1382. doi: 10.1038/nm.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]