

Abstract
Rationale
Myocardial infarction (MI) leads to heart failure (HF) and premature death. The respective roles of myocyte death and depressed myocyte contractility in the induction of HF after MI have not been clearly defined and are the focus of this study.
Objectives
We developed a mouse model in which we could prevent depressed myocyte contractility after MI and used it to test the idea that preventing depression of myocyte Ca2+-handling defects could avert post-MI cardiac pump dysfunction.
Methods and Results
MI was produced in mice with inducible, cardiac-specific expression of the β2a subunit of the L-type Ca2+ channel. Myocyte and cardiac function were compared in control and β2a animals before and after MI. β2a myocytes had increased Ca2+ current; sarcoplasmic reticulum Ca2+ load, contraction and Ca2+ transients (versus controls), and β2a hearts had increased performance before MI. After MI, cardiac function decreased. However, ventricular dilation, myocyte hypertrophy and death, and depressed cardiac pump function were greater in β2a versus control hearts after MI. β2a animals also had poorer survival after MI. Myocytes isolated from β2a hearts after MI did not develop depressed Ca2+ handling, and Ca2+ current, contractions, and Ca2+ transients were still above control levels (before MI).
Conclusions
Maintaining myocyte contractility after MI, by increasing Ca2+ influx, depresses rather than improves cardiac pump function after MI by reducing myocyte number.
Keywords: myocardial infarction, cardiac contractility, heart failure, Ca2+ handling
Vascular disease can lead to interruption of blood flow to the heart (myocardial infarction [MI]). The resulting loss of contractile mass causes an acute reduction in cardiac pump function. Cardiac output and blood pressure are initially maintained by activation of sympathetic reflex responses that increase myocyte contractility in regions of the heart where blood flow is maintained. Soon after MI, the ventricles begin to remodel (dilate), which further increases the work demands (systolic wall stress) on the surviving myocardium.
Myocyte contractility is increased after MI through activation of adrenergic signaling pathways that increase Ca2+ influx and sarcoplasmic reticulum (SR) uptake, storage, and release. The increased myocyte Ca2+ transients that enhance contractile function after MI are also thought to induce myocyte hypertrophy,1–3 increasing contractile mass and partially normalizing the increased systolic wall stress. However, persistent pathological stress in the remodeled, post-MI heart is associated with abnormal myocyte contractile properties4,5 and increases in the rate of myocyte death.6–8 These two changes develop with congestive heart failure (CHF) and increase during its progression.6,9 The respective contributions of myocyte contractile abnormalities (weak myocytes) and myocyte death (not enough myocytes) in the induction and progression of HF after MI is still not clearly defined. We studied this issue by preventing depression of myocyte contractile function in genetically modified mice subjected to MI.
Persistent pathological stress (MI and hypertension) induces abnormalities in myocyte Ca2+ handling5,10 and sympathetic signaling cascades.11 SR Ca2+ uptake rates are slowed; Ca2+ transient and action potential durations are prolonged5,12; and inotropic responses to sympathetic agonists are blunted.13 These changes are centrally involved in the slowing of contraction and relaxation rates, prolongation of contractile duration, and depressed contractility reserve.14,15 What is still not clear is whether these Ca2+ handling alterations induce cardiac decompensation and cause its progression or whether they are secondary to the ever increasing demand for enhanced myocyte function in the pathological heart.
The depressed myocyte contractility hypothesis predicts that HF therapies that increase myocyte contractility will improve cardiac pump function, HF symptoms, and survival. Unfortunately, most inotropic therapies that have been tested in patients over the past few decades have either had no effect on survival16 or have increased death rates.17,18 Therapies19 that have clinical benefit in HF, inhibitors of excess renin–angiotensin and β-adrenergic signaling, reduce rather than enhance myocyte contractility,19,20 suggesting that myocyte contractile abnormalities in HF might be the consequence of the excessive contractility demands rather than the cause of HF per se.
HF induction and progression is also associated with an increased rate of myocyte death from apoptosis and necrosis.8 Excessive activation of sympathetic and renin–angiotensin signaling pathways in HF,21 as well as oxidative stress, cytokine accumulation,22 and persistent activation of Ca2+ signaling,3,23,24 are all thought to contribute.25,26 Our working hypothesis is that excessive demands for contractility in HF increase cell death and it is cell death rather than reduced myocyte contractile function that causes HF progression.
The objective of our present study was to determine whether preventing depressed myocyte contractility after MI, without activating the sympathetic nervous system, improves cardiac pump function and slows CHF progression. To explore this topic we developed a genetically modified mouse in which we could increase Ca2+ influx by conditional, cardiac specific overexpression of the β2a subunit of the L-type Ca2+ channel (termed β2a mice).8 This approach increases Ca2+ influx and myocyte contractility26,27 without requiring sympathetic nerve activity. If depressed myocyte contractility causes abnormal pump function and CHF after MI, then we would expect better cardiac pump function in our β2a mice in which depressed myocyte contractility is prevented. If on the other hand the remodeling of myocyte Ca2+ handling after MI is a response to the excessive demands for enhanced function, and in part this remodeling protects the myocyte from Ca2+-mediated damage, then maintaining high Ca2+ might increase myocyte death and exacerbate the depression of cardiac pump function.
Methods
All methods have been described in detail in previous reports and are described in detail in the Online Data Supplement, available http://circres.ahajournals.org. Briefly, myocardial infarction was induced in wild-type and β2a transgenic mice by permanent occlusion of the left anterior descending coronary artery. Cardiac function was measured with echocardiography. At euthanasia, hearts were removed and used for functional studies (isovolumic hearts and isolated single ventricular myocytes [VMs]), tissue was used for Western or biochemical analyses, or hearts were fixed and processed for histological studies.
Results
β2a-LTCC Hearts Are Hypercontractile
Mice with conditional, cardiac-specific, low-level expression of Cav1.2 β2a were used (Figure 1A).8 A modified α-myosin heavy chain promoter was used for cardiac-specific expression of the β2a gene, and mice containing both tTA (tetracycline-controlled transactivator) and β2a transgenes (double transgenic) allowed for doxycycline (Dox) suppression of β2a expression (Dox-off system) during development. Mice were taken off Dox-containing chow after weaning (at 21 days) and used at 4 to 6 months of age, because at this age, the β2a gene was fully expressed and mice showed enhanced cardiac performance (Figure 1B), with no signs of cardiomyopathy.8 β2a mice had increased ejection fraction (EF) (Figure 1C) and fractional shortening (FS) (Figure 1D) versus controls. Posterior wall thickness (PWT) and septal wall thickness were greater and left ventricular (LV) internal diameter was smaller (LVID) in β2a animals (Figure 1E, 1F, and 1G). These data show that β2a hearts were hypercontractile with modest LV hypertrophy (Figure 1H).
Figure 1. β2a hearts are hypercontractile with mild hypertrophy.
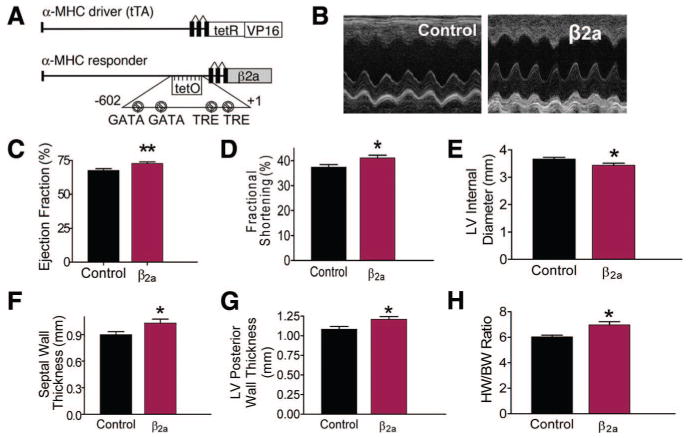
A, Bitransgenic inducible expression system to induce β2a expression. tTA is the tetracycline-controlled transactivator. B, Representative echocardiography M-mode image in control and β2a hearts. C through G, EF, FS, PWT, and septal wall thickness were greater, and left ventricle internal diameter (LVID) was smaller in β2a vs control mice (control, n=46; β2a, n=36). H, HW/BW ratio in β2a hearts (n=51) was greater than in controls (n=18). *P<0.05; **P<0.01.
Infarct Size Is Increased in β2a Hearts Because of Increased Myocyte Death
MI was induced by interruption of blood flow to the LV via left anterior descending coronary artery (LAD). The area of the heart with interrupted blood flow was identical in control and β2a hearts (area at risk [AAR]; control versus β2a: 45.1±3.2% versus 50.4±6.8%; Figure 2A; Online Figure I, A). However, myocardial infarct size (as a % of AAR) was significantly larger in β2a mice than in controls (control versus β2a: 45.3±6.1% versus 83.8±14.4%) (Figure 2B; Online Figure I, B). Infarction length measured 6 weeks after MI was also significantly greater in β2a than in control mice (control versus β2a: 21.7±4.9% versus 45.8±6.8%) (Figure 2C; Online Figure I, C). These data suggest that more myocytes in the AAR die in β2a mice. We used the TUNEL assay to measure apoptotic cell death in the MI border and in remote zones. TUNEL + myocyte number was significantly greater in β2a hearts 3 days (β2a versus WT: 59 015 versus 16 600 in border and 3407 versus 1984 labeled/106 nuclei in the remote zone) and 3-week after MI (959 versus 647 in border and 1415 versus 709 labeled/106 nuclei in remote zone) (Figure 2D through 2F; and Online Figure II, A and B). Caspase 3 activity was also greater in β2a hearts (remote zone) 3 weeks after MI; indicative of cells undergoing apoptosis (Figure 2G). Myocyte death is associated with replacement fibrosis28 and the collagen content of β2a hearts was significantly greater than in controls and increased to a greater extent with time after MI (Figure 2H). Ki67 (proliferative marker) labeling was greater in β2a versus WT hearts after acute (4223 versus 2621 labeled/106) or 3 weeks MI (1974 versus 1542 labeled/106) (Figure 2I; Online Figure II, C). Collectively these results show that there is an increased rate of death of β2a myocytes after MI stress, both at the infarct border zone (larger infarct size) and in remote areas of the heart.
Figure 2. MI and ischemia/reperfusion (I/R) causes more cell death in β2a hearts.
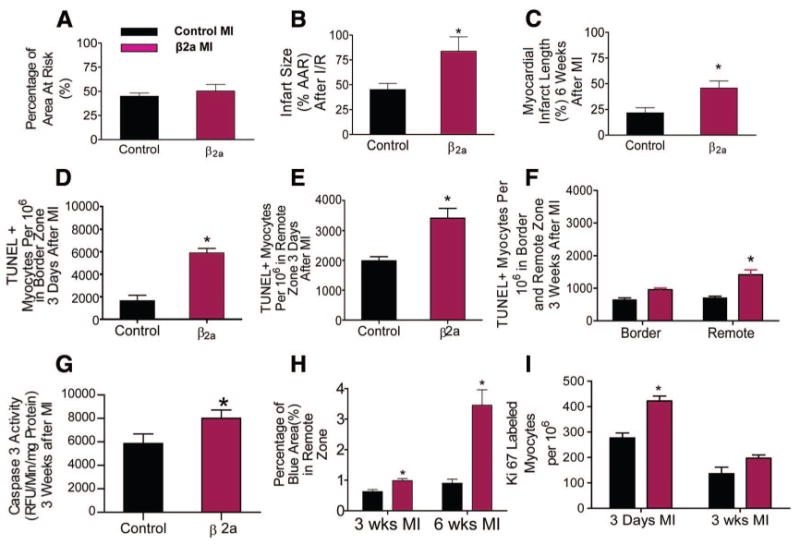
A, AAR was not different in control (n=15) and β2a hearts (n=6). B, Infarct size (% of AAR) in hearts with 30 minutes of ischemia followed by 24 hours of reperfusion (I/R) was greater in β2a (n=6) than in control hearts (n=8). C, Infarct length after permanent occlusion was greater in β2a (n=6) than in controls (n=7) and. D through F, Myocytes per 106 undergoing apoptosis in the border and remote zones of control (n=3) and β2a hearts (n=4). G, Caspase 3 activity in remote zone tissues (control, n=9; β2a, n=10). H, Fibrotic area (blue) in remote zones of trichrome-stained cardiac histological sections from control (n=3) and β2a hearts (n=3). I, Ki67+ myocytes per 106 in control (n=3) and β2a hearts (n=4) after MI. *P<0.05; **P<0.01.
β2a Hearts Have Depressed Function After Ischemia/Reperfusion Stress
Our MI experiments suggest that the β2a heart is more prone to acute ischemic injury. To more directly test this idea we compared the effects of in vitro ischemia-reperfusion (IR) stress on ventricular performance in isovolumic hearts subjected to 15 minutes of global no flow ischemia, followed by 30 minutes of reperfusion. Before IR, LV developed pressure (LVDP) and ±dP/dT (mm Hg/s) in β2a hearts were significantly greater than in controls (Figure 3A; Online Figure III, A and B). During the ischemic period, end diastolic pressure (EDP) became significantly greater in β2a hearts versus control, suggesting they develop Ca2+ overload-related increases in diastolic pressure (Figure 3B through 3C). β2a hearts also had greater disruption of contractile function after reperfusion (Figure 3B). Recovery of LVDP was significantly reduced in β2a versus control hearts (Figure 3B) and the EDP was significantly greater in β2a hearts than in control hearts (Figure 3D). The increased EDP and reduced LVDP are consistent with enhanced Ca2+ stress in the β2a that results in SR Ca2+ overload.26 These results document that β2a hearts are prone to ischemia-related injury, without the neuohormonal stressors present in the post-MI heart.
Figure 3. Ischemia/reperfusion depresses cardiac function in β2a hearts.

A, Representative recordings of LV pressure in control and β2a isolated hearts. B through D, LVDP and LV end diastolic pressure (EDP) during and after ischemia/reperfusion in control (n=8) and β2a (n=5) isolated hearts. *P<0.05.
More β2a Animals Die After MI and Survivors Have More Pathological Hypertrophy
Animals were studied over a 6 week period after MI. Representative examples of heart and lung morphology in control and β2a mice after MI surgery are shown in Online Figure I (D). β2a animals were significantly more likely to die after MI than were controls (Figure 4A), with only 19% alive after 6 weeks, versus 60% of controls.
Figure 4. Mortality and cardiac remodeling is increased in β2a mice after MI.
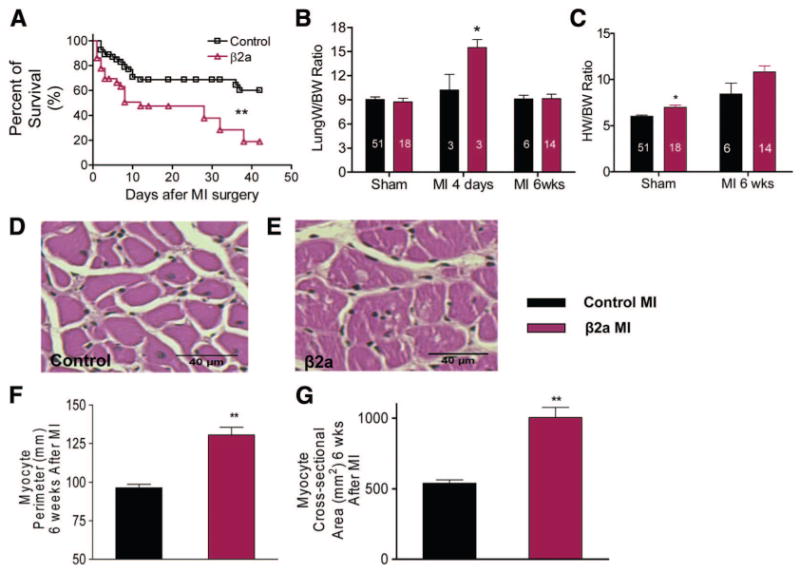
A, Kaplan–Meier survival curves during 6 weeks after MI in control (n=54) and β2a (n=36) mice. B and C, HW and lung weight (Lung W) were normalized to BW in control and β2a mice. D and E, Representative hematoxylin/eosin-stained sections from control and β2a hearts. F and G, Myocyte cross sectional area and perimeter in control (n=3) and β2a hearts (n=3). Numbers in the bars are the numbers of animals examined. **P<0.01; *P<0.05.
Lung weight was significantly increased in β2a animals early after MI, documenting more severe acute HF, and returned toward normal in those animals that survived for longer times (Figure 4B). Both groups of animals had increased heart weight/body weight (HW/BW) ratio, but in β2a mice, HW/BW was increased more than in controls (Figure 4C).
To define myocyte hypertrophy, we measured indices of myocyte size in tissue sections of animals euthanized 6 weeks after MI. Myocyte hypertrophy was significantly greater in β2a than in control hearts after MI (Figure 4D through 4G). These data show that β2a animals have exacerbated pathological responses to MI, with enhanced acute heart failure and increased pathological myocyte hypertrophy.
Cardiac Pump Function After MI Was Depressed More in β2a Hearts Than in Controls
Echocardiography was used to measure changes in LV function and chamber dimensions after MI (see examples in Online Figure IV, A and B). In sham animals, LV contractile function was greater in β2a versus controls (Figures 1 and 5). Importantly, cardiac structure and function were stable in sham β2a mice throughout the time period of this study (Figure 5). After MI, all animals had reduced pump function and significant enlargement in LV chamber dimensions (dilation) (Figure 5). LV FS and EF were significantly decreased 1 week after MI in control mice (FS, pre-MI versus after MI: 37.4% versus 17.6%; EF, 67.7% versus 38.2%) and β2a mice (FS, pre-MI versus after MI: 41.1% versus 19.9%; EF, 72.7% versus 40.8%), and there were now no differences in these functional parameters between β2a and control hearts. Over the next few weeks, FS and EF continued to decrease in β2a hearts (FS, after MI 2, 4, 6 weeks: 15.5%, 11.5%, 13.2%; EF, 32.7%, 24.7%, 25.4%), whereas in control mice, these functional parameters were stable (FS, after MI 2, 4, 6 weeks: 20.6%, 21.2%, 19.6%; EF: 41.9%, 42.6%, 40.4%) (Figure 5). At the end of 6-week study interval, cardiac function was significantly more depressed in β2a versus control hearts.
Figure 5. More severe cardiac failure in β2a mice after MI.
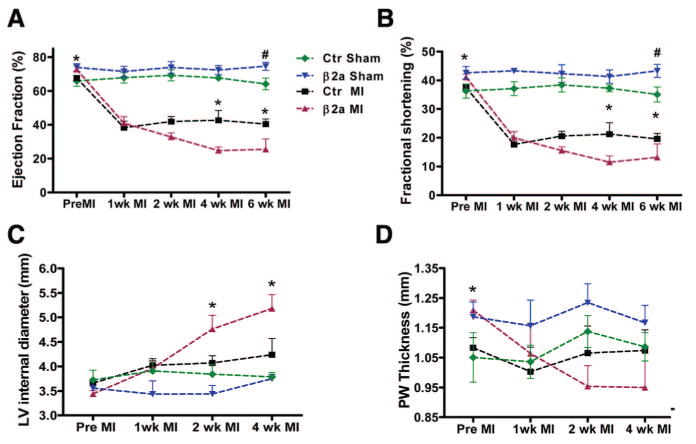
Average cardiac EF (A), FS (B), LV internal diameter (C), PWT (D) were measured in control (n=46) and β2a (n=36) mice before and 1, 2, 4, or 6 weeks after MI and in sham-operated mice (β2a, n=11; control, n=11). *P<0.05 control MI vs β2a MI; #P<0.05 control sham vs β2a sham.
There were significant pathological changes in ventricular geometry and wall thickness after MI (Online Figure IV, A and B). The magnitude of these changes was greater in β2a than in control hearts. LV wall thickness was greater in β2a versus control hearts before MI. After MI, PWT was decreased in both β2a and control hearts (control pre-MI versus post-MI: 1.08 versus 1.00 mm; β2a: 1.21 versus 1.06 mm). Over the next 3 weeks, PWT returned to values near pre-MI levels in control hearts, whereas PWT continued to decrease in β2a hearts (Figure 5D). All hearts showed some evidence of dilation after MI; however, LVID increased significantly more in β2a than in control hearts in the first week after MI (control pre-MI versus post-MI: 3.7 versus 4.0 mm; β2a: 3.4 versus 4.0 mm). By 4 weeks after MI, LVID was significantly more dilated in β2a hearts than in controls (4.2 versus 5.2 mm, Figure 5C). These results show that MI causes significantly greater deterioration of cardiac function and structure (lower EF, FS, enlarger chamber) in β2a mice.
LTCC Current (ICa,L) Remains Increased in β2a Mice After MI
We measured LTCC currents, cell contractions, and Ca2+ transients in myocytes from control and β2a hearts with or without MI. ICa,L density was significantly larger in sham β2a versus control myocytes (peak ICa,L in β2a versus control: − 24.5±1.7 [n=13] versus −11.98±3.1 pA/pF [n=11], P<0.05) (Figure 6A through 6C). The voltage dependence of ICa,L activation was shifted to negative potentials in β2a myocytes, consistent with the known function of this subunit29,30 and with what we have shown previously.8 After MI, ICa,L density was decreased in all myocytes but remained significantly larger in β2a than in control myocytes (β2a versus control: −17.6±1.4 n=15 versus −10.4±0.8 pA/pF n=12 P<0.05) (Figure 6A through 6C). ICa,L density in β2a after MI was significantly greater than in control myocytes before MI.
Figure 6. ICa,L is greater in β2a VMs from sham or MI hearts.
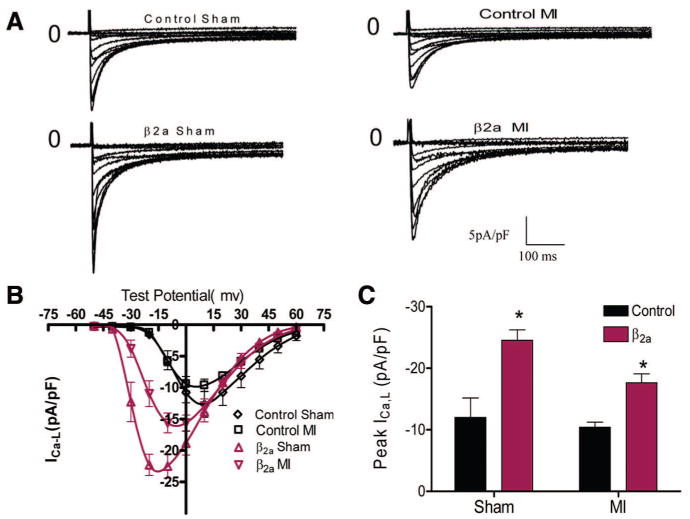
A, Representative ICa,L recordings in control and β2a VMs±MI. B, Average voltage–current relationship of ICa,L in control (sham, n=11; MI, n=12) and β2a VMs (sham, n=13; MI, n=15). C, Peak ICa,L was significantly greater in β2a than control myocytes before and after MI. *P<0.05.
ICa,L is increased by protein kinase A–mediated phosphorylation in normal myocytes.31 This regulation is altered in myocytes from diseased hearts.32,33 We measured the effects of isoproterenol (Iso) on ICa,L in myocytes from sham and post-MI hearts. In control (sham) myocytes, Iso increased ICa,L density (pre-Iso versus after Iso: −13.3±1.95 to −17.9±3.9 pA/pF, n=4, P<0.05) and caused a significant negative shift in the voltage dependence of ICa,L activation. Iso had no significant effect on ICa,L in β2a (sham) myocytes (−22.9±2.7 versus −20.2±0.9 pA/pF, n=7) (Figure 7A, 7C, and 7E; and Online Figure V [A, B, and E]). After MI, baseline ICa,L density was reduced in both control and β2a myocytes (Figure 6). In control MI myocytes Iso increased ICa,L but less so than under control conditions (control MI pre-Iso versus after Iso: −9.4±1.8 versus −12.6±2.7 pA/pF, n=6, P<0.05) and Iso shifted the voltage dependence of activation to more negative potential (Figure 7B, 7D, and 7F; and Online Figure V [C, D, and F]). Surprisingly, Iso caused a significant increase in ICa,L in β2a MI myocytes (pre-Iso versus after Iso: −17.0±1.3 versus −23.9±2.4 pA/pF, n=5, P<0.05) (Figure 7 B, 7D, and 7F; and Online Figure V [C, D, and F]), increasing ICa,L to values measured in control β2a myocytes. These results show that ICa,L in β2a myocytes remains significantly greater than in controls after MI.
Figure 7. Effects of Iso (1μmol/L) on ICa,L in sham or post-MI myocytes.
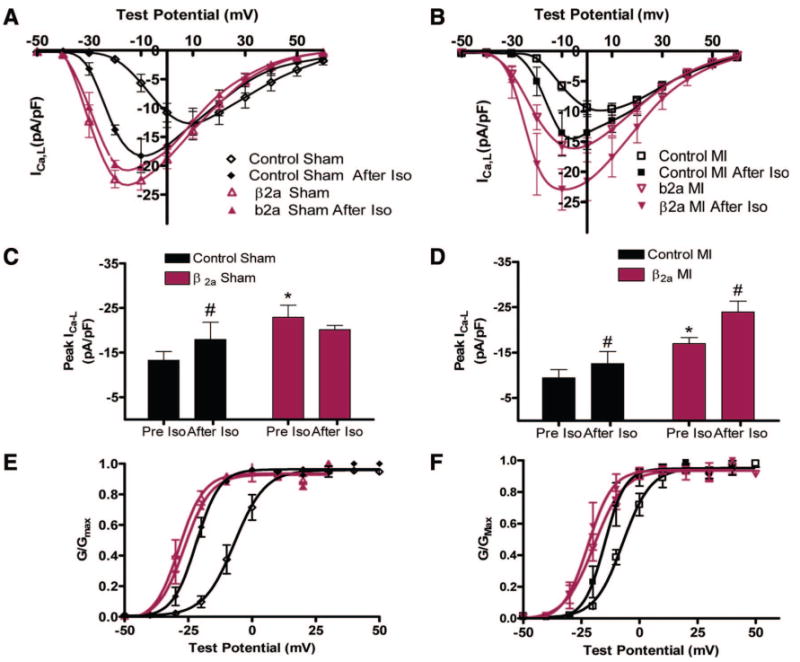
A and B, Current–voltage relationships in sham and MI (β2a and controls) with or without Iso. C, Iso increased peak ICa,L in control (n=4) but not in β2a ventricular myocytes (VMs) (n=7). D, Iso increased ICa,L in both control-MI (n=6) and β2a-MI VMs (n=5). E and F, Voltage dependence of ICa,L activation in sham or post-MI VMs with or without Iso. *P<0.05 between control and β2a VMs; #P<0.05 with or without Iso.
Contractions and [Ca2+]i Transients in β2a Myocytes After MI
We confirmed8 that β2a myocytes have larger contractions and Ca2+ transients than controls (Figure 8). FS of β2a was significantly greater than control (sham) myocytes (β2a versus control: 12.1±1.2% n=14 versus 7.8±1.0% n=19 P<0.05). After MI, contractions remained significantly greater in β2a than in control myocytes (β2a versus control: 12.3±0.7% n=28 versus 8.2±0.8% n=12 P<0.05).
Figure 8. Contractions, [Ca2+]i transients, and SR load with or without Iso.
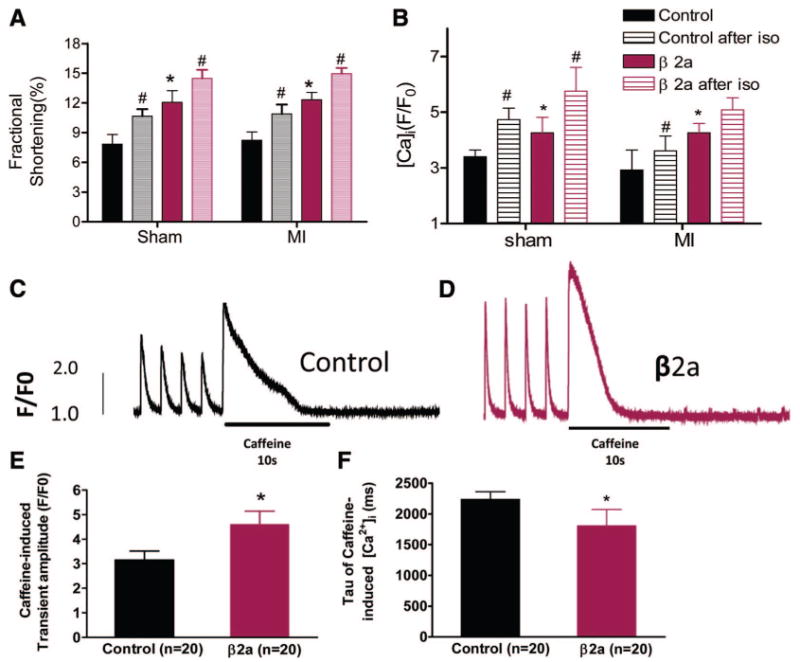
A, Comparison of FS between control and β2a in sham or post-MI myocytes with or without Iso (sham control, n=19 and β2a, n=9; post-MI control, n=9 and β2a, n=21). B, Comparison of [Ca2+]i transients between control and β2a in sham or post-MI myocytes with or without Iso (sham control, n=8 and β2a, n=9; post-MI control, n=5 and β2a, n=9). C and D, Representative example of caffeine-induced [Ca2+]i transients in control and β2a myocytes. E and F, Average data of peak caffeine-induced [Ca2+]i transients and rate of decay in control and β2a myocytes. *P<0.05 between control and β2a VMs; #P<0.05 with or without Iso.
Myocyte contractions in both control and β2a increased with Iso (control sham pre-Iso versus after Iso: 7.8±0.97% versus 10.7±0.70%, n=19, P<0.05; β2a sham: 12.1±1.2% versus 14.5±0.9%, n=9, P<0.05), with contractions in β2a being greater than in controls. Contractions of MI myocytes were increased by Iso, and again β2a myocytes had greater contractions than controls (post-MI control with or without Iso: 8.2±0.8% versus 10.9±0.95% n=9 P<0.05; post-MI β2a with or without Iso: 12.3±0.7% versus 14.9±0.6% n=21 P<0.05) (Figure 8A; Online Figure VI, A through D).
[Ca2+]i transients and SR Ca2+ content in β2a (sham) myocytes were significantly greater than in controls (Figure 8B through 8F), consistent with our previous results.8 After MI, [Ca2+]i transients in control myocytes were decreased, whereas those in β2a myocyte were similar to what was measured under basal conditions (F/F0 ratio in β2a sham versus control sham: 4.3±0.6 versus 3.4±0.2; post-MI β2a versus control: 4.3±0.3 versus 2.9±0.7) (Figure 8B; Online Figure VI, E through H). After MI, Iso increased Ca2+ transient amplitude in control and β2a myocytes, with Ca2+ transient amplitude being significantly greater in β2a myocytes (control [sham]) pre-Iso versus post-Iso: 3.4±0.2 versus 4.7±0.4 n=8 P<0.05; β2a [sham]: 4.3±0.6 versus 5.8±0.9, n=9, P<0.05; post-MI control: 2.9±0.7 versus 3.6±0.5, n=5, P<0.05; post-MI β2a: 4.3±0.3 versus 5.1±0.4, n=9, P<0.05).
Collectively, these data show that myocytes from β2a MI hearts with cardiac pump function that was worse than in control MI hearts remained hypercontractile.
Discussion
In the present study, we tested the hypothesis that preventing depression of myocyte contractility after MI improves cardiac pump function and slows heart failure progression. The major findings of this study are that increasing Ca2+ influx through the L-type Ca2+ channel after MI prevents depressed myocyte contractility but increases the risk of ischemic injury, precipitates sudden death, and exacerbates depressed cardiac pump function.
β2a Hearts Have Depressed Pump Function After Ischemia/Reperfusion
Our studies in isolated hearts show that hypercontractile β2a hearts have more depressed contractile function after IR (Figure 3). EDP was elevated during ischemia and after reperfusion in β2a hearts, and systolic function after reperfusion was more depressed than in controls. These results suggest that the higher Ca2+ influx in β2a myocytes precipitates SR Ca2+ overload26 resulting in diastolic and systolic dysfunction and Ca2+-mediated injury.34,35
β2a Hearts Have Larger Infarcts
The AAR after myocardial infarction was not different in control and β2a mice; however, infarct size (as a % of AAR) was significantly greater in β2a hearts than in controls (Figure 2). Not all portions of the AAR die after MI because the border of the ischemic region receives some blood flow and some myocytes in this region can survive. Our results show that a greater number of myocytes in the border zone died in the β2a hearts. The mechanism of enhanced myocyte death is not entirely clear, but we did show an increase in TUNEL+ myocytes in the β2a infarct border and remote zone, suggesting that apoptotic cell death is increased. We cannot rule out the possibility that necrotic cell death is also increased in β2a hearts subjected to MI.8
The factors that influence myocyte death in the MI border zone are complex and involve ischemia and systemic sympathetic reflex responses. After MI, cardiac pump function decreases and this elicits a reflex sympathetic nervous system response to maintain cardiac output and systemic blood pressure. Because β2a hearts have enhanced basal contractility, less sympathetic reflex response might be expected after MI, and this could reduce any damaging effects of sympathetic stimulation on myocyte survival. Because we found larger infarcts in β2a hearts we conclude that this primarily results from excess myocyte Ca2+ rather than excessive sympathetic activity.
Cardiac Dysfunction, Wall Thinning, and Chamber Dilation Are Enhanced in β2a Hearts After MI
MI causes cardiac structural remodeling (wall thinning and dilation).36 These changes in ventricular size and shape increase systolic wall stress, further increasing the contractile demands on a heart with a reduced number of working myocardial cells. This persistent elevation in wall stress causes progressive decay of myocardial performance and induces heart failure and premature death.36 Our results show that β2a hearts develop more dilation and poorer pump function than controls after MI (Figure 5) and that this was associated with increased rates of myocyte death in the remote zone of the β2a heart (Figure 2F). The most important finding in our study is that these detrimental changes take place in hearts in which we prevented depressed myocyte contractility (Figures 6 and 7), documenting that depressed myocyte contractile function is not a prerequisite for poor cardiac pump function after MI.
Why Is Cardiac Pump Function Depressed in the Dilated Heart?
In patients with heart failure, the dilated heart has a reduced EF, and blood is expelled slowly.37 The afterload (systolic wall stress) in the failing heart is significantly increased and correlates closely with heart failure severity and prognosis.38 Many groups,4 including ours,8,39 have shown that Ca2+ handling is deranged in failing myocytes and this depresses myocyte contractility reserve.5 Pathologically high systolic wall stress and depressed myocyte contractility both reduce the pump function of these hearts. However, it is still not clear whether either or both mechanisms represent the primary cause(s) of heart failure progression. Our study shows that heart failure progresses at a faster rate in mice in which myocyte contractility is maintained after MI (Figure 5).
Heart failure progression is also associated with an increased rate of myocyte death.8,25,40,41 Our experiments showed that apoptotic myocyte death was increased in remote zones of the β2a heart (versus control) after MI (Figure 2 and Online Figure II). We conclude that MI-induced heart failure progression in β2a mice results from a vicious cycle of myocyte loss, ventricular dilation, increased systolic wall stress (and Ca2+ signaling cascades; Online Figure VII), increased sympathetic responses (which preserves myocyte contractility at a very high level in β2a myocytes; Figures 6 through 8), and myocyte death mediated by excessive Ca2+ (and/or sympathetic nervous system) stress.
Could Depressed Myocyte Contractility Be the Effect of Heart Failure?
Ca2+ handling is deranged in heart failure.5 L-type Ca2+ channel density and phosphorylation are altered to limit Ca2+ influx.33 SERCA abundance is reduced and phospholamban phosphorylation is reduced to slow the rate of SR Ca2+ uptake and reduce SR Ca2+ loading.12,42 The phosphorylation of the SR Ca2+ release channel (ryanodine receptor) is abnormal and this leads to arrhythmias and SR Ca2+ leak.43 Collectively, theses defects produce a failing heart that must develop high systolic stress with a reduced ability to increase Ca2+ influx and SR Ca2+ uptake and release.
These changes in Ca2+ handling have led some to conclude that myocyte force development in the failing heart is less than in the normal heart. This is clearly not the case, because systolic wall stress (force being generated per unit myocardium) is significantly elevated.38 Although contractility reserve is reduced in these myocytes, the activity of neuroendocrine regulatory mechanisms must provide the stimulus to induce high force generation in vivo. Our present hypothesis is that the persistent need to develop high wall stress (requiring high Ca2+) induces the deranged Ca2+ handling that reduces contractility reserve.5 Therefore, deranged Ca2+ handing may be the effect of rather than the cause of heart failure progression. Our data suggest that downregulation of Ca2+ handling in the diseased heart might provide some protection from high Ca2+-related injury.
Relevance of the Study
Heart failure is a major health problem in our society. Available therapies provide only modest prolongation of life44; therefore, novel therapies are clearly needed. Importantly, those therapies44 that show clinical utility are negative inotropic agents (β-adrenergic antagonists and angiotensin antagonists). Conversely, those therapies that increase contractility have actually increased death rates of heart failure patients.17,18 Therefore, increasing Ca2+ stress (contractility) in failing hearts may not be the best strategy for long-term improvement in cardiac pump function. However, we forced Ca2+ handling to be increased by augmenting the most proximal portion of the Ca2+-handling cascade, Ca2+ influx. Approaches that improve Ca2+ handling by manipulation of more distal elements, such as increasing SERCa245 or blocking SR Ca2+ leak46 might rebalance cardiac contractility without predisposing the cell to Ca2+ overload and cell death.
Novelty and Significance.
What Is Known?
Persistent hemodynamic stress (from high blood pressure or after a myocardial infarction [MI]) requires an increase in the contractile strength of cardiac myocytes.
Increased contractile demand induces a sympathetic response that increases cell Ca2+ and contractility.
With the development of heart failure, Ca2+ handling becomes abnormal with a reduced ability to maintain high levels of contractile strength (muscle weakening), and there is an increase in myocyte death rate.
Most pharmacological approaches that increase contractility in human heart failure have not been improved patient outcome.
What New Information Does This Article Contribute?
Expressing the β2a subunit of the L-type Ca2+ channel prevents depression of myocyte contractility after myocardial infarction.
Maintaining high levels of contractility after MI was associated with an increase in infarction size.
Maintaining high levels of contractility with β2a expression was associated with exacerbation of depressed cardiac pump function and cardiac dilation. This study showed that MI-induced heart failure could be seen in hearts with hypercontractile myocytes.
Maintaining high Ca2+ and contractility after MI was associated with increased myocyte apoptosis and ventricular dilation.
Myocardial infarction results in a heart with fewer myocytes that must increase their force development to maintain systemic blood pressure. Over time, the heart dilates and fails. Although myocytes in the dilated heart must develop high stress in vivo, they develop contractile defects that limit their peak contractile performance. These changes may also protect myocytes from negative aspects (activation of cell death signaling) of sustained high Ca2+ states. This study tested whether preventing depression of myocyte contractility after MI improves cardiac performance or exacerbates MI related cardiac injury. Our results show that animals with genetically induced increases in myocyte contractility were more prone to stress related injury and death. Maintaining high contractility caused expansion of infarct size and exacerbated cardiac dilation and depressed pump function, even though the β2a myocytes remained hypercontractile. These studies suggest that positive inotropic therapy should be performed cautiously because in addition to the increased arrhythmogenic risk of this type of therapy (if it leads to enhanced myocyte Ca2+), it has the potential to induce myocyte death, further reducing myocyte number to exacerbate depressed cardiac pump function.
Supplementary Material
Online Figure I. Area at risk (AAR) and infarction size in hearts subject to I/R or MI. (A). Representative images of area at risk (AAR) in cardiac tissue sections stained with Evans blue. (B). Infarction area measurement in tissue sections stained with TTC from hearts after 30 minutes ischemia and 24 hours reperfusion. (C). Infarct length measurement in cardiac tissue sections stained with TTC from hearts at 6 weeks after MI. (D). Representative hearts and lungs from control and β2a mice at 6 weeks after MI
Online Figure II. TUNEL and Ki67 staining in cardiac tissue sections after MI. A-B. Representative images of TUNEL+ nuclei in border (A) and remote zone (B) of myocardium in control and β2a hearts. TdT is white, alpha-sarcomeric actin is red and DAPI is blue. (C). Representative Ki67 staining (shown as green) in tissue sections from control and β2a hearts. Scale bar = 10um.
Online Figure III. Baseline level of LVDP (A) and ±dp/dt (B) were greater in β2a isolated hearts (n=5) versus control (n=8). * p< 0.05
Online Figure IV (A). Representative LV B-mode echocardiographic images in a β2a mouse before and 2, 4 weeks after MI. The area between the red arrow heads indicate myocardial infarction zone. (B). Representative LV M-mode echocardiogrphic images in control and β2a mice before and 2,4 weeks after MI.
Online Figure V. (A-B). Representative recordings of ICa,L before and after the application of Iso in sham control and β2a VMs. (C-D). Representative recordings of ICa,L before and after application of Iso in post-MI control and β2a VMs. (E). Average half-activation potential (V0.5) in sham VMs before and after Iso treatment. (F). Average half-activation potential (V0.5) in post-MI VMs before and after Iso treatment.
Online Figure VI. (A-D). Representative recordings of myocyte shortening before and after Iso in sham or post-MI myocytes from control and β2a hearts. (E-H). Representative recordings of [Ca2+]i transients before and after Iso in sham or post-MI VMs from control and β2a hearts.
Online Figure VII. Western Blot analysis of PLBt and PLBPThr17 protein levels in control and β2a hearts after 3 weeks MI. (A). Representative western blot of PLBt and PLBPThr17 protein from control and β2a hearts. (B). Analysis of abundance of PLBt and PLBPThr17 protein levels normalized to GAPDH from control (n=3) and β2a hearts (n=3). * p < 0.05.
Acknowledgments
Sources of Funding: Supported by NIH grants HL089312, HL033921, and HL091799 (to S.R.H.) and HL088243 (to X.C.).
Non-standard Abbreviations and Acronyms
- AAR
area at risk
- β2a
β2a subunit of the L-type Ca2+ channel
- BW
body weight
- CHF
congestive heart failure
- Dox
doxycycline
- EDP
end diastolic pressure
- EF
ejection fraction
- FS
fractional shortening
- HF
heart failure
- HW
heart weight
- Iso
isoproterenol
- LAD
left anterior descending coronary artery
- LV
left ventricular
- LVDP
left ventricular developed pressure
- LVID
left ventricle internal diameter
- MI
myocardial infarction
- PWT
posterior wall thickness
- SR
sarcoplasmic reticulum
- VM
ventricular myocyte
Footnotes
Disclosures: None.
References
- 1.Houser SR. Ca(2+) signaling domains responsible for cardiac hypertrophy and arrhythmias. Circ Res. 2009;104:413–415. doi: 10.1161/CIRCRESAHA.109.193821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 3.Wilkins BJ, Molkentin JD. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun. 2004;322:1178–1191. doi: 10.1016/j.bbrc.2004.07.121. [DOI] [PubMed] [Google Scholar]
- 4.Gomez AM, Guatimosim S, Dilly KW, Vassort G, Lederer WJ. Heart failure after myocardial infarction: Altered excitation-contraction coupling. Circulation. 2001;104:688–693. doi: 10.1161/hc3201.092285. [DOI] [PubMed] [Google Scholar]
- 5.Houser SR, Piacentino V, III, Weisser J. Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol. 2000;32:1595–1607. doi: 10.1006/jmcc.2000.1206. [DOI] [PubMed] [Google Scholar]
- 6.Sam F, Sawyer DB, Chang DL, Eberli FR, Ngoy S, Jain M, Amin J, Apstein CS, Colucci WS. Progressive left ventricular remodeling and apoptosis late after myocardial infarction in mouse heart. Am J Physiol Heart Circ Physiol. 2000;279:H422–H428. doi: 10.1152/ajpheart.2000.279.1.H422. [DOI] [PubMed] [Google Scholar]
- 7.Abbate A, Biondi-Zoccai GG, Bussani R, Dobrina A, Camilot D, Feroce F, Rossiello R, Baldi F, Silvestri F, Biasucci LM, Baldi A. Increased myocardial apoptosis in patients with unfavorable left ventricular remodeling and early symptomatic post-infarction heart failure. J Am Coll Cardiol. 2003;41:753–760. doi: 10.1016/s0735-1097(02)02959-5. [DOI] [PubMed] [Google Scholar]
- 8.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BH, Hewett TE, Robbins J, Houser SR, Molkentin JD. Ca2+ -and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gill C, Mestril R, Samali A. Losing heart: the role of apoptosis in heart disease–a novel therapeutic target? FASEB J. 2002;16:135–146. doi: 10.1096/fj.01-0629com. [DOI] [PubMed] [Google Scholar]
- 10.Yue P, Long CS, Austin R, Chang KC, Simpson PC, Massie BM. Post-infarction heart failure in the rat is associated with distinct alterations in cardiac myocyte molecular phenotype. J Mol Cell Cardiol. 1998;30:1615–1630. doi: 10.1006/jmcc.1998.0727. [DOI] [PubMed] [Google Scholar]
- 11.Naga Prasad SV, Nienaber J, Rockman HA. Beta-adrenergic axis and heart disease. Trends Genet. 2001;17:S44–S49. doi: 10.1016/s0168-9525(01)02487-8. [DOI] [PubMed] [Google Scholar]
- 12.Piacentino V, III, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, Houser SR. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92:651–658. doi: 10.1161/01.RES.0000062469.83985.9B. [DOI] [PubMed] [Google Scholar]
- 13.Choi DJ, Koch WJ, Hunter JJ, Rockman HA. Mechanism of beta-adrenergic receptor desensitization in cardiac hypertrophy is increased beta-adrenergic receptor kinase. J Biol Chem. 1997;272:17223–17229. doi: 10.1074/jbc.272.27.17223. [DOI] [PubMed] [Google Scholar]
- 14.Ito K, Yan X, Tajima M, Su Z, Barry WH, Lorell BH. Contractile reserve and intracellular calcium regulation in mouse myocytes from normal and hypertrophied failing hearts. Circ Res. 2000;87:588–595. doi: 10.1161/01.res.87.7.588. [DOI] [PubMed] [Google Scholar]
- 15.Quaile MP, Rossman EI, Berretta RM, Bratinov G, Kubo H, Houser SR, Margulies KB. Reduced sarcoplasmic reticulum Ca(2+) load mediates impaired contractile reserve in right ventricular pressure overload. J Mol Cell Cardiol. 2007;43:552–563. doi: 10.1016/j.yjmcc.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 16.Amidon TM, Parmley WW. Is there a role for positive inotropic agents in congestive heart failure: focus on mortality. Clin Cardiol. 1994;17:641–647. doi: 10.1002/clc.4960171203. [DOI] [PubMed] [Google Scholar]
- 17.Landmesser U, Drexler H. Update on inotropic therapy in the management of acute heart failure. Curr Treat Options Cardiovasc Med. 2007;9:443–449. doi: 10.1007/s11936-007-0039-9. [DOI] [PubMed] [Google Scholar]
- 18.Curfman GD. Inotropic therapy for heart failure–an unfulfilled promise. N Engl J Med. 1991;325:1509–1510. doi: 10.1056/NEJM199111213252111. [DOI] [PubMed] [Google Scholar]
- 19.Eichhorn EJ. Restoring function in failing hearts: the effects of beta blockers. Am J Med. 1998;104:163–169. doi: 10.1016/s0002-9343(97)00171-x. [DOI] [PubMed] [Google Scholar]
- 20.Reiken S, Wehrens XH, Vest JA, Barbone A, Klotz S, Mancini D, Burkhoff D, Marks AR. Beta-blockers restore calcium release channel function and improve cardiac muscle performance in human heart failure. Circulation. 2003;107:2459–2466. doi: 10.1161/01.CIR.0000068316.53218.49. [DOI] [PubMed] [Google Scholar]
- 21.Ferrari R, Ceconi C, Curello S, Visioli O. The neuroendocrine and sympathetic nervous system in congestive heart failure. Eur Heart J. 1998;19(Suppl F):F45–F51. [PubMed] [Google Scholar]
- 22.Shan K, Kurrelmeyer K, Seta Y, Wang F, Dibbs Z, Deswal A, Lee-Jackson D, Mann DL. The role of cytokines in disease progression in heart failure. Curr Opin Cardiol. 1997;12:218–223. doi: 10.1097/00001573-199705000-00002. [DOI] [PubMed] [Google Scholar]
- 23.Engelhardt S, Hein L, Dyachenkow V, Kranias EG, Isenberg G, Lohse MJ. Altered calcium handling is critically involved in the cardiotoxic effects of chronic beta-adrenergic stimulation. Circulation. 2004;109:1154–1160. doi: 10.1161/01.CIR.0000117254.68497.39. [DOI] [PubMed] [Google Scholar]
- 24.Bers DM, Guo T. Calcium signaling in cardiac ventricular myocytes. Ann N Y Acad Sci. 2005;1047:86–98. doi: 10.1196/annals.1341.008. [DOI] [PubMed] [Google Scholar]
- 25.Nishida K, Otsu K. Cell death in heart failure. Circ J. 2008;72(Suppl A):A17–A21. doi: 10.1253/circj.cj-08-0669. [DOI] [PubMed] [Google Scholar]
- 26.Chen X, Zhang X, Kubo H, Harris DM, Mills GD, Moyer J, Berretta R, Potts ST, Marsh JD, Houser SR. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ Res. 2005;97:1009–1017. doi: 10.1161/01.RES.0000189270.72915.D1. [DOI] [PubMed] [Google Scholar]
- 27.Jaleel N, Nakayama H, Chen X, Kubo H, MacDonnell S, Zhang H, Berretta R, Robbins J, Cribbs L, Molkentin JD, Houser SR. Ca2+ influx through T- and L-type Ca2+ channels have different effects on myocyte contractility and induce unique cardiac phenotypes. Circ Res. 2008;103:1109–1119. doi: 10.1161/CIRCRESAHA.108.185611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jugdutt BI. Remodeling of the myocardium and potential targets in the collagen degradation and synthesis pathways. Curr Drug Targets Cardiovasc Haematol Disord. 2003;3:1–30. doi: 10.2174/1568006033337276. [DOI] [PubMed] [Google Scholar]
- 29.Bodi I, Mikala G, Koch SE, Akhter SA, Schwartz A. The L-type calcium channel in the heart: the beat goes on. J Clin Invest. 2005;115:3306–3317. doi: 10.1172/JCI27167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Striessnig J. Pharmacology, structure and function of cardiac L-type Ca(2+) channels. Cell Physiol Biochem. 1999;9:242–269. doi: 10.1159/000016320. [DOI] [PubMed] [Google Scholar]
- 31.Miriyala J, Nguyen T, Yue DT, Colecraft HM. Role of Cavbeta subunits, and lack of functional reserve, in protein kinase A modulation of cardiac Cav1.2 channels. Circ Res. 2008;102:e54–e64. doi: 10.1161/CIRCRESAHA.108.171736. [DOI] [PubMed] [Google Scholar]
- 32.Mukherjee R, Spinale FG. L-type calcium channel abundance and function with cardiac hypertrophy and failure: a review. J Mol Cell Cardiol. 1998;30:1899–1916. doi: 10.1006/jmcc.1998.0755. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Piacentino V, III, Furukawa S, Goldman B, Margulies KB, Houser SR. L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res. 2002;91:517–524. doi: 10.1161/01.res.0000033988.13062.7c. [DOI] [PubMed] [Google Scholar]
- 34.Cheung JY, Bonventre JV, Malis CD, Leaf A. Calcium and ischemic injury. N Engl J Med. 1986;314:1670–1676. doi: 10.1056/NEJM198606263142604. [DOI] [PubMed] [Google Scholar]
- 35.Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovasc Res. 2008;77:334–343. doi: 10.1093/cvr/cvm005. [DOI] [PubMed] [Google Scholar]
- 36.Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. Lancet. 2006;367:356–367. doi: 10.1016/S0140-6736(06)68074-4. [DOI] [PubMed] [Google Scholar]
- 37.Isaaz K, Ethevenot G, Admant P, Brembilla B, Pernot C. A new Doppler method of assessing left ventricular ejection force in chronic congestive heart failure. Am J Cardiol. 1989;64:81–87. doi: 10.1016/0002-9149(89)90657-7. [DOI] [PubMed] [Google Scholar]
- 38.Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81:1161–1172. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- 39.Houser SR, Piacentino V, III, Mattiello J, Weisser J, Gaughan JP. Functional properties of failing human ventricular myocytes. Trends Cardiovasc Med. 2000;10:101–107. doi: 10.1016/s1050-1738(00)00057-8. [DOI] [PubMed] [Google Scholar]
- 40.Khoynezhad A, Jalali Z, Tortolani AJ. A synopsis of research in cardiac apoptosis and its application to congestive heart failure. Tex Heart Inst J. 2007;34:352–359. [PMC free article] [PubMed] [Google Scholar]
- 41.Gustafsson AB, Gottlieb RA. Autophagy in ischemic heart disease. Circ Res. 2009;104:150–158. doi: 10.1161/CIRCRESAHA.108.187427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kubo H, Margulies KB, Piacentino V, III, Gaughan JP, Houser SR. Patients with end-stage congestive heart failure treated with beta-adrenergic receptor antagonists have improved ventricular myocyte calcium regulatory protein abundance. Circulation. 2001;104:1012–1018. doi: 10.1161/hc3401.095073. [DOI] [PubMed] [Google Scholar]
- 43.Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foody JM, Farrell MH, Krumholz HM. Beta-blocker therapy in heart failure: scientific review. JAMA. 2002;287:883–889. doi: 10.1001/jama.287.7.883. [DOI] [PubMed] [Google Scholar]
- 45.Byrne MJ, Power JM, Preovolos A, Mariani JA, Hajjar RJ, Kaye DM. Recirculating cardiac delivery of AAV2/1SERCA2a improves myocardial function in an experimental model of heart failure in large animals. Gene Ther. 2008;15:1550–1557. doi: 10.1038/gt.2008.120. [DOI] [PubMed] [Google Scholar]
- 46.Wehrens XH, Lehnart SE, Reiken S, van der Nagel R, Morales R, Sun J, Cheng Z, Deng SX, de Windt LJ, Landry DW, Marks AR. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc Natl Acad Sci U S A. 2005;102:9607–9612. doi: 10.1073/pnas.0500353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Figure I. Area at risk (AAR) and infarction size in hearts subject to I/R or MI. (A). Representative images of area at risk (AAR) in cardiac tissue sections stained with Evans blue. (B). Infarction area measurement in tissue sections stained with TTC from hearts after 30 minutes ischemia and 24 hours reperfusion. (C). Infarct length measurement in cardiac tissue sections stained with TTC from hearts at 6 weeks after MI. (D). Representative hearts and lungs from control and β2a mice at 6 weeks after MI
Online Figure II. TUNEL and Ki67 staining in cardiac tissue sections after MI. A-B. Representative images of TUNEL+ nuclei in border (A) and remote zone (B) of myocardium in control and β2a hearts. TdT is white, alpha-sarcomeric actin is red and DAPI is blue. (C). Representative Ki67 staining (shown as green) in tissue sections from control and β2a hearts. Scale bar = 10um.
Online Figure III. Baseline level of LVDP (A) and ±dp/dt (B) were greater in β2a isolated hearts (n=5) versus control (n=8). * p< 0.05
Online Figure IV (A). Representative LV B-mode echocardiographic images in a β2a mouse before and 2, 4 weeks after MI. The area between the red arrow heads indicate myocardial infarction zone. (B). Representative LV M-mode echocardiogrphic images in control and β2a mice before and 2,4 weeks after MI.
Online Figure V. (A-B). Representative recordings of ICa,L before and after the application of Iso in sham control and β2a VMs. (C-D). Representative recordings of ICa,L before and after application of Iso in post-MI control and β2a VMs. (E). Average half-activation potential (V0.5) in sham VMs before and after Iso treatment. (F). Average half-activation potential (V0.5) in post-MI VMs before and after Iso treatment.
Online Figure VI. (A-D). Representative recordings of myocyte shortening before and after Iso in sham or post-MI myocytes from control and β2a hearts. (E-H). Representative recordings of [Ca2+]i transients before and after Iso in sham or post-MI VMs from control and β2a hearts.
Online Figure VII. Western Blot analysis of PLBt and PLBPThr17 protein levels in control and β2a hearts after 3 weeks MI. (A). Representative western blot of PLBt and PLBPThr17 protein from control and β2a hearts. (B). Analysis of abundance of PLBt and PLBPThr17 protein levels normalized to GAPDH from control (n=3) and β2a hearts (n=3). * p < 0.05.