

Abstract
In this study we gain insight into the structural and functional characterization of the Aeropyrum pernix oligopeptide-binding protein (OppAAp) previously identified from the extracellular medium of an Aeropyrum pernix cell culture at late stationary phase. OppAAp showed an N-terminal Q32 in a pyroglutamate form and C-terminal processing at the level of a threonine-rich region probably involved in protein membrane anchoring. Moreover, the OppAAp protein released into the medium was identified as a “nicked” form composed of two tightly associated fragments detachable only under strong denaturing conditions. The cleavage site E569-G570 seems be located on an exposed surface loop that is highly conserved in several three-dimensional (3D) structures of dipeptide/oligopeptide-binding proteins from different sources. Structural and biochemical properties of the nicked protein were virtually indistinguishable from those of the intact form. Indeed, studies of the entire bacterially expressed OppAAp protein owning the same N and C termini of the nicked form supported these findings. Moreover, in the middle exponential growth phase, OppAAp was found as an intact cell membrane-associated protein. Interestingly, the native exoprotein OppAAp was copurified with a hexapeptide (EKFKIV) showing both lysines methylated and possibly originating from an A. pernix endogenous stress-induced lipoprotein. Therefore, the involvement of OppAAp in the recycling of endogenous proteins was suggested to be a potential physiological function. Finally, a new OppA from Sulfolobus solfataricus, SSO1288, was purified and preliminarily characterized, allowing the identification of a common structural/genetic organization shared by all “true” archaeal OppA proteins of the dipeptide/oligopeptide class.
The controlled uptake of molecules by cells is essential for life, representing the way by which nutrients from the surrounding medium are imported and communication with the environment is maintained. One of the largest and most diverse protein families functioning as a transport system is the protein-dependent ATP-binding cassette (ABC) transporter (15, 16, 22, 25). It is generally composed of two multitransmembrane permeases constituting the translocator component and two intracellular ATP-binding proteins, expressed either as separate polypeptides or fused to each other in any possible combination (3, 30). Unlike their eukaryotic counterparts, bacterial ABC import systems involve a further component, the substrate-binding protein (SBP), which determines the uptake selectivity of their cognate ABC transporters, acting as the initial receptor to bind and deliver the substrates to the permease domain (16, 22).
Bacterial ABC uptake systems are divided into two main classes: the carbohydrate transporter (CUT) and the dipeptide/oligopeptide (DOP) uptake transporters (3, 30). At a genetic level, genes encoding the protein components of these systems are typically organized into an operon structure, which is strongly conserved despite the extensive shuffling of gene locations driving bacterial evolution.
The SBP component of ABC uptake systems shows great differences in terms of amino acid sequence (25). In Gram-negative bacteria, SBPs reside in the periplasmic space (14, 33), while in Gram-positive bacteria, they are anchored to the outer surface of the cell membrane via N-terminally linked lipids (4, 18, 33). The archaeal SBPs belonging to the two transporter classes, the CUT and DOP classes, differ in size, domain organization, and type of signal peptides (2, 3). SBPs of the CUT family exhibit an N-terminal signal peptide followed by a stretch of hydroxylated amino acids, and it is assumed that these proteins are anchored to the cytoplasmic membrane via N-terminal lipid modifications or N-terminal transmembrane (TM) domains (TMDs) (2, 10, 19). On the other hand, SBPs of the DOP class contain a N-terminal bacterium-like signal peptide and a transmembrane domain at the C-terminal end, which is preceded by a long stretch of hydroxylated amino acids. The substrate-binding domain of these SBPs is believed to be membrane anchored by the carboxy-terminal membrane-spanning domain (2).
So far, different ABC extracellular-binding proteins typically involved in carbohydrate transport have been studied mostly for the genera Sulfolobus (11), Thermococcus, and Pyrococcus (20), showing that they have a very high affinity for their carbohydrate substrates.
However, analyses of the archaeal sequenced genomes revealed the presence of several ABC systems originally designed as oligopeptide transporters, but investigations of the SBP components showed that oligosaccharides were their likely substrates (3, 11, 20).
Recently reported data (13) referred to the isolation and characterization of two archaeal OppA proteins from Sulfolobus solfataricus, SSO2619 and SSO1273. These binding proteins showed different peptide selectivities but the same structural/genomic organizations.
In this paper, we report a more detailed structural/functional study of the first archaeal oligopeptide-binding protein of an ABC-type transporter, Aeropyrum pernix OppA (OppAAp), previously isolated from A. pernix culture medium at late stationary phase (26). A. pernix is an obligate aerobic hyperthermophilic archaeon isolated from deep-sea hydrothermal vents, which derives its energy primarily from the aerobic degradation of peptides and amino acids (29).
The purified OppAAp protein evidenced the occurrence of protein processing that did not seem to affect the secondary structure and the peptide-binding activity compared to those of the intact bacterially expressed recombinant form. In addition, OppAAp was found as an unprocessed membrane-associated protein during the exponential growth phase of A. pernix, and functional investigations supplied new and interesting clues into the involvement of OppAAp in the recycling of endogenous proteins. Finally, an appealing notation emerged, analyzing the OppAAp primary structure and its ABC operon architecture, revealing the occurrence of common features in structural/genetic organization also observed for the two OppA proteins isolated from S. solfataricus and useful for a possible protein function prediction of SBPs in Archaea. These findings were soundly confirmed by the identification of a novel OppA protein, SSO1288, purified from S. solfataricus and structurally and functionally characterized.
MATERIALS AND METHODS
Enzymes and reagents.
Trypsin, endoproteinase V8, CNBr, dithiothreitol, and alpha-cyano-4-hydroxycinnamic acid were purchased from Sigma. High-performance liquid chromatography (HPLC)-grade trifluoroacetic acid (TFA) was obtained from Carlo Erba (Italy). [3H]bradykinin (B3H) was obtained from Perkin-Elmer, and the EKFKIV synthetic peptide with the two lysines in either the methylated or unmethylated form was obtained from Primm (Italy). All other reagents and solvents of the highest purity were available from J. T. Baker (Holland).
Strains and growth conditions.
Aeropyrum pernix K1 (JCM 9820) was grown at 90°C in a 10-liter fermentor, and OppAAp was purified as described previously by Palmieri et al. (26). The preparation of the membrane surface protein fraction from A. pernix was performed as described previously by Palmieri et al. (27). Escherichia coli strain BL21(DE3)-CodonPlus-RIL (Stratagene) was used for gene expression. Plasmids pMOS-Blue (Promega) and pET-22b (Novagen) were used for cloning and expression experiments. S. solfataricus cultures were performed according to methods described previously by Gogliettino et al. (13).
Protein isolation and purification.
Proteins were precipitated from 10 liters of filtered culture medium by the addition of (NH4)2SO4 up to 90% saturation, and OppAAp was purified as described previously by Palmieri et al. (26). The protein concentration was determined according to the Bradford assay method (7).
Electrophoresis.
Native PAGE was performed on a 9% polyacrylamide gel (pH 9.5) as a stacking gel and a 12.5% acrylamide gel (pH 7.5) as a running gel, at 15 mA for 3 h.
SDS-PAGE analysis was carried out on 12.5% polyacrylamide gels according to methods described previously by Laemmli (21). For molecular mass determinations, the gel was calibrated with prestained protein markers (New England Biolabs). Prior to electrophoresis the protein sample was diluted in standard denaturing sample buffer (12.5 mM Tris-HCl [pH 6.8], 0.4% SDS, 0.14 M β-mercaptoethanol) and heated in a boiling-water bath for 5 min (Bio-Rad). Alternatively, the protein was precipitated with trichloroacetic acid (TCA) (20% [vol/vol] final concentration), washed twice with ethyl ether and acetone, and then resuspended in denaturing sample buffer.
In situ digestions.
Protein bands stained with Coomassie brilliant blue were excised from preparative SDS electrophoresis gels on a 12.5% polyacrylamide gel and subjected to in situ digestions (24). Enzymatic digestions were carried out with trypsin or endoproteinase V8 (12.5 ng/μl) in 50 mM ammonium bicarbonate buffer (pH 8.5).
CNBr cleavage.
Peptide bond cleavage at the methionine residues was performed by incubating purified OppAAp with CNBr (Fluka, Switzerland). The lyophilized protein (0.1 mg) was dissolved in 0.1 ml of 70% TFA and hydrolyzed with CNBr at room temperature for 18 h in the dark. The sample was diluted five times with water (at 4°C), lyophilized, and then dissolved in 0.02 ml of 0.2% TFA for matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) analysis.
MALDI-mass spectrometry (MS) analysis.
MALDI-TOF mass spectra were recorded by using a Voyager STR instrument (Applied Biosystems). Peptide solution in 0.3% TFA was mixed with either 10 mg/ml alpha-cyano-hydroxycinnamic acid in acetonitrile (0.1% TFA [2.5:1, vol/vol]) or 50 mg/ml 4-hydroxycinnamic acid in acetonitrile (0.1% TFA [1:1, vol/vol]) and applied onto the metallic sample plate and dried under a vacuum. Mass calibration was performed by using external standards. Raw data were analyzed by using computer software provided by the manufacturer and are reported as monoisotopic.
Nano-LC-ESI-MS/MS analysis.
An LC-tandem MS (MS/MS) experiment on the trypsin digest was carried out with an Agilent 1100 system (Agilent Technologies) coupled with a 4000 Q Trap mass spectrometer (Applied Biosystems). Peptides were separated with a Zorbax 300SB-C18 nanocolumn (3.5 μm; 0.075 by 150 mm) at a flow rate of 0.2 μl/min using the following gradient: 5% solvent B to 65% solvent B over 45 min (solvent A contained 0.1% formic acid and 2% acetonitrile in water; solvent B contained 0.1% formic acid and 2% water in acetonitrile). Each peptide was loaded onto Agilent Zorbax 300SB-C18 trap columns (5 μm; 0.3 mm by 5 mm). The interface temperature was kept at 150°C. For LC-MS coupling, the Agilent column was connected with a nanospray source, equipped with two cameras, and focused on the electrospray ionization (ESI) needle and the curtain plate hole, using a liquid junction with distal coated silica tips with an outer diameter (OD) of 360 μm and an inner diameter (ID) of 20 μm. The needle voltage was set at 2 kV. Enhanced resolution (ER), enhanced product ion (EPI), and time-delayed fragmentation (TDF) spectra of the selected doubly charged ions were acquired. For MS/MS scan modes, a scan rate of 4,000 Da/s was used, with Q trapping activated to achieve maximum sensitivity. The trap fill time was set to 100 ms, and the collision energy (CE) was set to 33 V for doubly charged ions and 26 V for triply charged ions. The TDF CE was set to 18 V. The declustering potential was set at 50 V. Nitrogen was used as a curtain gas (at a value of 10 liters/h) and collision gas (set to high). Two independent nano-LC-ESI-MS/MS experiments were performed for each sample.
Sequence and structural analysis.
Automated N-terminal sequence analysis was performed by using a Perkin-Elmer Applied Biosystems 477A pulsed-liquid protein sequencer equipped with a model 120A phenylthiohydantoin analyzer. The sequence database was searched by using the BLAST-PSI program (5). Multiple-sequence alignments were generated with the ClustalW program (32). All experiments were performed in triplicate with two different protein preparations.
Peptide-binding assays.
The peptide-binding activity of purified OppAAp was assayed by using xenopsin or bradykinin (both from Sigma). The purified protein (0.8 nmol) was added to 50 mM sodium phosphate buffer (pH 7.0) containing different amounts of each peptide substrate (0.2 to 2.0 nmol) in a final volume of 0.2 ml. The mixtures were incubated at 90°C for 30 min and successively kept in ice to stop the reaction. Each sample was filtered through a Microcon YM filter (Millipore) with a cutoff of 10,000 Da at 4°C and 12,000 × g. The eluate recovered after each filtration (0.18 ml) was analyzed by reverse-phase HPLC (BioLC; Dionex) on a μBondapak C18 column (3.9 by 300 mm; Waters) eluted with a linear gradient (0 to 60% acetonitrile in 0.1% TFA) at a flow rate of 1 ml/min. For each binding assay a control peptide sample incubated in the absence of OppAAp was run in parallel, and the peak area was used to calculate the relative amount of bound peptide. Mixtures of the peptide substrates incubated with an equimolar concentration of bovine serum albumin (BSA) or trehalose-binding protein from S. solfataricus were used as negative controls for peptide binding.
The peptide-binding activity was also evaluated, using the radioligand B3H as described previously by Gogliettino et al. (13), with some modifications. B3H (0.25 pmol) was incubated with OppAAp (1 pmol) in 50 mM sodium phosphate buffer (pH 7.0) in a final volume of 0.1 ml. The mixture was incubated at 90°C for 30 min and successively put on ice to stop the reaction. All experiments were performed in triplicate with two different protein preparations.
Cloning and expression of recombinant protein.
Chromosomal DNA, used as a template in PCR assays, was prepared from A. pernix cells according to methods described previously by Yoshida et al. (35). The following oligonucleotide primers were synthesized: 1583f (5′-GATATCGTGGACAAGGGGCCCGTGG-3′) and 1583r (5′-GCCGCGGCCGCTGTTGTGGTGGTCG-3′). Extra bases (underlined) were added to create ends compatible with the cloning vector. The PCR amplifications were performed on a PCR thermal cycler (Hybaid) in a 0.05-ml volume of reaction mixture containing 1× PCR buffer GC (Finnzymes), 0.2 mM deoxynucleoside triphosphates, 1 μM (each) primers, 2.5 U of Phusion polymerase (Finnzymes), and 100 ng of genomic DNA template. The amplification conditions used were 95°C for 5 min (1 cycle), followed by 95°C for 1 min, 60°C for 1 min, and 72°C for 1 min for 40 cycles and a final extension step at 72°C for 10 min. The amplified product was cloned into the pMOS-Blue vector (Promega) according to the manufacturer's instructions and then sequenced on both strands by using the ABI Prism system. All restriction enzymes and T4 DNA ligase were obtained from New England Biolabs. The 2.2-kb NdeI/NotI DNA fragment excised from the pMOS-Blue cloning vector was then subcloned into the NdeI/NotI-digested pET-22b expression vector (Novagen). The resulting plasmid was designated pET-1583 and was used to transform E. coli strain BL21(DE3)-CodonPlus-RIL for the expression assays.
Recombinant cells harboring plasmid pET-1583 were grown in LB medium (2.25 liters) containing ampicillin (0.1 mg ml−1) until the A600 reached 0.7 OD units. Protein expression was induced by the addition of 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG), followed by further cultivation for 3 h. The cells were harvested by centrifugation at 9,000 × g for 30 min at 4°C, suspended in 50 mM Tris-HCl buffer (pH 8.0) containing 2 mM phenylmethylsulfonyl fluoride (PMSF), and disrupted by two cycles with a French press (American Instrument Company).
The cell debris was removed by centrifugation at 18,000 × g for 30 min at 4°C, and the supernatant fraction was recovered. After a 3-fold dilution, the proteic solution was incubated at 70°C for 30 min under conditions of continuous agitation. After centrifugation at 18,000 × g for 30 min at 4°C, the supernatant phase containing the protein of interest was extensively dialyzed against 1 M ammonium sulfate in 20 mM Tris-HCl buffer (pH 8.0) at 4°C overnight. The sample was then loaded onto a phenyl-Sepharose column (1.6 by 2.5 cm) connected to a fast protein liquid chromatography (FPLC) system (Amersham) equilibrated with 1 M ammonium sulfate in 20 mM Tris-HCl (pH 8.0) (buffer A) at a flow rate of 0.5 ml/min. The fractions containing the protein of interest were eluted with a linear gradient (0 to 100%) of buffer A in distilled water and then dialyzed against 20 mM Tris-HCl buffer (pH 8.0). The collected sample was purified by gel filtration chromatography on a Superdex 200 column (3.2 mm by 300 mm; Pharmacia) connected with a Smart system (Pharmacia Biotech) preequilibrated with 50 mM Tris-HCl buffer (pH 8.0) containing 0.15 M NaCl at a flow rate of 0.05 ml/min. The purified recombinant protein was stored at 4°C in 50 mM Tris-HCl buffer (pH 7.0) for further characterization.
Isolation and identification of a natural peptide bound to OppAAp.
Purified OppAAp (1 nmol) was loaded onto a Superose 12 HR 10/30 gel filtration chromatography column (Amersham, United Kingdom) connected to an FPLC system. The column was eluted with 50 mM sodium phosphate buffer (pH 7.0) containing 0.15 M NaCl (flow rate, 1 ml/min). The calibration of the column was performed by using the following standard gel filtration calibration kit (Amersham) standards: thyroglobulin (670 kDa), IgG (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (1.3 kDa). All fractions containing OppAAp were pooled, desalted, and further analyzed by reverse-phase HPLC on a Phenomenex Jupiter C18 column (250 by 2.1 mm; 300-Å pore size) with a linear gradient of 15 to 65% acetonitrile (0.1% TFA) in water (0.1% TFA) over 55 min at a flow rate of 200 μl/min.
Individual fractions were collected and analyzed by MALDI-TOF analysis. MS/MS experiments with samples eluted from the reverse-phase HPLC column described above were also carried out with an HCT Bruker ion trap system (Bruker). Lyophilized samples were dissolved in a solution containing 20% formic acid and 80% acetonitrile and were introduced into the source by using a syringe pump system at a flow rate of 10 μl/min; the interface temperature was kept at 350°C. The needle voltage was set at 4.5 kV. The instrument acquired full-scan spectra of the singly charged ions. For MS/MS scan modes, a scan rate of 8,000 m/z/s was used. The maximum value of the trap fill time was set to 100 ms, and the CE was changed from 30% to 200% of the amplitude value. Nitrogen was used as the curtain gas (at a value of 7 liters/h), and helium was used as the collision gas (set to a high value). The amount of the endogenous ligands copurified with OppAAp was determined by reverse-phase HPLC with a Phenomenex Jupiter C18 column, building calibration curves with the synthetic peptides. All experiments were performed in triplicate with two different protein preparations.
Circular dichroism spectroscopy.
Circular dichroism (CD) spectra were obtained with a Jasco J-715 spectropolarimeter with 400 μl of 8.0 × 10−7 M protein in 5 mM Tris-HCl (pH 7.5). Hellma quartz cells of a 0.1-cm path length were used for far-UV (190 to 250 nm). The temperature of the sample cell was regulated by a PTC-348 WI thermostat. Thermal CD experiments were performed by raising the cell temperature from 25°C to 90°C in the 250- to 195-nm UV range. They were signal averaged by adding three scans and baseline corrected by subtracting a buffer spectrum. The sample was then cooled back to 25°C in order to monitor the final folding of the protein.
Purification of SSO1288 from S. solfataricus.
Extracellular proteins from a 10-liter S. solfataricus culture in TYS medium (13) were precipitated by the addition of (NH4)2SO4 up to 90% saturation as previously described (13). The precipitate was resuspended in 50 mM Tris-HCl (pH 7.0) and extensively dialyzed against the same buffer. The protein solution was then loaded onto an anionic-exchange chromatography column of DEAE-cellulose (DE52, 1 by 24 cm; Whatman) equilibrated with 50 mM Tris-HCl buffer (pH 7.0). Bound proteins were eluted by a 400-ml linear gradient from 0.0 to 1.0 M NaCl in the equilibration buffer at a flow rate of 0.8 ml/min. Fractions containing proteins migrating at approximately 90 kDa on SDS-PAGE gels were pooled, dialyzed against 50 mM Tris-HCl buffer (pH 8.0) containing 1.0 M ammonium sulfate, and applied onto a HiTrap phenyl-Sepharose HP column (Amersham) preequilibrated with the same buffer. The column was washed at a flow rate of 1 ml/min, and elution was performed with a linear gradient of ammonium sulfate from 1 M to 0 M in 50 mM Tris-HCl buffer (pH 8.0). The collected fractions containing the SSO1288 protein (identified by the N-terminal sequence) were concentrated on an Amicon PM-10 membrane and loaded onto an anionic-exchange chromatography MonoQ column (Pharmacia) in a Smart system (Pharmacia Biotech) equilibrated with 50 mM Tris-HCl buffer (pH 7.0). The column was washed at a flow rate of 0.05 ml/min, and a linear gradient of 0.0 to 1.0 M NaCl in the equilibration buffer (1 ml) was applied. Fractions containing the protein of interest were pooled, desalted, and concentrated for further characterization.
RESULTS AND DISCUSSION
Mapping of the N- and C-terminal ends of OppAAp.
An ABC transporter oligopeptide-binding protein from the hyperthermophilic archaeon Aeropyrum pernix (OppAAp) was previously purified and preliminarily characterized (26).
In this study the results of new investigations of the properties of OppAAp allowed the recognition of some basic common structural features of the archaeal proteins among the DOP class of transporters.
As previously reported, the N terminus of OppAAp was supposed to be Q32 converted into the pyroglutamate form (26). Therefore, OppAAp was digested by trypsin, and a portion of the tryptic peptide mixture was analyzed by LC-MS/MS. The ion corresponding to the doubly charged pQ32-K43 peptide (Fig. 1) was selected and fragmented, and the y ion series (y8, 934.7; y7, 837.7; y6, 738.6; y5, 609.7; y4, 508.7; y3, 395.7; y2, 294.7; y1, 147.7) clearly indicated that the peptide 32QAGQPVETITFK43 was the N terminus (Fig. 1) of the exoprotein OppAAp, being the first residue Q32 in a pyroglutamate form. Hence, the ape1583-translated product was processed for translocation at the A31-Q32 site that is highly conserved in several archaeal substrate-binding proteins (3), cleaving off a 31-amino-acid signal peptide. Indeed, the enzymatic conversion of the N-terminal glutamine into pyroglutamic acid is known to be catalyzed by glutaminyl cyclase (QC), which has been isolated from many sources but never found in members of the Archaea (34). Thus, the N-terminal glutamine residue of OppAAp could undergo a nonenzymatic spontaneous cyclization resulting in the formation of pyroglutamic acid, as described previously by Welker et al. (34).
FIG. 1.

Amino acid sequence deduced from the A. pernix open reading frame (ORF) APE1583.1. Bold red letters identify the N and C termini of the OppAAp protein (pQ32 and T819). The N-terminal sequence, included in a green box, was obtained by LC-MS/MS analysis. The nicked peptide bond (E570-G571) is marked by a red asterisk. The N-terminal sequence of the 27-kDa fragment, obtained by Edman degradation analysis, is black boxed. The coverage sequence obtained by proteolysis is shown underneath the amino acid sequence as follows: blue lines indicate the peptides obtained by endoproteinase V8 hydrolysis, and red lines indicate peptides obtained by CNBr hydrolysis. Regions in gray and blue correspond to the 57-kDa and 27-kDa fragments, respectively.
Since the tryptic digestion did not cover the C-terminal region of OppAAp, endoproteinase V8, in situ hydrolysis of the protein was performed, followed by MALDI-MS analysis (see Table S1 in the supplemental material). The results allowed us to obtain further sequence information (Fig. 1), but no mass values corresponding to peptides sited in the C-terminal threonine-rich region (S/T rich) were detected, thus suggesting that the exoprotein OppAAp could have a ragged C-terminal end.
To achieve more information on the OppAAp C-terminal end, a chemical digestion with CNBr (see Table S2 in the supplemental material) was performed under strong acidic conditions to favor the formation of homoserine lactone over open homoserine at the C terminus of the originated peptides. Therefore, only homoserine lactone derivatives were observed. The results further validated the OppAAp amino acid sequence and allowed a possible recognition of the C terminus at position T819 (Fig. 1). In fact, the mass signals at m/z 4,166.2, 4,267.9, 4,368.1, and 4,479.3 (Table S2), associated with the peptides at residues 780 to 816, 780 to 817, 780 to 818, and 780 to 819, respectively, all contained a threonine residue at the C terminus. In addition, these assignments were confirmed by submitting a portion of the digest to a single step of Edman degradation followed by MALDI-MS analysis (Table S2). Therefore, the occurrence of C-terminal processing at the level of the S/T-rich region of OppAAp released into the extracellular medium was most likely proved, suggesting the involvement of the C-terminal region in protein membrane anchoring. Indeed, the long stretch of hydroxylated amino acids may function as a flexible linker region to couple the substrate-binding domain to the transmembrane-anchoring region (11).
Molecular properties of the exoprotein OppAAp.
The amino acid sequence of the purified exoprotein OppAAp, covered by mass-mapping experiments (spanning from Q32 to T819), has a theoretical mass value of 88.3 kDa, which is in agreement with the 90-kDa value estimated by gel filtration chromatography (26) but considerably larger than the 42-kDa value determined by SDS-PAGE (see Fig. S1A, lane 2, in the supplemental material) (26). On the other hand, OppAAp subjected to a strong denaturing procedure by TCA precipitation migrated as two electrophoretic bands with apparent molecular masses of 57 kDa and 27 kDa (Fig. S1A). Similar species were observed after boiling OppAAp for 30 min in 2% SDS (data not shown). Thus, it appeared that the 42-kDa form represented an anomalously migrating species, a frequently observed phenomenon occurring in hyperthermophilic proteins, exhibiting a high level of resistance to thermal and/or SDS denaturation (12, 13). The two fragments were investigated by in situ digestions with trypsin and endoproteinase V8, followed by MALDI-MS or LC-MS/MS analysis. Results showed that the 57-kDa digests included the peptides spanning from the N-terminal pQ32 (glutamine in a pyroglutamate form) to residue E569 (Fig. S1B), while the mass signals identified from the 27-kDa digests corresponded to the region between G570 and T819 (Fig. S1B). Noticeably, the sequence coverage from the two complementary polypeptide fragments obtained by mass spectrometry analyses (Fig. S1A, lane 3) was the same as that for the single band at 42 kDa and that previously reported (26). Moreover, Edman degradation analysis of the electroblotted 27-kDa band showed the N-terminal sequence (GAQASAKVTF) (Fig. 1) starting from G570, as revealed from the 42-kDa form corresponding to an incompletely denatured protein. On the other hand, no sequence was detected on the native protein in solution, possibly due to some interference factors in the N-terminal sequencing reaction. Therefore, we propose that the OppAAp released into the medium represents a “nicked isoform” of the protein most likely generated by a proteolytic event at the E569-G570 site (Fig. 1) producing two complementary fragments that are tightly associated (17, 28). The high level of resistance of this active complex to the denaturing conditions could be due to the extensive contacts with each other, forming a tight interlocked network of interactions according to the principle of cooperativity governing protein structure (28).
The susceptibility of the putative site (E569-G570) to proteolytic cleavage was explored by aligning the OppAAp sequence with those of the cognate proteins whose crystal structures are available (Fig. 2 A). According to this multiple alignment, the region including the peptide bond E569-G570 bore some similarity both in length and in sequence (Fig. 2B) to an exposed loop of domain III in the crystal structures of AppA proteins from Bacillus subtilis (31), Lactococcus lactis, and Thermotoga maritima (Fig. 2A). Interestingly, our data suggested that the E569-G570 site represented a cleavable “permissive site” located away from the binding pocket in a highly flexible, solvent-exposed region of OppA.
FIG. 2.

(A) Cartoon rocket representation of the three-dimensional (3D) structures of three periplasmic binding proteins: Lactococcus lactis OppA (OppALl) (Protein Data Bank [PDB] accession number 3DRF), AppA from Bacillus subtilis (AppABs) (PDB accession number 1XOC), and Thermotoga maritima OppA (OppATm) (PDB accession number 1VR5). The peptide bond which in OppAAp is subjected to proteolysis is marked by yellow arrows. (B) Multiple-sequence alignment of OppAAp with the binding proteins mentioned above. The alignment was generated by ClustalW and colored according to the ClustalX coloring scheme. The bar consensus graph represents the alignment quality using a BLOSUM62 score based on observed substitutions. The black arrow indicates a common sequence region corresponding to the external loop, which is nicked in OppAAp.
Membrane surface localization of OppAAp.
To date, ABC substrate-binding components have been found as membrane-associated proteins. In a previous work we carried out a proteomic analysis to identify exoproteins and outer surface proteins extracted from A. pernix cells harvested at the middle exponential growth phase (0.5 OD at 600 nm [OD600] units) (27). The results indicated that OppAAp was present mainly within the membrane-surface protein fraction, and it was found largely in the electrophoretic band migrating at 200 kDa (Fig. 3), as revealed by SDS-PAGE and mass spectrometry analyses (27). Similar investigations of the extracellular protein fraction (27) showed that at this exponential stage of cell growth, OppAAp was appreciably released into the culture medium, as it was predominantly identified in the 200-kDa electrophoretic band (Fig. 3). However, neither the extracellular nor the membrane-associated protein was proteolysed at the E569-G570 site, since the tryptic peptide 558DYIPGVGSVVSEGAQASAK576, including the E569-G570 bond, was identified in both OppAAp forms (data not shown) (27). Conversely, at the late stationary phase, OppAAp was purified as the exoprotein nicked form (see Fig. S1A, lane 2, in the supplemental material), thus suggesting that proteolysis and other posttranscriptional modifications occurred after the protein was released into the medium. The large relative amount of OppAAp in both the surface membrane and the extracellular fractions and its resistance to SDS denaturing conditions (26), which has been reported frequently for other archaeal proteins (12, 13), could explain the distribution in several SDS-PAGE bands.
FIG. 3.
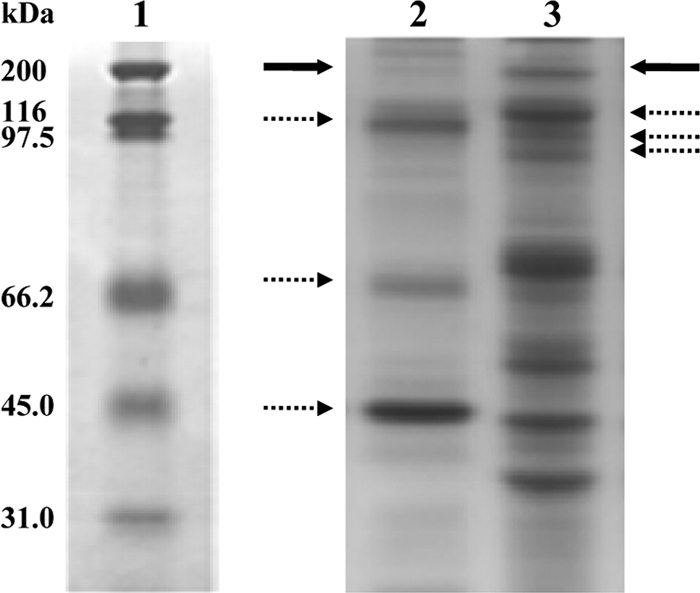
SDS-PAGE analysis of extracellular and outer surface protein extracts from Aeropyrum pernix cells harvested at the middle exponential growth phase. Lane 1, molecular mass markers (broad range); lane 2, extracellular protein fraction; lane 3, membrane-surface protein fraction. OppAAp was identified from tryptic digests followed by LC-MS/MS analysis of several electrophoresis bands. A large amount of OppAAp was revealed in the 200-kDa band, indicated by bold arrows; the protein was also identified from different electrophoretic bands (dashed arrows).
Expression and purification of a recombinant truncated form of OppAAp.
To assess a possible structure-function relationship of the nicked exoprotein form with respect to the membrane-anchored unproteolysed protein, OppAAp was expressed in E. coli cells as a monomer. Since the N and C termini of native OppAAp mapped to the Q32 and T819 residues, respectively, a pair of oligonucleotides was designed to match the corresponding genomic region on the ape1583.1 gene. Upon an amplification assay, a single fragment of 2.2 kb was yielded from A. pernix genomic DNA, cloned into the pMOS-Blue vector, and sequenced. After restriction analyses, the gene was cloned into pET-22b to construct plasmid pET-1583 to obtain the recombinant protein fused to the 6His tag at the C-terminal end. The OppAAp protein was expressed in E. coli BL21(DE3)-CodonPlus-RIL cells and isolated from the crude extracts as a soluble protein with a final yield of 44% (Table 1).
TABLE 1.
Purification of the recombinant oligopeptide-binding protein OppAAp based upon 2.25-liter cultures of E. colia
| Purification step | Total amt of protein (mg) | % binding protein | Amt of binding protein (mg) | Yield (%) | Fold purification |
|---|---|---|---|---|---|
| Extract | 1,025 | 11.6 | 119 | 100 | 1 |
| Thermoprecipitate | 307 | 29.2 | 90 | 75.6 | 2.5 |
| Phenyl Sepharose | 180 | 50 | 90 | 75.6 | 4 |
| Superdex 200 | 58 | 90 | 52.2 | 44 | 7.8 |
The percentage of oligopeptide-binding protein was determined by subjecting samples from each purification step to SDS-PAGE and by scanning individual lanes by using a Bio-Rad ChemiDOC laser scanning densitometer instrument. Due to the presence of a minor protein component which migrates close to the oligopeptide-binding protein, the percentage of the first three steps should be taken as an upper limit.
Recombinant OppAAp subjected to standard denaturing procedures associated with SDS-PAGE analysis showed two bands migrating at 83 kDa and 42 kDa (see Fig. S1C, lane 3, in the supplemental material). However, by TCA precipitation, a unique electrophoretic band with a relative molecular mass of about 83.0 kDa was revealed by SDS-PAGE analysis (Fig. S1C, lane 2). Moreover, the native and recombinant OppAAp proteins had the same molecular mass values determined under native conditions (about 90 kDa) and comparable electrophoretic mobilities under native PAGE conditions (Fig. S1D), thus suggesting similar quaternary structures.
Peptide-binding activity of the native and recombinant forms of OppAAp.
The binding activity of the purified native (exoprotein) and recombinant OppAAp forms was assayed by using the peptide substrates xenopsin and bradykinin. Results obtained with the native protein indicated an approach toward binding saturation in both instances (Fig. 4 A), and Scatchard plot analyses (data not shown) allowed the determination of the dissociation constants (Kd), which were in the submicromolar range, together with the Bmax values corresponding to a binding stoichiometry of 1:1 (mol/mol) (Fig. 4 A). Comparable binding experiments were performed by using the same peptide substrates and recombinant OppAAp as the binding protein (Fig. 4 B). Notably, the Kd and Bmax values were in the same order of magnitude as those observed for the native exoprotein, showing little variations.
FIG. 4.

Peptide-binding activity of native and recombinant forms of OppAAp. (A) Representative saturation curve of the native exoprotein OppAAp using bradykinin (•) or xenopsin (○) as a substrate. (Insert) Binding assay performed using the radioligand B3H (▵) as a substrate and native OppAAp. Control experiments with the incubation of B3H alone (○) and with BSA (dashed line) were carried out. (B) Saturation curve of recombinant OppAAp using bradykinin (•) or xenopsin (○) as a substrate.
The high affinity that OppAAp displayed toward the tested peptides was in agreement with that reported previously for the cognate archaeal carbohydrate-binding proteins and the homologous OppA proteins from S. solfataricus so far characterized (1, 13). These data confirmed that the SBP components of ABC systems in members of the Archaea are characterized by a very high affinity for their substrates, allowing the cells to efficiently utilize carbon sources in substrate-poor environments such as hydrothermal vents in the deep sea or in hot volcanic sulfuric pools.
A direct binding assay was also performed by using the radioligand B3H and native OppAAp as the binding protein and monitoring complex formation by gel filtration chromatography (13). The results showed that when B3H was gel filtered alone or with BSA (10 pmol), used as a control protein, the entire radioactivity was recovered in a peak between 0.8 and 2.2 ml of the elution volume. In the presence of the native OppAAp protein (1 pmol), 67% of radioactivity was found in an excluded peak eluted within a volume of 0.2 to 0.8 ml; the decreasing net elution volume of the cpm peak validated the bradykinin-OppAAp complex formation (Fig. 4A, insert).
These results evidenced that the processed nicked isoform of OppAAp was able to bind oligopeptide substrates with a high affinity. In addition, the functional properties of recombinant OppAAp, expressed as a unique polypeptide, suggested that the proteolytic nick could not influence the biological activity of the protein. Finally, both the native and recombinant forms, lacking the S/T-rich and TM domains in the C-terminal end, retained their biological activity, thus suggesting no contribution of these structural regions to the protein function.
Identification of a peptide ligand copurified with the native exoprotein OppAAp.
Oligopeptide-binding proteins are frequently purified in their liganded form, carrying a population of peptides differing in size and amino acid composition (23, 33). This phenomenon is interpreted as being a consequence of the moderate to high affinity that OppA proteins usually display for their substrates and their binding slow off rates.
On the basis of these observations, a further study was performed to eventually identify the liganded peptides carried by the purified transporter. As shown in Fig. S2 in the supplemental material, purified OppAAp eluted from a gel filtration column was further subjected to C18 reverse-phase chromatography to isolate the bound peptides.
Results from MALDI-MS analyses of all the collected fractions revealed a signal only at m/z 791.5, associated with the predominant peak, eluted from the reverse-phase column. These samples were further analyzed by LC-MS/MS, which showed that the main peak was a mixture of a hexapeptide (EKFKIV) with one and/or two lysine residues in the methylated form. A small amount (about 30%) of this hexapeptide (EKFKIV) was found to be unmodified as also revealed by Edman degradation analysis. In order to assess a specific recognition of the native OppAAp protein toward EKFKIV, binding assays were carried out under the conditions optimized for the peptide control, bradykinin (see Fig. S3 in the supplemental material) and using OppAAp/substrate molar ratios of 1:50 and 1:80 mol/mol. Preliminary results indicated that the amounts of bound peptides were 87% and 83% for the methylated and unmethylated forms of EKFKIV, respectively, and 50% for bradykinin, proving that the recovery of the peptide copurified with OppAAp could not be ascribed to artifacts of the protein preparation. However, the OppAAp fraction purified in complex with EKFKIV accounted for about 6%, as evaluated by determining the amount of the peptide (0.06 nmol) released from 1 nmol OppAAp (Fig. S2). Therefore, the protein was largely in a noncomplexed form and suitable for analyses of functional properties.
It is quite interesting that the above-described peptide is a fragment of a putative stress-induced membrane lipoprotein sequence encoded by the ape2211 gene of A. pernix. This finding may lead one to suppose that OppAAp and the cognate ABC transporters could be involved in the recycling of peptides arising from proteins of the same organism released from cells at late stationary phase. The occurrence of the bound peptide in the methylated form supported this working hypothesis, since a substoichiometric N methylation of lysine residues is a very common posttranslational modification in members of the Archaea (36).
Secondary structural analysis of the native and recombinant forms of OppAAp.
In order to obtain information about protein secondary structure, circular dichroism spectroscopy analyses of the native and recombinant OppAAp proteins were performed. The CD spectra obtained in a 195- to 250-nm-wavelength range (Fig. 5) were performed by covering a temperature range of 25°C to 90°C at increasing steps of 5°C, with a heating rate of 40°C per h. The plots obtained from the native and recombinant OppAAp proteins were nearly identical in the near-UV region; thus, only those of the native protein were reported. An analysis of the spectra showed that by increasing the temperature up to 90°C, no significant gain in molar ellipticity values was revealed in the region close to the minimum at 210 nm, and the same behavior was observed by cooling the sample back to 25°C, indicating a great stability of the proteins. Since molar ellipticity is an optical property of the protein molecules closely related to their conformational state, our results evidenced that temperature caused null or low structural changes in the OppAAp protein. Moreover, the virtually superimposable CD spectra of the native and recombinant forms suggested that the proteolytic cleavage occurring at E570-G571 did not affect the secondary and tertiary structures of the protein.
FIG. 5.

CD spectra of native OppAAp. Analysis was performed in the temperature range of 25°C to 90°C and by setting the wavelength coverage to 195 to 250 nm. Continuous and dashed-line curves represent CD spectra at 25°C and 90°C, respectively. Mol. Ellip., molar ellipticity.
Basic structural/genetic motifs of archaeal OppA proteins.
As described previously, the ABC uptake transporter systems in the Archaea, as in the Bacteria, are divided into two main classes: the CUT and the DOP classes (2, 30). This is mostly clear when the domain organization of the SBP components is examined (Fig. 6). Members of the CUT class show an unusual N-terminal cleavage site followed by a hydrophobic stretch of amino acids (TMD) that function as an N-terminal membrane anchor in the processed protein. The flexibility is provided by a domain of hydroxylated (S/T-rich) amino acids (11). These signal sequences resemble class III signal peptides, and indeed, an archaeal type IV prepilin signal peptidase can process the precursor of binding proteins, especially in S. solfataricus (Fig. 6) (2). Moreover, the classical lipid motif for attaching the binding protein on the cell surface, as found in Gram-positive bacteria, can be observed (4, 18, 33). In S. solfataricus and A. pernix, some members of the CUT class exhibit a TMD in the C-terminal region (Fig. 6). Conversely, binding proteins of the DOP class reveal a classical cleavable N-terminal bacterial secretion signal sequence. The processed and secreted proteins achieve their membrane attachment via a hydrophobic C-terminal TMD that is thought to be integrated into the membrane and is connected to the soluble part of the protein with a flexible linker region (S/T-rich) that allows mobility (Fig. 7 A).
FIG. 6.

Schematic domain organization of SBPs from Sulfolobus solfataricus and Aeropyrum pernix. DOP and CUT indicate the dipeptide/oligopeptide and carbohydrate uptake transporter classes. SP sp, classical bacterium-like signal peptide; SP TIVP, archaeal type IV pilin signal peptide; SP TatP, Tat signal peptide; BD, binding domain; S/T-rich, serine/threonine-rich domain; TMD, transmembrane domain. ST-rich+ and ST-rich−, CUT subfamilies including SBP members with or without an S/T-rich domain. The characterized oligopeptide-binding proteins are shown in green, while SBPs with carbohydrate-binding activity are in red.
FIG. 7.

Structural and functional characteristics of the four “real OppA” proteins belonging to the newly defined class. (A) Alignment of the C-terminal amino acid sequences of the ORFs SSO1273, SSO2619, SSO1288, and APE1583.1. The gray and black boxes indicate the highly conserved threonine-rich ST linker and the transmembrane region (TM), respectively. (B) All the N-terminal amino acid sequences were determined by Edman degradation analysis (SSO2619, first reported in this work); the hypothetical archaeal consensus processing site of the OppA proteins was also reported. (C) Oligopeptide-binding activity of the OppA proteins from S. solfataricus and Aeropyrum pernix. The specificity of the four isolated proteins toward oligopeptide ligands was assayed with bradykinin and xenopsin; data are reported as a percentage versus the maximum binding activity showed by SSO1288 on bradykinin (100%); the trehalose (TreS)-binding protein was used as a negative control (0% activity).
As recently reported, SBPs showing the typical structural features of the DOP class were later revealed to be sugar-binding proteins (SSO2669, SSO3043, and SSO3053) (3, 11, 20), and these divergences made the prediction of the functional specificity of ABC transporters slightly unsuccessful.
In this paper we further characterized an OppA protein from A. pernix displaying the same domain organization as that of two more OppA proteins recently studied from S. solfataricus and belonging to the archaeal DOP class. Moreover, the three isolated archaeal OppA proteins are SBP components of ABC uptake systems that are not surrounded by genes encoding sugar-metabolizing enzymes, suggesting that all these structural/genetic motifs could be useful for a possible protein function prediction of SBPs in members of the Archaea (Fig. 6). We set out to test this hypothesis by reassigning all the putative or characterized ABC-type SBPs from S. solfataricus and A. pernix to the DOP or CUT class in view of these findings to select the possible true OppA candidates from the two microorganisms (Fig. 6). With this analysis, only OppA from A. pernix and SSO1273, SSO2619, and SSO1288 from S. solfataricus were revealed to be members of the DOP class, with the latter being the unique component not yet characterized as an OppA protein. All the remaining SBP members of the CUT class exhibit a large assortment of domain organizations (Fig. 6), suggesting that the structural requirements needed to play its sugar-capturing/transporter role in the cell membrane can be less restrictive than those required for oligopeptide binding.
Therefore, to support our hypothesis, we decided to purify and functionally characterize the SSO1288 protein. The protein was isolated from the extracellular medium of S. solfataricus cell cultures by three chromatographic steps (Table 2). Purified SSO1288 shows a molecular mass of 90 kDa under denaturing conditions (see Fig. S4, lane 3, in the supplemental material) and an N-terminal sequence of YALITNFQTP, which perfectly matched the polypeptide encoded by the sso1288 gene starting from position +26, indicating secretion signal processing.
TABLE 2.
Purification of the native oligopeptide-binding protein SSO1288 from 10 liters of Sulfolobus solfataricus cell culturea
| Purification step | Total amt of protein (mg) | % binding protein | Amt of binding protein (mg) | Yield (%) | Fold purification |
|---|---|---|---|---|---|
| Broth | 21.64 | 100 | 1.19 | 100 | 1 |
| Ammonium sulfate (90%) | 18.01 | 97 | 1.17 | 98.32 | 1.03 |
| DEAE-52 | 2.65 | 33.15 | 0.90 | 76.92 | 2.93 |
| Phenyl-Sepharose | 0.30 | 21.5 | 0.07 | 7.78 | 1.54 |
| MonoQ | 0.07 | 90 | 0.06 | 85.71 | 4.18 |
The percentage of oligopeptide-binding protein was determined by subjecting samples from each purification step to SDS-PAGE and by scanning individual lanes using a Bio-Rad ChemiDOC laser scanning densitometer instrument.
Binding assays using bradykinin and xenopsin as the ligands (13) were performed for comparisons with the other three OppA proteins (OppAAp, SSO1273, and SSO2619) in order to demonstrate that the isolated SSO1288 protein was able to specifically bind short peptides. All the assays were carried out with a 1:1 molar ratio and identical concentrations of the specific protein and ligands. As shown in Fig. 7C, 100% capture of both ligands was obtained by SSO1288, while OppAAp revealed 70% to 80% binding toward both peptide substrates. The binding activities of SSO2619 and SSO1273 were somehow intermediate. Structural (Fig. 7A and B) and functional (Fig. 7C) data not only confirmed the membership of SSO1288 to the DOP family but also contributed to defining the archaeal consensus processing site of OppA as being [AT]−3 [SI]−2 [AG]−1 [QY]+1 [AST]+2 (Fig. 7B). This signal peptide cleavage site basically matched the motif specifically recognized by prokaryotic (P)-type SPase I (2), while the consensus pattern identified for the three sugar-binding proteins (SSO3043, SSO3053, and SSO2669) of [VAI]−3 [NL]−2 [S]−1 [QA]+1. [S]+2 of the DOP class (Fig. 6) could be recognized by eukaryotic (ER)-type SPases (2, 8); therefore, the consensus sequence can be used as a tool for the discrimination of new “real OppA” proteins.
Conclusions.
An enhancement of the peptide transport in two of the most studied archaeal species, A. pernix and S. solfataricus, has been presented, and implications of findings from this study were discussed. DOP-binding proteins in bacteria (4, 6, 9) were usually membrane anchored; by extension, in members of the Archaea, these proteins were supposed to bind the external side of the cellular membrane via a TMD present in the carboxyl-terminal region (3). Conversely, OppAAp, when purified from cells at stationary phase, was found as a soluble active form in the extracellular medium (26), proteolytically processed at the C terminus. In fact, the C-terminal threonine-rich region, the TMD, and the putative consensus sequence for glycosylphosphatidylinositol (GPI) lipid modification (10, 26) were removed, but the OppAAp protein preserved its function, suggesting no contribution of these structural regions to the protein activity. Furthermore, OppAAp was specifically processed at the level of the E569-G570 bond, producing two fragments tightly associated in an extracellular heterodimeric complex able to efficiently bind oligopeptide substrates. In addition, the sequence of the nicked loop showed a reasonable degree of conservation throughout the divergent archaeal oligopeptide-binding family. Since the unnicked recombinant protein was able to bind the same oligopeptides tested with the native OppAAp protein with similar affinities, it can therefore be hypothesized that the loop in which cleavage occurred was not directly involved in protein function, although further analyses were necessary to better clarify a possible functional role of this region.
Finally, the two newly investigated OppA proteins, OppAAp and SSO1288, in addition to the already characterized SSO1273 and SSO2619 proteins, gave us the tools for planning new hypotheses. Usually, the SBPs of both the CUT and DOP classes were supplied with similar structural motifs but different domain organizations; nevertheless, in some cases sugar-binding proteins have revealed the same structural architecture as that found for OppA proteins, further complicating the interpretation of the binding data. Moreover, an additional discriminatory tool was the check for the gene neighborhoods in the ABC transporter operon. Our goal was to identify four archaeal SBPs experimentally characterized as being “true” OppA proteins (Fig. 6), having common structural motifs and no genes encoding sugar-metabolizing enzymes in the operon's surrounding regions. These data allow the proposal of new guidelines for the field of SBP classification. Databank screening for putative OppA proteins from other archaeal organisms using these structural/genetic motifs could provide new important insights into the DOP class.
Supplementary Material
Footnotes
Published ahead of print on 19 November 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Albers, S. V., et al. 1999. Glucose transport in the extremely thermoacidophilic Sulfolobus solfataricus involves a high-affinity membrane-integrated binding protein. J. Bacteriol. 181:4285-4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albers, S. V., and A. J. Driessen. 2002. Signal peptides of secreted proteins of the archaeon Sulfolobus solfataricus: a genomic survey. Arch. Microbiol. 177:209-216. [DOI] [PubMed] [Google Scholar]
- 3.Albers, S. V., S. M. Koning, W. N. Konings, and A. J. Driessen. 2004. Insights into ABC transport in Archaea. J. Bioenerg. Biomembr. 36:5-15. [DOI] [PubMed] [Google Scholar]
- 4.Alloing, G., P. de Philip, and J. P. Claverys. 1994. Three highly homologous membrane-bound lipoproteins participate in oligopeptide transport by the Ami system of the Gram-positive Streptococcus pneumoniae. J. Mol. Biol. 241:44-58. [DOI] [PubMed] [Google Scholar]
- 5.Altschul, S. F., et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borezee, E., E. Pellegrini, and P. Berche. 2000. OppA of Listeria monocytogenes, an oligopeptide-binding protein required for bacterial growth at low temperature and involved in intracellular survival. Infect. Immun. 68:7069-7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 8.Dalbey, R. E., and G. von Heijne. 1992. Signal peptidases in prokaryotes and eukaryotes—a new protease family. Trends Biochem. Sci. 17:474-478. [DOI] [PubMed] [Google Scholar]
- 9.Detmers, F. J., F. C. Lanfermeijer, and B. Poolman. 2001. Peptides and ATP binding cassette peptide transporters. Res. Microbiol. 152:245-258. [DOI] [PubMed] [Google Scholar]
- 10.Eisenhaber, B., P. Bork, and F. Eisenhaber. 2001. Post-translational GPI lipid anchor modification of proteins in kingdoms of life: analysis of protein sequence data from complete genomes. Protein Eng. 14:17-25. [DOI] [PubMed] [Google Scholar]
- 11.Elferink, M. G., S. V. Albers, W. N. Konings, and A. J. Driessen. 2001. Sugar transport in Sulfolobus solfataricus is mediated by two families of binding protein-dependent ABC transporters. Mol. Microbiol. 39:1494-1503. [DOI] [PubMed] [Google Scholar]
- 12.Gentile, F., et al. 2002. SDS-resistant active and thermostable dimers are obtained from the dissociation of homotetrameric β-glycosidase from hyperthermophilic Sulfolobus solfataricus in SDS. Stabilizing role of the A-C intermonomeric interface. J. Biol. Chem. 277:44050-44060. [DOI] [PubMed] [Google Scholar]
- 13.Gogliettino, M., et al. 2010. A highly selective oligopeptide binding protein from the archaeon Sulfolobus solfataricus. J. Bacteriol. 192:3123-3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodell, E. W., and C. F. Higgins. 1987. Uptake of cell wall peptides by Salmonella typhimurium and Escherichia coli. J. Bacteriol. 169:3861-3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Higgins, C. F. 1992. ABC transporters: from microorganisms to man. Annu. Rev. Cell Biol. 8:67-113. [DOI] [PubMed] [Google Scholar]
- 16.Higgins, C. F. 2001. ABC transporters: physiology, structure and mechanism—an overview. Res. Microbiol. 152:205-210. [DOI] [PubMed] [Google Scholar]
- 17.Hutchens, T. W., J. F. Henry, and T. T. Yip. 1991. Structurally intact (78-kDa) forms of maternal lactoferrin purified from urine of preterm infants fed human milk: identification of a trypsin-like proteolytic cleavage event in vivo that does not result in fragment dissociation. Proc. Natl. Acad. Sci. U. S. A. 88:2994-2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jenkinson, H. F., R. A. Baker, and G. W. Tannock. 1996. A binding-lipoprotein-dependent oligopeptide transport system in Streptococcus gordonii essential for uptake of hexa- and heptapeptides. J. Bacteriol. 178:68-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi, T., R. Nishizaki, and H. Ikezawa. 1997. The presence of GPI linked protein(s) in an archaeobacterium, Sulfolobus acidocaldarius, closely related to eukaryotes. Biochim. Biophys. Acta 1334:1-4. [DOI] [PubMed] [Google Scholar]
- 20.Koning, S. M., M. G. Elferink, W. N. Konings, and A. J. Driessen. 2001. Cellobiose uptake in the hyperthermophilic archaeon Pyrococcus furiosus is mediated by an inducible, high-affinity ABC transporter. J. Bacteriol. 183:4979-4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 22.Lee, S. J., A. Bohm, M. Krug, and W. Boos. 2007. The ABC of binding-protein-dependent transport in Archaea. Trends Microbiol. 15:389-397. [DOI] [PubMed] [Google Scholar]
- 23.Levdikov, V. M., et al. 2005. The structure of the oligopeptide-binding protein, AppA, from Bacillus subtilis in complex with a nonapeptide. J. Mol. Biol. 345:879-892. [DOI] [PubMed] [Google Scholar]
- 24.Medugno, L., et al. 2003. A novel zinc finger transcriptional repressor, ZNF224, interacts with the negative regulatory element AldA-NRE. and inhibits gene expression. FEBS Lett. 534:93-100. [DOI] [PubMed] [Google Scholar]
- 25.Monnet, V. 2003. Bacterial oligo-peptide binding proteins. Cell. Mol. Life Sci. 60:2100-2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palmieri, G., et al. 2006. Identification of the first archaeal oligopeptide binding protein from the hyperthermophile Aeropyrum pernix. Extremophiles 10:393-402. [DOI] [PubMed] [Google Scholar]
- 27.Palmieri, G., R. Cannio, I. Fiume, M. Rossi, and G. Pocsfalvi. 2009. Outside the unusual cell wall of the hyperthermophilic archaeon Aeropyrum pernix K1. Mol. Cell. Proteomics 8:2570-2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ray, S. S., H. Balaram, and P. Balaram. 1999. Unusual stability of a multiply nicked form of Plasmodium falciparum triosephosphate isomerase. Chem. Biol. 6:625-637. [DOI] [PubMed] [Google Scholar]
- 29.Sako, Y., et al. 1996. Aeropyrum pernix gen. nov., sp. nov., a novel aerobic hyperthermophilic archaeon growing at temperatures up to 100 degrees C. Int. J. Syst. Bacteriol. 46:1070-1077. [DOI] [PubMed] [Google Scholar]
- 30.Schneider, E. 2001. ABC transporters catalyzing carbohydrate uptake. Res. Microbiol. 152:303-310. [DOI] [PubMed] [Google Scholar]
- 31.Tame, J. R., E. J. Dodson, G. Murshudov, C. F. Higgins, and A. J. Wilkinson. 1995. The crystal structures of the oligopeptide-binding protein OppA complexed with tripeptide and tetrapeptide ligands. Structure 3:1395-1406. [DOI] [PubMed] [Google Scholar]
- 32.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The Clustal_X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4867-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tynkkynen, S., et al. 1993. Genetic and biochemical characterization of the oligopeptide transport system of Lactococcus lactis. J. Bacteriol. 175:7523-7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Welker, E., et al. 2007. Oxidative folding and N-terminal cyclization of onconase. Biochemistry 46:5485-5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshida, K., et al. 2001. Combined transcriptome and proteome analysis as a powerful approach to study genes under glucose repression in Bacillus subtilis. Nucleic Acids Res. 29:683-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zappacosta, F., G. Sannia, L. A. Savoy, G. Marino, and P. Pucci. 1994. Post-translational modifications in aspartate aminotransferase from RT Sulfolobus solfataricus. Detection of N-epsilon-methyllysines by mass RT spectrometry. Eur. J. Biochem. 222:761-767. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.