

Abstract
Evolving lineages face a constant intracellular threat: most new coding sequence mutations destabilize the folding of the encoded protein. Misfolded proteins form insoluble aggregates and are hypothesized to be intrinsically cytotoxic. Here, we experimentally isolate a fitness cost caused by toxicity of misfolded proteins. We exclude other costs of protein misfolding, such as loss of functional protein or attenuation of growth-limiting protein synthesis resources, by comparing growth rates of budding yeast expressing folded or misfolded variants of a gratuitous protein, YFP, at equal levels. We quantify a fitness cost that increases with misfolded protein abundance, up to as much as a 3.2% growth rate reduction when misfolded YFP represents less than 0.1% of total cellular protein. Comparable experiments on variants of the yeast gene orotidine-5′-phosphate decarboxylase (URA3) produce similar results. Quantitative proteomic measurements reveal that, within the cell, misfolded YFP induces coordinated synthesis of interacting cytosolic chaperone proteins in the absence of a wider stress response, providing evidence for an evolved modular response to misfolded proteins in the cytosol. These results underscore the distinct and evolutionarily relevant molecular threat of protein misfolding, independent of protein function. Assuming that most misfolded proteins impose similar costs, yeast cells express almost all proteins at steady-state levels sufficient to expose their encoding genes to selection against misfolding, lending credibility to the recent suggestion that such selection imposes a global constraint on molecular evolution.
Keywords: proteomics, stability, heat shock, evolutionary rate
Most new genetic mutations arising in protein-coding sequences decrease the likelihood that the encoding protein will fold properly (1, 2). Misfolding reduces the concentration of functional proteins, squanders cellular time and energy on production of useless proteins (3), and generates misfolded proteins that may harm cells (4). Given the high probability and diverse effects of failed protein folding, isolating and quantifying the influences of misfolding on fitness are essential for the development of mechanistic answers to fundamental questions in evolutionary biology: the distribution of fitness effects of new mutations, the interpretation of varying rates of molecular evolution, and the significance of compensatory mutations. Understanding how misfolding affects cell fitness may also illuminate the molecular basis of human diseases, particularly neurological disorders linked to misfolded protein toxicity (4, 5).
Patterns of coding sequence evolution across taxa depend strongly on gene expression, with apparently limited contribution from protein function (6, 7). This has led to the misfolding hypothesis: that within-genome variation in purifying selection on coding sequences is predominantly shaped by the fitness cost of misfolded proteins and therefore, correlates with gene expression and protein abundance (8, 9). It predicts that selection against protein misfolding purges most destabilizing mutations from high-expression genes but allows accumulation of destabilizing mutations in low-expression genes, because the latter produces many orders of magnitude fewer proteins.
Cells contend with overabundant misfolded proteins by synthesizing interacting chaperone and degradation systems to sequester, refold, and degrade misfolded substrates (10). In the eukaryotic endoplasmic reticulum (ER), misfolded proteins provoke the unfolded protein response (UPR) (11), whereas in the cytosol, a broader heat-shock response combats protein misfolding, oxidative stress, altered membrane integrity, and reduced growth rate (12). Recent work suggests that the heat-shock response may mask a milder and more targeted response to counteract cytosolic protein misfolding (13–15), dubbed the UPR-Cyto (15), but the molecular components of this response remain unclear.
Despite evidence that protein misfolding imposes a cellular cost, the magnitude and origin of this cost remain unknown. Misfolding can impose costs in three distinct ways: loss of function, diversion of protein synthesis resources away from essential proteins, and toxicity of the misfolded molecules. Studies quantifying the fitness cost incurred because of misfolded protein toxicity are particularly sparse and inconclusive (5, 16–18). However, many studies have measured the growth rate cost of losing protein function (19–21) and expressing gratuitous proteins (3, 18, 20, 22). No study has experimentally isolated and quantified a fitness cost attributable to the folding state of an intracellularly synthesized protein.
Here, we experimentally isolate the toxic effects of misfolded proteins in budding yeast, Saccharomyces cerevisiae. Using flow cytometry to precisely assay relative growth rates, we measure the fitness cost attributable to the folding state of a cytosolic protein. We then survey the global proteomic response to misfolded protein overabundance to explore the defenses activated by this specific intracellular threat.
Results
Strains Expressing Misfolded Gratuitous Proteins Isolate the Fitness Cost of Misfolding.
To isolate the cost of protein misfolding, we compared the growth of yeast strains expressing gratuitous wild-type (WT) or mutated protein variants at equal levels under the control of a galactose-titratable GAL1 promoter (23). We studied two proteins: YFP and orotidine-5′-phosphate decarboxylase (URA3). YFP, derived from jellyfish, confers no fitness advantage in yeast, and therefore, no functional benefit is lost on misfolding. URA3 encodes the yeast protein (Ura3p) that catalyzes the final step in uracil synthesis and confers no benefit when uracil is provided in the growth medium. By eliminating the cost associated with loss of function and standardizing the cost of protein expression, we isolate the fitness cost of misfolding.
Mutant YFPs Have Decreased Solubility Relative to WT YFP.
We isolated putatively misfolded YFP mutants by screening for mutations that resulted in temperature-sensitive fluorescence. Combining these mutations resulted in four mutant YFPs (YFPm1–4) (Table S1) with decreased solubility (Fig. 1 A and B), characteristic of disrupted folding (24). We speculate that these mutations hinder the folding pathway of YFP, biasing proteins to the formation of insoluble aggregates and away from proper folding. An imperfect correlation between solubility and fluorescence (Fig. 1 A and B) suggests that these mutations also perturb fluorophore maturation.
Fig. 1.

Protein variants display characteristic misfolding phenotypes. (A) Western blotting reveals partitioning of YFP variants into the insoluble fraction of a two-phase lysis. Blots of total (n = 2), soluble (n = 5), and insoluble (n = 4) cell fractions were probed from strains expressing 3×FLAG-YFP variants for 2 h, with a representative of each shown. Strains carried an rpn4 deletion (a regulator of the proteasome) and were treated with 100 μM of the proteasome inhibitor bortezomib during induction (43) to reduce misfolded protein degradation. A reference protein (PGAL1-3×Myc-YFPref) present in each strain was used to normalize galactose induction and loading between strains. Error bars show 95% confidence intervals about the mean reference-normalized fold change over YFPwt. (B) YFP variants fluoresce at different levels. Densities show single-cell variability across >10,000 cells, with arbitrary heights set to visually separate distributions. (C) YFP-Ura3wt dispersed throughout the cytosol, whereas YFP-Ura3m1 often localized in a single bright focus. Scale bar is estimated.
To estimate how often each mutant YFP adopts a native or misfolded state, we measured the fraction detected in total, insoluble, and soluble cell fractions by quantitative Western blotting. All strains express similar total amounts of YFP (Fig. 1A).
Mutant YFP-URA3 Forms Fluorescent Foci in Yeast Cells.
We created a putatively misfolded Ura3 protein mutant (Ura3m1) by combining eight single-nucleotide mutations known to confer temperature sensitivity individually (25) and N-terminally tagging the WT and misfolded variants with YFP. Microscopy revealed that YFP-Ura3wt dispersed throughout the cytosol, whereas YFP-Ura3m1 usually localized in a single bright focus (Fig. 1C), consistent with protein aggregation and sequestration observed with other misfolded proteins in yeast (26). Unlike YFP-Ura3wt, YFP-Ura3m1 was unable to grow on a medium lacking uracil (Fig. S1).
Misfolded Proteins Slow Exponential Growth.
We quantified the relative growth rate of YFP and YFP-Ura3p test strains by competing each against an isogenic reference strain (KG007), which constitutively expressed RFP, and using flow cytometry to count relative numbers of RFP-labeled and -unlabeled cells for roughly 17 generations. By keeping cells at low density with periodic dilutions, we attempted to maintain exponential growth throughout the experiment, in which case we expect and observe (Fig. 2) a linear relationship between the generations of the reference strain and the logarithm of the proportion of RFP-labeled and -unlabeled cells. Competitive growth rates show no density-dependence (Fig. S2).
Fig. 2.

YFP variants display different competitive growth rates. YFP (test strains) vs. RFP (reference strain) cell count ratios, assessed by flow cytometry and normalized to an initial mean YFP/RFP ratio of 1, changed log-linearly with the number of reference strain generations. Lines show least-squares regression fits.
With YFP expression fully induced, the test strains grew at reproducibly different rates (Fig. 2). The change in labeled/unlabeled cell proportion over time quantifies the instantaneous exponential growth rate (r = generations/time) of each test strain as a proportion of the reference strain's growth rate (rYFP/rref). Differences between slopes quantify the additive selective advantage s of one strain over another, again normalized to the reference strain [i.e., sm/wt = (rYFPm* − rYFPwt)/rref], which are multiplied by 100 to express selective advantage/disadvantage as a percentage of reference strain growth rate.
Each of the strains expressing a misfolded YFP variant, YFPm1–4, grew significantly slower than the strain expressing YFPwt, with sm/wt ranging from 0.7% to 3.2% per reference strain generation (Fig. 3A, induced), whereas uninduced strains showed no significant differences in fitness (Fig. 3A, uninduced). The fitness disadvantage attributable to misfolded proteins, sm/wt, increased roughly linearly with the relative amount of insoluble YFP in each test strain (Fig. 3B). To further probe this apparent linear relationship, we titrated the amount of the galactose inducer (23) in the growth competitions between mutant YFPm3 and the reference strain, monitoring protein expression by YFPm3 fluorescence. This experiment revealed a linear dependence of fitness cost on expression of the misfolded protein down to the limit of detection (Fig. 3C).
Fig. 3.

Misfolded proteins impose a fitness disadvantage. (A) Fitness disadvantage of YFP-variant–expressing strains relative to a strain expressing YFPwt as a percentage of RFP-expressing reference strain growth rate, when fully induced with 27.5 mM galactose (induced) and 0 mM galactose (uninduced). Error bars show 95% confidence intervals about the mean. (B) Fitness disadvantage relative to strains expressing YFPwt correlates with the relative amount of insoluble YFP (Fig. 1A). (C) The fitness cost of YFPm3 increases linearly with protein level monitored by average YFPm3 fluorescence across all four time points. (D) Misfolded YFP-Ura3m1 confers an induction-dependent fitness cost relative to folded YFP-Ura3wt.
Strains expressing YFP-Ura3m1 also display a growth disadvantage (sm/wt = 1.2%) relative to those expressing YFP-Ura3wt (Fig. 3D, induced). Consistent with the YFP-variant results, uninduced strains harboring URA3 variants show a fitness advantage over the induced strains (Fig. 3D, uninduced).
The observed induction-dependent growth defects do not seem to result from any specific amino acid substitution or perturbation of protein function. Similar folding-state-dependent growth defects are observed for two unrelated proteins, YFP and Ura3p. The YFPm2 variant shares no substitutions with YFPm1 or YFPm3 (Table S1), suggesting that the substitutions’ effects, rather than their identity, cause the growth defect. The YFPm1 variant harbors the substitution V163A, which was found to confer improved folding, as monitored by increased solubility and thermal stability, in two independent studies of GFP (24, 27). Reversion of this single substitution to 163V, creating variant YFPm3, decreased fluorescence eightfold, increased insoluble protein production by one-third (Fig. 1A), and resulted in a threefold increase in the growth rate defect relative to the YFPm1 variant (Fig. 3 A and B).
Comparing the Costs of Expression and Misfolding.
The growth rate costs of expressing YFPwt or YFP-Ura3wt, given by the difference between induced and uninduced competitions, were roughly the same, about 1.4% (Fig. 3 A and D, uninduced; slight correction for slowed reference strain growth in induced conditions is discussed in SI Appendix, SI Materials and Fig. S3). This observed cost is less than the cost of misfolding for YFP mutants YFPm3 and YFPm4, neither of which is fully misfolded, suggesting that protein misfolding imposes a stronger selective pressure than protein synthesis on a per-protein basis. The ∼1.4% cost of expressing a single gene from the GAL1 promoter is likely too high, because it includes any cost contributed by misfolding of YFPwt and YFP-Ura3wt.
Cells Mount a Specific Proteomic Response to Misfolded YFP.
To assess the proteome-level response to chronic misfolding of a single gene's products, we quantified differences in total protein levels between stable isotope-labeled yeast strains KG079 and KG071, respectively expressing a misfolded (YFPm3) and folded (YFPwt) variant of YFP, using liquid chromatography and tandem MS (LC-MS/MS) followed by quantitation using MaxQuant (28). We captured the proteomic environment cells experience during our growth rate experiments by harvesting cells after 20 h of low-density, exponential-phase growth.
We quantified protein-level differences for 3,031 of 4,840 genes for which a protein product has been detected during log-phase growth by at least one of four recent proteome-wide studies (29–32). To identify significant differences, the SD of log ratios below which 99.5% of detected proteins fell (2.06), combined with the number of times each ratio was measured, was used to construct an envelope of expected protein variability (Fig. 4, gray lines). The 26 proteins with at least two peptides detected and ratios having 95% confidence intervals (CI) completely outside this envelope (Fig. 4A and Table S2) were scored as significantly changed. Repeating the experiment without galactose induction revealed no significant protein ratio differences by these criteria (Fig. 4B).
Fig. 4.

Chronic low-level expression of a misfolded protein induces a cytosolic unfolded protein response. (A) Mean protein ratios for 3,031 proteins, plotted against the number of measurements used to generate each mean ratio, between strains KG079 (expressing misfolded YFPm3) and KG071 (expressing folded YFPwt). Gray envelope shows expected variation for the most variable proteins as a function of number of measurements (in the text). Error bars show 95% confidence interval on the mean assuming log ratios are drawn from a normal distribution. Other protein quality control proteins are highlighted for comparison (open squares), such as cytosolic proteins SSB1/SSB2/ZUO1/SSZ1, which associate with nascent polypeptide chains at the ribosome, and JEM1/LHS1/SCJ1/ERJ5, ER chaperones up-regulated in the UPR. (B) Without galactose induction of YFP variants, no protein levels differ significantly.
The majority of significantly induced proteins form an interacting set of cytosolic chaperones and cochaperones. Ssa1p and Ssa2p, closely related heat-shock proteins (HSPs) in the HSP70 family, proliferate along with their activator Sti1p, cochaperone Sis1p, regulator Ydj1p (HSP40 family), and interaction partner Hsp104p (itself a prominent disaggregating chaperone of the HSP100 family) (33). Hsp82p and Hsc82p (abundant HSP90-family chaperones) rise along with their activator Aha1p, associated isomerase Cpr6p, and interacting ATPase Sse1p (HSP70 family); 20 of 25 differentially regulated proteins (excluding YFP) have promoters known to be bound by the heat shock-reactive transcription factor Hsf1p encoded by HSF1 (34).
Not all cytosolic chaperones respond, as shown by the unchanged levels of cytosolic ribosome-associated chaperone complex members (Fig. 4A). Protein folding-related ER proteins identified as up-regulated in the UPR (11) also show no significant response (Fig. 4A).
The only significantly repressed protein, Hsp12p, is also a heat-shock protein and a target of Hsf1p, although one whose HSF1-dependent induction depends on the stress-related transcription factors MSN2/4 (35, 36). We detected minimal ribosomal protein repression (no significant changes; mean fold change of ribosomal proteins = 0.962 induced and = 1.00 uninduced), contradicting claims that ribosomal gene repression is a hallmark of yeast's response to misfolded proteins (35).
The polypeptide encompassing the C-terminal amino acids 41–238 shared by YFPwt and YFPm3 showed a threefold increase. Because Western blots showed equal protein levels after 2 h and the misfolded YFP-Ura3m1 variant aggregated into fluorescent foci (Fig. 1 A and C), we speculate that the ratio difference at 20 h arises because of deposition of YFPm3 in aggregates rather than differential expression.
To ascertain whether YFPwt alone sufficed to trigger a similar response, we quantified protein levels between KG071 cultures grown in the presence or absence of the inducer, galactose. The only significant changes were in YFP and several proteins, such as Gcy1p, known to be induced by galactose (Fig. S4), confirming that the chaperone-related response is caused primarily by differences—presumably in folding state—between YFPwt and YFPm3.
Misfolded YFP Represents a Small Fraction of Total Cellular Protein.
Following ref. 32, total protein abundance was estimated from LC-MS/MS signal intensities by linear regression on a dataset that quantified molecules per cell (29), normalizing for the total abundance of all proteins observed in both datasets. With full induction, we estimate that steady-state YFP abundance during exponential growth represents 0.1% of total cellular protein, ∼47,000 molecules per cell. In contrast, the most abundant protein that we detect, the glycolytic enzyme enolase Eno2p, is present at an estimated 1.3 million copies per cell, and the estimated total protein content of a haploid yeast cell is ∼50 million molecules per cell (29).
Discussion
We have isolated the growth rate cost imposed by the intracellular misfolding of a single gene product. The measured growth defect results from the presence of misfolded proteins in a dosage-dependent manner and not from loss of biological function or a burden on protein synthesis machinery. In one case, a single amino acid substitution known to compromise the thermostability and solubility of other fluorescent protein variants triples the growth rate defect associated with the misfolded variant. Cells respond to steady-state protein misfolding by synthesizing an interacting set of cytosolic chaperone and adjuvant proteins consistent with the regulatory action of a single transcription factor.
Fitness Cost of Misfolded Proteins.
The fitness cost of steady-state expression of a mostly misfolded cytosolic protein at less than 0.1% of total protein is s = 3.2%. For comparison, natural selection efficiently acts on fitness effects exceeding the inverse of the effective population size, which in yeast is ∼107 (37). This implies that mutations resulting in s ∼ 0.00001% will be efficiently purged from the yeast genome. Our measured values for sm/wt suggest that most of the yeast genome is expressed at levels sufficient to expose misfolding to selection. In the simplest case, where the cost from misfolded proteins is additive (as suggested by Fig. 3 B and C) addition of a single steady-state misfolded molecule per cell will decrease fitness by s ∼ 0.00008%, almost an order of magnitude above the threshold of detection by selection.
Our experimental results support hypotheses suggesting that selection against misfolded protein production imposes a general constraint on coding sequence evolution (6, 9). In using results from two yeast proteins to speculate about the general cost of misfolding, we do not intend to downplay the unique attributes of individual proteins, such as differential toxicity on misfolding (9). Neither do we discount possible differences in sensitivity between species (18), although, given the remarkable conservation of protein quality-control machinery across taxa (10), claims of such differences require strong evidence.
Comparing the Cost of Expression and Misfolding.
Recent work on the lac operon in Escherichia coli led to the conclusion that the cost of protein expression in that system “is in the process, not in the products” (3). Our results suggest that both process and products can be costly in S. cerevisiae and offer a chance to compare these costs. We observe that the cost from misfolded proteins, a costly product of gene expression, exceeds 3% upon misfolding of <0.1% of total steady-state yeast protein. This is two times the ∼1.4% cost resulting from expression of the same quantity of folded protein. The observed production cost may overestimate the cost of expression because of hidden costs from misfolding of WT proteins.
Contrary to these findings, a very recent study found that WT lacZ (β-galactosidase) overexpressed in E. coli resulted in a large (13.5%) growth rate decrease and that misfolded lacZ variants often mitigated this cost, leading to the conclusion that “misfolded proteins are no more toxic than wild-type proteins” (18). In this study, lacZ was expressed from a multicopy plasmid, producing a prominent band visible by Coumassie staining of total protein (figure 1b in ref. 18) characteristic of overexpressed protein. Expression of WT lacZ induced the chaperone GroEL to levels approaching that of heat-shocked cells, suggesting that lacZ overexpression severely influenced protein homeostasis. A previous study showed that lacZ overexpression has potent effects, such as ribosome destruction, induction of heat-shock proteins, and growth inhibition (22). A separate study showed that, similar to our study, when folded and misfolded variants of the N-terminal domain of λ-repressor were expressed in E. coli, only the misfolded variant triggered a chaperone response (17). Together, our study and previous results underscore the necessity of avoiding overexpression when attempting to study the effects of physiologically and evolutionarily relevant changes in protein misfolding.
Defining the Steady-State Cytosolic Unfolded Protein Response.
The unfolded protein response in the lumen of the endoplasmic reticulum, dubbed the UPR (38), UPR-L (15), or UPRer (39), is well-studied, and responses in other compartments, such as the mitochondrial UPRmt (39), have recently been identified. Here, we identify the proteins involved in the UPR's cytosolic counterpart, the UPR-Cyto (15), which represent a subset of proteins involved in the heat-shock response.
Several previous studies have concluded that misfolded proteins induce a subset of the heat-shock response in bacterial (17), fungal (35), plant (13, 14), and vertebrate cells (40) by either monitoring transcript or protein changes in response to widespread misfolding (13, 35) or monitoring only a handful of heat-shock proteins (13, 14, 17, 40). Recent studies have led to the proposal that the UPR-Cyto is a specific HSF1-mediated module of the eukaryotic heat-shock response (13, 15). Our study provides key evidence supporting this proposal by quantifying the global protein-level response to chronic misfolding of a single nonessential protein species in the cytosol of exponentially growing cells. Most induced proteins are HSF1 targets. We further show that protein misfolding provokes a chaperone response with minimal ribosomal protein repression, contra (35), and that key folding-related proteins induced in the UPR-L are not induced in the UPR-Cyto.
Our observation that small amounts of misfolded protein induce a chaperone response may shed light on the normal level of misfolded protein in the cytosol. The data suggest three possibilities: (i) misfolding is very infrequent (e.g., much less than 1% of total steady-state cytosolic protein is misfolded), and the small amount contributed by YFP misfolding constitutes a substantial change that induces a response; (ii) misfolding is common (e.g., 1–10% of total protein is misfolded), but cells are hypersensitive to even tiny changes in this amount; or (iii) misfolded YFP forms an unusually potent inducer of the response. Our data cannot distinguish these possibilities. However, to the extent that our growth rate measurements reveal the sensitivity of cells to misfolding, three observations—the linear response to misfolded protein expression at very low levels (Fig. 3 B and C), the similar costs incurred by misfolding of the yeast protein Ura3p, and the expectation of strong selection against misfolding given the magnitude of the measured costs—lead us to speculate that case i is most likely: folding is highly efficient in S. cerevisiae.
That yeast is capable of mounting a modular response to protein misfolding, apparently without activating other stress responses, suggests that misfolding is an authentic independent stimulus and does not always co-occur with other stressors such as heat shock or oxidative stress. Remodeling the proteome for a full-bore stress response might be unduly costly under conditions in which deploying a compartment-specific protein quality control module would suffice.
Stimulus or Response?
Is the fitness cost that we observe caused by the stimulus, expression of a heterologous, putatively toxic, misfolded protein, or the response, induction of the UPR-Cyto? It is possible that misfolded YFP imposes no cost in the absence of a response but instead acts as a pharmaceutical, merely simulating the authentic signal that awakens the UPR-Cyto. Note that our central result, a quantitative fitness cost caused specifically by proteins in the misfolded state, does not depend on whether the cost is because of stimulus or response; cells bear the cost either way. Rather, we now seek the meaning of that response.
The most obvious explanation for our results is that the cells react appropriately, mounting a costly response (the UPR-Cyto) to address an even costlier problem (misfolded protein toxicity). An alternative explanation holds that misfolded YFP is harmless but, by mimicking the molecular signature of another stress such as heat shock, stimulates a costly and inappropriate response. These hypotheses make opposing predictions about the fitness of yeast cells in which the UPR-Cyto is suppressed: in the former case, cells should grow slower as toxic misfolded proteins run amok, and in the latter case, cells should grow faster as their overreaction is calmed.
We leave tests of these hypotheses to a future study. Nevertheless, substantial evidence for cytotoxicity of misfolded proteins independent of their function (5) motivates us to make a strong prediction: suppression of chaperone induction will exacerbate rather than alleviate the competitive fitness defects that we measure.
Conclusion
Our study illustrates the value in isolating and quantifying the consequences of protein misfolding to understand their relative contributions to molecular evolution and cell biology. The results support hypotheses that assume that misfolded proteins impose a selective cost independent of protein function and a model of protein quality control in which a small interacting set of proteins responds specifically to misfolded proteins in the eukaryotic cytosol.
Methods
Yeast Strains and Transforming DNA.
All strains used in this study (Table S3) are derived from BY4741 and BY4742 (41). All yeast transformations were performed using the lithium-acetate method (42), were confirmed by PCR on genomic DNA, and were genomic sequence-confirmed. Details of all yeast strains constructed for this study, oligonucleotides used in construction (Table S4), plasmids with sequence information (Table S5), and cultures used for protein experiments are available in SI Appendix, SI Materials. All Western and fitness measurements were replicated using two isogenic independently isolated transformants, except for fitness measurements of YFPm2.
Soluble, Insoluble, and Total Protein Isolation and Quantification.
Approximately 1.25 × 108 cells were harvested by centrifugation at 3,000 × g for 1 min from mid-log cultures. Insoluble and soluble protein fractions and total protein extracts were extracted as described in SI Appendix, SI Materials. All Western-analyzed strains (MD729, MD732, KG030, KG032, and KG035) contained two distinct epitope-tagged copies of YFP: PGAL1-3×Myc-YFPref and one of the PGAL1-3×FLAG-YFPwt/m1–4 variants (Table S3). We used the reference protein YFPref as a control to ensure that differences in signal intensities were not the result of differences in culture densities, expression resulting from galactose induction, or loading volume. Both strains analyzed by MS, KG071 and KG079, were auxotrophic for lysine (lys2Δ0). KG079 grown on media containing 13C-15N-lysine and KG071 grown on 12C-14N-lysine or vice versa were harvested for total protein, which was mixed 1:1, digested in solution by the endoproteinase LysC, and analyzed by LC-MS/MS using an Orbitrap Velos (Thermo Fisher) and MaxQuant (28). Additional details of protein quantification can be found in SI Appendix, SI Materials.
Competitive Growth Rate Measurements.
We measured fitness using flow cytometry to monitor growth competitions of fluorescently tagged strains (KG001, KG006, KG039, KG041, KG046, KG007, SW001, and SW004) as in ref. 21 but with controlled maximum culture densities maintained through frequent dilution and without high-throughput automation. Relevant modifications are listed in SI Appendix, SI Materials. Growth experiments were performed in synthetic complete media (Sunrise Biosciences) with 2% sucrose, 1% raffinose, and 27.5 mM galactose (induced) or no galactose (uninduced).
Competitive Fitness Quantification.
Let nx (ti) be the number of cells of type x at time point i (time ti). If rx measures the growth rate in generations per unit time (the inverse of the doubling time), then 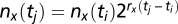 , and the number of generations that pass between time point i and j is
, and the number of generations that pass between time point i and j is 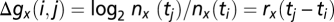 . To eliminate absolute time, we pace each strain using a reference strain growing in the same vessel, yielding
. To eliminate absolute time, we pace each strain using a reference strain growing in the same vessel, yielding 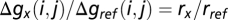 and cumulative generations
and cumulative generations 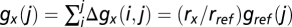 , such that reference-relative growth rate measurements rx/rref can be extracted by fitting straight lines as in Fig. 2. The difference in growth rates between two strains, the selective advantage of strain x over strain y, is then
, such that reference-relative growth rate measurements rx/rref can be extracted by fitting straight lines as in Fig. 2. The difference in growth rates between two strains, the selective advantage of strain x over strain y, is then 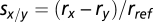 .
.
Supplementary Material
Acknowledgments
We thank J. Koschwanez, C. Marx, J. Wakeley, and N. Ingolia for helpful discussions and T. Kortemme, M. Eames, C. Wilke, M. Laub, B. Stern, A. Murray, and V. Denic for discussions and critical input on the manuscript. We are grateful to the MaxQuant team for their efforts. This work was supported by National Institutes of Health Grants 1R01GM088344-01, P50GM068763, GM079536, and GM065169.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1017570108/-/DCSupplemental.
References
- 1.Pakula AA, Sauer RT. Genetic analysis of protein stability and function. Annu Rev Genet. 1989;23:289–310. doi: 10.1146/annurev.ge.23.120189.001445. [DOI] [PubMed] [Google Scholar]
- 2.Tokuriki N, Tawfik DS. Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature. 2009;459:668–673. doi: 10.1038/nature08009. [DOI] [PubMed] [Google Scholar]
- 3.Stoebel DM, Dean AM, Dykhuizen DE. The cost of expression of Escherichia coli lac operon proteins is in the process, not in the products. Genetics. 2008;178:1653–1660. doi: 10.1534/genetics.107.085399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J Mol Med. 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 5.Bucciantini M, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 6.Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci USA. 2005;102:14338–14343. doi: 10.1073/pnas.0504070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drummond DA, Raval A, Wilke CO. A single determinant dominates the rate of yeast protein evolution. Mol Biol Evol. 2006;23:327–337. doi: 10.1093/molbev/msj038. [DOI] [PubMed] [Google Scholar]
- 8.Pál C, Papp B, Hurst LD. Highly expressed genes in yeast evolve slowly. Genetics. 2001;158:927–931. doi: 10.1093/genetics/158.2.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parsell DA, Lindquist S. The function of heat-shock proteins in stress tolerance: Degradation and reactivation of damaged proteins. Annu Rev Genet. 1993;27:437–496. doi: 10.1146/annurev.ge.27.120193.002253. [DOI] [PubMed] [Google Scholar]
- 11.Travers KJ, et al. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 12.Brauer MJ, et al. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol Biol Cell. 2008;19:352–367. doi: 10.1091/mbc.E07-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sugio A, Dreos R, Aparicio F, Maule AJ. The cytosolic protein response as a subcomponent of the wider heat shock response in Arabidopsis. Plant Cell. 2009;21:642–654. doi: 10.1105/tpc.108.062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aparicio F, et al. Virus induction of heat shock protein 70 reflects a general response to protein accumulation in the plant cytosol. Plant Physiol. 2005;138:529–536. doi: 10.1104/pp.104.058958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Metzger MB, Michaelis S. Analysis of quality control substrates in distinct cellular compartments reveals a unique role for Rpn4p in tolerating misfolded membrane proteins. Mol Biol Cell. 2009;20:1006–1019. doi: 10.1091/mbc.E08-02-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gidalevitz T, Krupinski T, Garcia S, Morimoto RI. Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet. 2009;5:e1000399. doi: 10.1371/journal.pgen.1000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parsell DA, Sauer RT. Induction of a heat shock-like response by unfolded protein in Escherichia coli: Dependence on protein level not protein degradation. Genes Dev. 1989;3:1226–1232. doi: 10.1101/gad.3.8.1226. [DOI] [PubMed] [Google Scholar]
- 18.Plata G, Gottesman ME, Vitkup D. The rate of the molecular clock and the cost of gratuitous protein synthesis. Genome Biol. 2010;11:R98. doi: 10.1186/gb-2010-11-9-r98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giaever G, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 20.Dekel E, Alon U. Optimality and evolutionary tuning of the expression level of a protein. Nature. 2005;436:588–592. doi: 10.1038/nature03842. [DOI] [PubMed] [Google Scholar]
- 21.Breslow DK, et al. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Methods. 2008;5:711–718. doi: 10.1038/nmeth.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong H, Nilsson L, Kurland CG. Gratuitous overexpression of genes in Escherichia coli leads to growth inhibition and ribosome destruction. J Bacteriol. 1995;177:1497–1504. doi: 10.1128/jb.177.6.1497-1504.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ingolia NT, Murray AW. Positive-feedback loops as a flexible biological module. Curr Biol. 2007;17:668–677. doi: 10.1016/j.cub.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siemering KR, Golbik R, Sever R, Haseloff J. Mutations that suppress the thermosensitivity of green fluorescent protein. Curr Biol. 1996;6:1653–1663. doi: 10.1016/s0960-9822(02)70789-6. [DOI] [PubMed] [Google Scholar]
- 25.Jakubowska A, Korona R. Lack of evolutionary conservation at positions important for thermal stability in the yeast ODCase protein. Mol Biol Evol. 2009;26:1431–1434. doi: 10.1093/molbev/msp066. [DOI] [PubMed] [Google Scholar]
- 26.Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454:1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crameri A, Whitehorn EA, Tate E, Stemmer WP. Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat Biotechnol. 1996;14:315–319. doi: 10.1038/nbt0396-315. [DOI] [PubMed] [Google Scholar]
- 28.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 29.Ghaemmaghami S, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 30.Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol. 2007;25:117–124. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- 31.Newman JR, et al. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006;441:840–846. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- 32.de Godoy LM, et al. Comprehensive mass spectrometry-based proteome quantification of haploid versus diploid yeast. Nature. 2008;455:1251–1254. doi: 10.1038/nature07341. [DOI] [PubMed] [Google Scholar]
- 33.Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 34.Teixeira MC, et al. The YEASTRACT database: A tool for the analysis of transcription regulatory associations in Saccharomyces cerevisiae. Nucleic Acids Res. 2006;34:D446–D451. doi: 10.1093/nar/gkj013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trotter EW, et al. Misfolded proteins are competent to mediate a subset of the responses to heat shock in Saccharomyces cerevisiae. J Biol Chem. 2002;277:44817–44825. doi: 10.1074/jbc.M204686200. [DOI] [PubMed] [Google Scholar]
- 36.Erkina TY, Tschetter PA, Erkine AM. Different requirements of the SWI/SNF complex for robust nucleosome displacement at promoters of heat shock factor and Msn2- and Msn4-regulated heat shock genes. Mol Cell Biol. 2008;28:1207–1217. doi: 10.1128/MCB.01069-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lynch M, Conery JS. The origins of genome complexity. Science. 2003;302:1401–1404. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- 38.Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 39.Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006;174:229–239. doi: 10.1534/genetics.106.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ananthan J, Goldberg AL, Voellmy R. Abnormal proteins serve as eukaryotic stress signals and trigger the activation of heat shock genes. Science. 1986;232:522–524. doi: 10.1126/science.3083508. [DOI] [PubMed] [Google Scholar]
- 41.Brachmann CB, et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 42.Amberg DC, Burke DJ, Strathern JN. Methods in Yeast Genetics: a Cold Spring Harbor Laboratory Course Manual. NY: Cold Spring Harbor; 2005. [Google Scholar]
- 43.Fleming JA, et al. Complementary whole-genome technologies reveal the cellular response to proteasome inhibition by PS-341. Proc Natl Acad Sci USA. 2002;99:1461–1466. doi: 10.1073/pnas.032516399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.