


Abstract
The DNA gyrase negative supercoiling mechanism involves the assembly of a large gyrase/DNA complex and conformational rearrangements coupled to ATP hydrolysis. To establish the complex arrangement that directs the reaction towards negative supercoiling, bacterial gyrase complexes bound to 137- or 217-bp DNA fragments representing the starting conformational state of the catalytic cycle were characterized by sedimentation velocity and small-angle X-ray scattering (SAXS) experiments. The experiments revealed elongated complexes with hydrodynamic radii of 70–80 Å. Molecular envelopes calculated from these SAXS data show 2-fold symmetric molecules with the C-terminal domain (CTD) of the A subunit and the ATPase domain of the B subunit at opposite ends of the complexes. The proposed gyrase model, with the DNA binding along the sides of the molecule and wrapping around the CTDs located near the exit gate of the protein, adds new information on the mechanism of DNA negative supercoiling.
INTRODUCTION
Topoisomerases are important enzymes present in most cells in all three domains of life that help to solve DNA topological entanglements associated with biological processes, such as replication, transcription, and recombination (reviewed in (1)). In order to maintain the topology of DNA, topoisomerases transiently cleave one or two DNA strands from the same or different DNA molecule so another single- or double-stranded region can pass through the break before religation. In this manner topoisomerases are capable of generating or removing DNA supercoils, catenates and knots. Interference with topoisomerase activity has led to the development of successful antibacterial and anticancer drugs (2,3). However, design of new chemotherapeutic agents based on the mechanism of topoisomerases necessitates a more complete mechanistic and structural understanding of these enzymes.
There are two types of topoisomerases: type I proteins cleave a single DNA strand, while type II proteins cleave the two strands of a DNA duplex in a concerted manner. While most type I and type II enzymes relax supercoiled DNA, all bacteria and some archaea have a type IIA topoisomerase, DNA gyrase (4), that is unique in its ability to introduce negative (−) supercoils into DNA in an ATP-dependent manner (5). In bacteria, the introduction of (−) supercoils is essential to initiate the replication fork formation (6), to relieve the positive (+) supercoils that form ahead of the replication fork (7), and to maintain a steady-state supercoiling level in the bacterial chromosome (8). Although the mechanism employed by gyrase has been extensively studied, many of the atomic details of this process remain vague.
All type IIA enzymes employ an ATP-dependent enzyme-bridged strand passage mechanism. In this proposed mechanism (reviewed in (1) and illustrated in Figure 4), ∼40-bp of duplex DNA, the G-segment, bind to the core of the enzyme and are cleaved by the active site tyrosines, while another DNA duplex, the T-segment, is captured through the ATP-induced dimerization of a protein gate, the N-gate. After passage through the transiently broken G-segment (DNA gate), the T-segment exits the protein through another protein gate, the C-gate. ATP hydrolysis and release reset the conformation of the enzyme and DNA to their initial state, poised for another strand-passage event or release of the DNA. Upon completion of one enzymatic cycle the linking number of the DNA substrate changes in steps of ±2 (9,10). The main difference between gyrase and all other enzymes of the same subfamily resides in the directionality of the reaction.
Figure 4.

Model of the DNA gyrase catalytic cycle. The different domains correspond to: GyrB ATPase domain (yellow), GyrB toprim domain (orange), GyrB tail1 domain (green), GyrB tail2 domain (pink), GyrA DNA breakage-reunion domain (blue), GyrA CTD (cyan), G-segment DNA (black) and T-segment DNA (red). step 1: DNA gyrase complex components as found independently in solution (red and white star symbol indicates active site location); step 2: Complex formation and initial binding of G-segment DNA to the gyrase core; step 3: Wrapping of the DNA around the GyrA CTDs; step 4: GyrB ATP binding, capture of the T-segment DNA, cleavage of the G-segment (indicated by the red and yellow star symbol), and closure of the N-gate; step 5: ATP hydrolysis, GyrB ATPase domain rotation and T-segment transfer through the cleaved G-segment; step 6: Second ATP hydrolysis, second GyrB ATPase domain rotation, transfer of the T-segment through the C-gate, ligation of the G-segment. From step 6, the reaction can continue on in a processive manner introducing multiple supercoils or it can end releasing the DNA substrate.
Gyrases are ∼350 kDa A2B2 heterotetramers formed by two A (GyrA) and B (GyrB) subunits (11). Structures of different domains of gyrases from several bacteria (12–19) provide a near complete atomic picture of the enzyme and suggest the location of the N- and C-gates that open or close to allow T-segment transport through both the protein and the cleaved G-segment. A structure of a GyrA 59 kDa N-terminal domain (16), termed the breakage-reunion domain, shows that this domain contains a winged-helix domain and a tower domain and forms a heart-shaped homodimer with two protein interfaces, the DNA- and C-gates. Crystal structures of the remaining 30–35 kDa comprising the C-terminal domain (CTD) of GyrA (13,17), show a domain forming a β-pinwheel with a positively charged amino-acid perimeter. The GyrB subunit contains an ATPase N-terminal domain followed by other domains necessary for DNA binding, named toprim and tail domains. Individual structures of the N- and C-terminal domains of GyrB demonstrate that these domains can also associate into dimers (12,18,19). Identical subunit organization has been found in other bacterial type IIA heterotetramers, such as topoisomerase IV (topoIV). Eukaryotic yeast topoisomerase II (topoII) displays marked similarities to gyrase, both at the sequence and structural level, with the major differences being that: (i) the enzyme is a homodimer where the A and B subunits have been fused together into a single chain and (ii) topoII lacks the DNA-binding CTDs.
The ability of gyrase to introduce (–) supercoils instead of relaxation, which is an energetically more favorable event, resides in its capacity to wrap around 128–140 bp of DNA (20–23) into a (+) toroidal supercoil (24). In contrast, other type IIA enzymes (25,26), including topoIV (27), bind only 28–40 bp without wrapping. This DNA wrapping characteristic has been attributed to the GyrA CTD, which by itself can bind DNA in a (+) supercoiled manner (28). Furthermore, removal of the CTDs causes gyrase to lose the ability to introduce (−) supercoils, although it can still relax DNA in a manner similar to other enzymes of the same subfamily (29). Right-handed wrapping may explain why the CTDs are necessary for the (−) supercoiling ability of gyrase (28) and its preference for acting on juxtaposed segments at left-handed crossings of DNA (30), which can be found in plectonemic regions of (+) supercoiled DNA. It has been suggested (23) that in the gyrase reaction mechanism, the G-segment binds to the core of gyrase while the flanking DNA region wraps around the CTD, thus positioning a closely spaced T-segment in an orientation that would strongly bias its capture before ATP binding and directing the subsequent strand passage event towards (−) supercoiling.
As the mechanistic differences between gyrase and other type IIA enzymes appear to depend largely on the path of the DNA around the enzyme, it is important to obtain structural information on a complex of the entire gyrase assembly bound to DNA. Here we present solution studies of intact DNA gyrase bound to large fragments of duplex DNA. Electrophoretic mobility shift assays (EMSAs) and analytical ultracentrifugation (AUC) sedimentation velocity experiments show a unique monodisperse species in solution with the expected molecular weight and biochemical characteristics for a DNA-bound gyrase complex. Small-angle X-ray scattering (SAXS) measurements, which agree well with the AUC experiments, allowed the generation of ab initio molecular envelopes that reveal the three-dimensional domain organization of the heterotetramer and offer the possibility of evaluating the current model for the beginning stage of the catalytic cycle of the enzyme. The SAXS-based model shows similarities and differences to the existing models regarding the domain arrangement of DNA gyrase and has implications for our understanding of the overall mechanism of the enzyme.
MATERIALS AND METHODS
A more detailed version is provided in the online Supplementary Data.
Enzymes and DNAs
Escherichia coli GyrA and GyrB pMCSG7 (31) clones were individually expressed in BL21(DE3) cells. The N-terminal His6 tagged proteins were purified using affinity and gel filtration columns and then frozen. A mutant GyrA subunit lacking the CTDs (GyrA-ΔCTD) [residues 1–523, (16)] was cloned and purified in the same manner. 137- and 217-bp oligonucleotides covering a major oxolinic acid-mediated cleavage site at position 991 of pBR322 (20), which included pBR322 positions 935–1065 and 895–1105 were purified from tandem repeat plasmid constructs (32). A 39-bp oligonucleotide (nt 974–1012 in pBR322) was purchased from IDT. Reconstitution parameters were optimized using EMSAs (28,33,34) run at 200 V at 4°C in 18 mM Tris–Borate, pH 8.0 and 5 mM SrCl2 following a protocol described previously (35,36) and adapted to a 491 Prep Cell system (BioRad) for complex purification.
Footprinting
Two picomoles of the radiolabeled 217-bp duplex DNA were incubated with reconstituted gyrase or the GyrA-ΔCTD gyrase, digested with 0.3 U of DNase I (NEB) for 1.5 min at 25°C and then quenched. Digested samples were run on a 4% native polyacrylamide gel followed by extraction of the DNA-bound complexes. Oxolinic acid-induced cleavage by DNA gyrase was performed under the same reaction conditions. Samples were extracted with phenol/chloroform/isoamyl alcohol (25:24:1), precipitated with ethanol, dried and resuspended. Using standard methods, samples were denatured and run on a 6% denaturing polyacrylamide gel which was transferred to filter paper, dried and exposed to PhosphorImager screens before quantification.
AUC
Purified DNA-bound gyrase complexes in 18 mM Tris-Borate, pH 8.0 and 5 mM SrCl2 were analyzed by sedimentation velocity experiments run at 20°C at 32 000 rpm. Cells were radially scanned every ∼3 min. Intensity data, collected at 260 and 280 nm, were converted into pseudo-absorbance data, edited and processed with Ultrascan II version 9.9 (37). The value of the hydrodynamic radius (RH) was derived from the Svedberg equation: s = M(1−ρ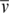 )/(6πηRHNA), where NA is the Avogadro constant. A variation of this equation was used to calculate the molecular masses of the samples: s = M(1−ρ
)/(6πηRHNA), where NA is the Avogadro constant. A variation of this equation was used to calculate the molecular masses of the samples: s = M(1−ρ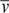 )/(fNA), where f is the frictional coefficient. All calculations assumed a temperature, partial specific volume (
)/(fNA), where f is the frictional coefficient. All calculations assumed a temperature, partial specific volume (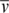 ), solvent viscosity (η), and solvent density (ρ) of 20°C, 0.73457 cm3/g, 1.0277 cp and 0.9995 g/cm3, respectively.
), solvent viscosity (η), and solvent density (ρ) of 20°C, 0.73457 cm3/g, 1.0277 cp and 0.9995 g/cm3, respectively.
SAXS
Scattering data of purified DNA-bound gyrase complex samples in 18 mM Tris-Borate, pH 8.0 and 5 mM SrCl2 were collected with SAXS and WAXS detectors covering the momentum transfer range of 0.005 < q < 1.8 Å−1 (q = 4πsinθ/λ = 2π/d, where 2θ is the scattering angle). The low- and high-angle scattering data were azimuthally integrated over a 60° section, averaged and normalized by the sample concentration, which was estimated based on the absorbance of the sample at 280 nm. Scattering due to the buffer was subtracted, the data were condensed and noise was removed. Low- and high-angle momentum transfer scattering data were merged together and cropped to 0.0063 < q < 0.9917 Å−1. The program GNOM (38) was used to calculate the radius of gyration and particle distance distribution function P(r).
Ab initio envelope calculation
DAMMIN (39) was used to create models with and without 2-fold symmetry. Top models were averaged with DAMAVER (40). Fitting of known crystal structures into the averaged SAXS envelopes was done manually and also using the program SASREF (41) to produce the final gyrase model. DNA modeling was all done manually. The final SAXS envelope and model were evaluated using COLACOR (42) and the AUC simulation software HydroPro (43).
RESULTS
Gyrase forms a stable complex with a 137 base pair DNA fragment
In order to characterize a complex of gyrase with DNA, it was necessary to purify a stable and homogeneous sample. Gyrase is known to bind roughly 128–140 bp of DNA; hence, experiments were carried out using a 137-bp DNA fragment containing a preferred E. coli gyrase oxolinic acid-mediated cleavage site (20) and purified E. coli enzyme subunits. Reconstitution of the complex required optimizing several parameters, including the molar ratios of the purified A and B subunits and the DNA. To study the complex and its behavior under varying reconstitution conditions, EMSAs were employed (28,33). Despite using highly purified proteins and DNA samples, trace contamination by nucleases and proteases produced degradation both of the protein and DNA. For this reason, different divalent cations were tested in lieu of magnesium knowing that different cations had been used previously and resulted in complexes of gyrase bound to DNA with a higher frequency of trapped complex (44–47). Although calcium prevented nuclease activity in the EMSA, it exacerbated protease activity in the purified DNA-bound complex. Strontium was found to be a suitable substitute (Figure 1A) that prevented both nuclease and protease activities as well as improved the stability of the DNA-bound gyrase complex. Surprisingly, strontium still supported gyrase activity, although not as efficiently as in the presence of magnesium (Supplementary Figure S1A). As the purpose of the substitution was to stabilize the complex and prevent degradation, strontium was deemed to be an excellent substitute for magnesium, which is involved in cleavage and religation of the DNA substrate (reviewed in (48)). Attempts to purify the DNA-bound complex from aggregates and free DNA using gel filtration or gel extraction methods were unsuccessful as the complex dissociated. For this reason, once the reconstitution reactions had been optimized by EMSAs, the complex was purified by continuous elution preparative gel electrophoresis (Figure 1B). The complex produced in this manner was stable, with no observed aggregation, degradation or disassociation of the protein or DNA even after several days of storage at 4°C as measured by EMSAs and AUC as described below.
Figure 1.

Preparation and biophysical characterization of DNA-bound gyrase complexes. (A) Example of a typical gel mobility shift assay used to determine the appropriate reaction conditions for complete DNA binding. Lane 1: Control DNA, lane 2: Empty, lanes 3–7: Reconstituted E. coli DNA gyrase heterotetramer mixed with a 217-bp DNA fragment in the following molar ratios; 1:1, 1:5, 1:10, 1:20, 1:35. In all cases, 1.5 μg (0.6 μM) of a 217-bp DNA fragment were used and no nalidixic acid was included. The gel shows that a ratio of around 1:10 DNA to protein was needed to ensure consistent complete complex formation; however a ratio of 1:4 was also found to be sufficient for other preparations. The protein to DNA molar ratio was optimized for each new protein preparation as it depends on the protein preparation, the length of the DNA substrate, and the cations used. Addition of nalidixic acid did not change the ratio of DNA to protein needed for complex formation, but aided in long-term stabilization of the purified complex. The need for a molar excess of protein over DNA has been observed before (20,67,70) and is probably due to the presence of inactive, aggregated or misfolded protein. (B) Native 4% polyacrylamide gel of fractions from continuous gel electrophoresis purification of the complex. The native gel was stained with ethidium bromide to locate the DNA. The inset shows the same gel stained with Coomassie Brilliant Blue and indicates the presence of the protein. Note that protein and DNA co-migrate, as expected. Lane 1 contains 137-bp of DNA as a marker. The bottom of the loading wells are indicated by a -w- label located between the gels, and shows that after purification there was no aggregation observed. (C) Modeling of the analytical ultracentrifugation sedimentation velocity profiles of a purified DNA-bound DNA gyrase complex bound to 137-bp of DNA in the presence of nalidixic acid. The sample included a purified E. coli gyrase complex bound to a 137-bp DNA fragment that had been reconstituted at a molar ratio of 1:10 with 1 mg/ml nalidixic acid. The purple curves represent the theoretical fits (bottom plot), and the residuals of the fit are shown in the top plot. For clarity, only every fifth scan (green curves) is shown. (D) Plot of the sedimentation and frictional coefficient distribution calculated based on the fitting to the data.
The conditions for the preparation of the complex were selected to trap a putative intermediate before any cleavage or strand passage event where the DNA is wrapped around the protein with the DNA in the active site. In order to ascertain whether the bound DNA is wrapped around the heterotetramer, as suggested by previous footprinting (20,22) and single molecule (23) experiments, footprinting experiments of the intact complexes and also a mutant E. coli gyrase enzyme lacking the CTDs of the GyrA subunits (GyrA–ΔCTD, residues 1–523) were performed using a larger DNA fragment (217-bp). The footprint of the intact E. coli complex (Supplementary Figure S2A) shows the 10-bp protection periodicity that was previously observed for gyrase (20,22). In contrast, the GyrA–ΔCTD complex does not show this periodic protection pattern (Supplementary Figure S2B). In addition, oxolinic acid-induced cleavage of the DNA showed that the DNA was positioned properly in the active site of the full-length E. coli enzyme and that the DNA/protein complex was bound as expected for a DNA with a preferred gyrase binding site. The 10-bp cleavage pattern observed by DNase I digestion was not altered in the presence of nalidixic acid, suggesting that it does not interfere with the conformational state where DNA is wrapped around the enzyme (data not shown). Taken together, these results indicate that DNA in the complex is wrapped around the gyrase tetramer and properly positioned in the active site.
To determine whether other conformational states were easily accessible and to establish further the state of the complex in the catalytic cycle, ATP, ADP or adenosine 5′-(β,γ-imido)triphosphate (AMP-PNP) were added to the complex reconstitution reactions and the complexes were analyzed by EMSAs. As expected, based upon the proposed mechanism, these experiments show near complete disassociation of the stabilized complex upon the addition of ATP or ADP (data not shown). Depending on the order of addition, AMP-PNP produced different results (Supplementary Figure S1B). The complex remained stable with no observable dissociation when AMP-PNP was added an hour after the DNA-bound complex was reconstituted whereas if AMP-PNP was added to the protein simultaneously with the DNA, a small fraction of the DNA remained unbound. One possible explanation for this observation is that AMP-PNP bound to the available free GyrB subunits inducing conformational changes (49) that prevented complex formation. Although AMP-PNP binding to an already reconstituted DNA-bound complex was not confirmed, homogeneous complexes prepared in this way were purified and showed the same shape and biophysical properties as complexes prepared in the absence of AMP-PNP (data not shown).
Analysis of the purified E. coli complex bound to 137-bp DNA by AUC sedimentation velocity experiments revealed that initial samples at concentrations ranging from 0.2 to 0.5 mg/ml contained two species and therefore were not homogeneous enough for structural studies (data not shown). The major species represented a molecule with a similar molecular weight and shape to what was expected for the gyrase complex. However, approximately 15% of the boundary fraction contained a second smaller species. This species was suspected to be free DNA, even though it was undetectable by ethidium bromide staining of EMSA gels, and presumed to be due to either the purification process or the complex dissociating slowly with time. The presence of the second species was significantly reduced and the stability of the complex increased throughout the purification procedure by the addition of nalidixic acid, a topoisomerase poison known to bind near the DNA cleavage site and stabilize the complex (46,50–52), during the complex reconstitution. In this manner, greater purity and long term stabilization of the complex was accomplished and addition of a cleavage site poison was included in some further experiments.
Sedimentation velocity experiments of the stabilized purified E. coli complex bound to 137-bp of DNA in the presence of nalidixic acid displayed a well-behaved, non-aggregated, single species with a sedimentation coefficient of 15.18 ± 0.25 S and a frictional ratio (f/f0) of 1.46 ± 0.04 (Figure 1C and D). For comparison, simulations of a spherical model with a molecular weight similar to that of the complex and under the same experimental conditions predicted a sedimentation coefficient of 20.9 S and frictional ratio of 1.0 at 20°C. Therefore, the experimental values indicate that the complex is slightly asymmetric with an overall elongated shape. The AUC measurements also indicate an observed hydrated molecular weight of 528 ± 19 kDa for the purified sample, in good agreement with the respective predicted molecular weight of 474 kDa for a heterotetrameric E. coli complex bound to 137-bp of DNA. The calculated hydrodynamic radius (RH) for the complex was 80 Å for the DNA-bound E. coli complex when the AUC-derived experimental molecular weight and sedimentation coefficient were used and ∼70 Å when the expected molecular weight was used. Similar values were obtained when purified E. coli complexes bound to a 217-bp DNA fragments were used instead.
Small-angle X-ray scattering data processing and analysis of DNA-bound gyrase complexes
Although there are known structures of different fragments of both GyrA and GyrB from several organisms (12–19) and also of other type IIA topoisomerases alone and in complex with DNA (50,53–59), there are no known structures of an intact type IIA topoisomerase in the presence or absence of DNA. In order to obtain low resolution information of the DNA-bound gyrase complex, and taking advantage of the purity and homogeneity of the preparations of gyrase bound to DNA, SAXS experiments of different complexes were performed. SAXS data were collected from dilution series of samples of E. coli DNA-bound gyrase complexes (bound to either 137-bp or 217-bp of DNA) and a GyrA-ΔCTD gyrase (bound to 39-bp of DNA) in the presence and absence of a cleavage site poison at 22°C at concentrations ranging from 0.3–6.0 mg/ml. Although data were collected on equipment covering a momentum transfer range q of 0.005–1.8 Å−1, primary data analyses were limited to a q range of 0.006–0.9917 Å−1 (or real space distance of 1047.20 > d > 6.35 Å) to remove minor parasitic scattering interference found at low-angles, as well as inaccurate high-angle measurements (Figure 2A).
Figure 2.

Small-angle X-ray scattering data and envelope modeling. (A) Normalized experimental SAXS curves of all final DNA-bound complexes that were used for further analysis. The horizontal and vertical axes correspond to q, the momentum transfer, and to the logarithm of the scattering intensity (arbitrary units). The lower left inset shows Guiner plots and RG values calculated from the final data sets using q ranges reported by AutoRg and Primus. The horizontal and vertical axis correspond to q2 (Å−2) and the log of the scattering intensity. The solid symbols indicate the q range used to determine RG. The upper right inset shows the pairwise distribution functions P(r) and RG values calculated from the final data sets. The horizontal and vertical axes correspond to the radius (Å) and to the calculated P(r). The curves in all three plots are colored coded according to the complex analyzed as indicated by the key at the top of panel A. Representative (B) P1 and (C) P2 bead models built by DAMMIN (39) calculated using data from samples of gyrase bound to a 137-bp DNA fragment in the presence of nalidixic acid. The best models from the calculations were averaged to build SAXS envelopes (40). (D) SAXS envelopes for five different DNA-bound gyrase complexes. Envelopes I-IV correspond to the complexes of E. coli gyrase with (I) 137-bp DNA fragment and nalidixic acid, (II) 137-bp DNA fragment without nalidixic acid, (III) 217-bp DNA fragment without nalidixic acid, (IV) 217-bp DNA fragment with nalidixic acid. Envelope V corresponds to an E. coli gyrase GyrA-ΔCTD truncation mutant bound to a 39-bp DNA fragment in the presence of ciprofloxacin, a potent quinolone. Note the same overall shape with four protruding domains amongst all envelopes calculated using the intact protein. The CTD truncated sample (V) forms a significantly smaller complex. The scale bar corresponds to ∼50 Å.
During data collection, Guinier plots for each full-length gyrase complex dilution series were evaluated using the recommended q range for a particle of this size, ∼0.00005–0.0003 Å−2 [qmaxRG < 1.3, (60), RG ∼70 Å based on previous AUC data]. The q ranges used in these Guinier plots, as well as the calculated RG and I0 values, were confirmed by analyzing the data with other programs: AutoRg (61), Primus (62) and GNOM (38) (Supplementary Data and Figure S4). These plots showed that the full-length complex samples were overall well-behaved with minimal variation in RG and I0 among the dilution series and similar samples, indicating that there were no concentration-dependent effects (Supplementary Figures S3 and S4). The qmax values for the final Guinier plot (Figure 2A) were selected based on the q ranges reported by AutoRg and Primus (Supplementary Figure S4). Furthermore, it should be noted that data normalization was based on sample concentrations estimated from the absorbance at 280 nm, which can be affected by the different ligands among the samples. Therefore, although the Guinier plot I0 values among dilutions of the same sample are consistent, the I0 values of different samples, while very close, are not comparable due to this estimation.
In order to estimate the maximum interatomic pairwise distance (Dmax) in the complexes, plots of Dmax versus the radius of gyration (RG) (63) (Supplementary Figure S5A) were calculated using the results from the program GNOM (38). The minimal value of Dmax was extracted from these plots by selecting the value of Dmax where RG starts to plateau (63), ∼75–80 Å for all full-length complexes, followed by an evaluation of the fit to the experimental data using GNOM. Once the Dmax value was established, pairwise distribution functions representing the distribution of interatomic distances were calculated with GNOM. The asymmetrical bell-shaped distributions, with tails extending towards a Dmax of 230–300 Å, revealed that the full-length E. coli gyrase complexes are elongated and have a reciprocal space RG of 70–82 Å (Figure 2A upper right inset), observations consistent with the AUC results. In contrast, SAXS data from the GyrA-ΔCTD complex show a significantly reduced RG of ∼52 Å, consistent with a truncated complex. Finally, due to the lack of a sharp plateau in the the Dmax versus RG plots, the sensitivity of the envelope calculations to the Dmax value employed was tested by computing and comparing models with larger Dmax values (Supplementary Figure S5C). The overall shape of these envelopes did not change (Supplementary Figure S5B), although the molecules become more elongated, as expected for fixed RG values and larger Dmax values.
DNA-bound gyrase complex SAXS envelope and model construction
To obtain an envelope for the complex based on the SAXS data, a series of independent ab initio calculations under different assumptions were performed with the program DAMMIN (39), which uses a simulated annealing algorithm to create a model of densely packed beads that best fits the scattering data (Supplementary Data and Figure S5C). Although this approach has the inherent limitation that all spheres have the same composition while the actual complex is formed by two species (DNA and protein) with different average electron densities, this method has been previously used successfully in reconstructions of protein complexes and large nucleic acids (60,64). Due to the relative mass of the protein and DNA in the gyrase complex, using uniform spheres only slightly overestimates the final volume. Furthermore, as crystal structure fragments consistently show that the gyrase heterotetramer and other type II enzymes have 2-fold symmetry (P2), the calculations were done both without imposing any symmetry (P1) and also imposing P2 symmetry. Finally, a qmax value of at least 0.3 Å−1 is required for a molecule with a Dmax of this size [q > 2π/Dmax, (60)]. As the high-angle intensity data do not contribute to the size and shape of the particle and can cause modeling problems in DAMMIN (39,65), different values of the q range were explored. The best range was found to be 0.006 Å−1 < q < 0.6 Å−1. Details of the modeling procedure are given in the Supplementary Data section.
The P1 (Figure 2B) and P2 (Figure 2C) models shared many features and fit the data well, as measured by the globally reduced χ2 of those models with an acceptable normalized spatial discrepancy (NSD). Given the P2 symmetry of the crystal structures and that these models were deemed better overall; this symmetry was imposed on the final models. Averaging of the best models with the program DAMAVER (40) resulted in envelopes that were approximately 200 × 190 × 170 Å3 for all full-length E. coli complexes (Figure 2D). The envelopes show an elongated molecule with two protuberances or domains at each end of the longest dimension. Each of these domains is large, about 70 × 60 × 70 Å3 and can easily accommodate a medium-sized protein domain.
Manual modeling of the gyrase heterotetramer within these envelopes utilized previously determined structures representing most regions of gyrase (12–16,18) and a large fragment of topoII in complex with a 30-bp DNA fragment (55). An example of this modeling is shown in Figure 3A using the envelope of the full-length gyrase complex bound to 137-bp of DNA. The full DNA, although present in the samples, was not included in the initial modeling of the gyrase heterotetramer as there is no structure of a type II topoisomerase or gyrase in complex with a very large DNA fragment. The yeast topoII structure (55) served as an excellent model for the core of gyrase as both molecules are very similar in this region. This structural homolog was initially manually placed in the main body of the envelope where it fits extremely well. This suggested that this region of the envelope corresponds to the gyrase core that comprises all of GyrA except for the CTD and a large fragment of the GyrB C-terminus including the toprim and tail domains. The remaining domains missing from this structure correspond to the GyrB ATPase domains and the GyrA CTDs. Conformational changes such as those proposed when comparing the structures of topoII and GyrA (16), the topoisomerase VI ATPase domains (49), and the GyrB C-terminal domains (19) could not be discerned at this level of resolution and hence one model was selected for each region. The placement of the topoII fragment strongly suggests that the two domains nearest to the ‘top’ of the envelope correspond to the ATPase domains, since they are closer to the homologous GyrB C-terminal domains found in the topoII core. This leaves the two ‘bottom’ domains as the location for the GyrA CTDs. During the placement of the structures, the 2-fold symmetric DNA-bound topoII dimer was fit as one object while the two ATPase domains (12) and the two CTDs (13) were fit as four independent monomers.
Figure 3.

Modeling of the DNA-bound gyrase based on SAXS envelopes. (A) Initial protein model of E. coli gyrase based on the final SAXS envelope of the full-length complex bound to a 137-bp DNA fragment in the presence of nalidixic acid. P2 symmetry was imposed on the envelope and the model was manually built to fit this envelope, as described in the text. The S. cerevisiae topoII crystal structure (55) (PDB ID 2RGR) was used to model the E. coli GyrA 59 kDa N-terminus (blue), the GyrB toprim domain (orange), the GyrB tail1 domain (green) and 16-bp of DNA (black) and corresponds to the main body of the complex. The structures of the E. coli GyrB ATPase domain (12) (PDB ID 1EI1) (yellow) and the E. coli GyrA 33 kDa CTD (13) (PDB ID 1ZIO) (cyan) were used to model the protruding domains. (B) Views of the E. coli envelope. The black line marks the approximate path of the DNA in the core of the enzyme. The yellow lines represent the orientation of the ATPase domains relative to the center of the DNA, while the teal lines pass through the CTDs. The rightmost view shows that the CTDs and the ATPase domains are not on the same plane with respect to the core of the enzyme, but rotationally displaced by around 60°. (C) Completed model and cartoon representing the DNA-bound gyrase samples analyzed by SAXS. In order to illustrate how the DNA would extend from the CTDs to the N-gate a much larger fragment than the one used in the experiments is shown in the cartoon representation. In the SAXS model, the DNA exits the core of the protein between the tower domain of one GyrA subunit and the toprim/tail domain of the GyrB subunit that is interacting with the opposing GyrA subunit, forming a clear gap where the necessary GyrB tail2 domain could interact with the DNA while maintaining the full-length GyrB tadpole structure. Modeling of the highly bent DNA as it leaves the core domain suggests that it is solvent accessible and hence probably flexible (indicated in the DNA model on the left by showing only the backbone path) before it wraps around the CTDs, although interactions with the tail2 domain that induce more bending could direct it to follow a path closer to the protein and keeping it more within the SAXS envelope. The different domains correspond to: GyrB ATPase domain (yellow), GyrB toprim domain (orange), GyrB tail1 domain (green), GyrB tail2 domain (pink), GyrA DNA breakage-reunion domain (blue), GyrA CTD (cyan), 138-bp of DNA (black), and DNA extension (grey). The scale bar in panels A and B corresponds to ∼50 Å.
The manual positioning of the domains was supported by parallel calculations using SASREF (41), which models quaternary structure arrangement of known structures against SAXS data. Note that this calculation is not necessarily accurate, as the DNA volume (∼20%) was not included in the modeling. Nevertheless, the final manually and SASREF-calculated placement of the domains was consistent, except for the switching between one CTD and one ATPase domain in the SASREF calculation. The swapping of these domains was not unexpected due to likeness of the protruding domains, but it created a model with the incorrect symmetry and connectivity and hence it was discarded (Supplementary Figure S5D). The correlation between the final arrangement of gyrase domains without DNA and the E. coli envelope was 0.724 as calculated by the program Situs (42). Finally, hydrodynamic simulations (43) using the final gyrase protein model, predict a sedimentation coefficient of 14.19 S at 20°C, an RG of 74.6 Å and an RH of 67.5 Å, values that are in good agreement with those obtained independently from the AUC and SAXS data.
Possible concerns over the effect of a cleavage site poison and the length of the DNA on the position of the CTDs were investigated through a series of additional SAXS experiments. Trials with a longer 217-bp DNA fragment or without nalidixic acid also produced elongated envelopes in which the CTDs were at opposite ends from the ATPase domains, indicating that neither the addition of a poison nor an additional 80-bp of DNA affected the overall shape of the molecule significantly at this resolution (Figure 2D). Additionally, a GyrA-ΔCTD complex was formed using a 39-bp DNA fragment spanning the same preferred gyrase binding site to help determine the position of the CTDs. SAXS measurements of the DNA-bound GyrA–ΔCTD complex, in which no DNA binding occurred outside the main core of the molecule, resulted in a truncated envelope (Figure 2D). The truncated envelope agrees well with the envelope for the full length molecule aside from the missing putative CTDs and further supports the placement of the CTDs in our model.
DISCUSSION
The mechanism employed by DNA gyrase enzymes to change the topology of DNA has been studied by a wide variety of methods (66). In the proposed reaction cycle, both protein and DNA pass through a series of conformational states coupled to ATP hydrolysis in order to achieve the topological changes in the DNA. A major difference between gyrase and most other type IIA topoisomerases resides in the DNA binding domain at the C-terminal end of the GyrA subunit. It has been suggested that this domain wraps the DNA forming a ( + ) supercoil and directs the reaction towards (–) supercoiling (28,29). Furthermore, single molecule gyrase experiments (23) suggest that in the absence of ATP, ADP or other cofactors, there is a favored equilibrium from unwrapped to wrapped DNA. In addition, the proposed mechanism suggests that only the presence of ATP would allow the cycle to progress with the concomitant rearrangements of both protein and DNA, a progression that could cause the complex to partially dissociate. These observations suggest that it may be possible to isolate and purify a well-defined state in the reaction cycle, where the DNA has entered the active site and is wrapped around the CTDs of the enzyme; poised for DNA cleavage and strand passage. While this state would represent a single snapshot of a complex cycle, its knowledge would help clarify the relative arrangement of the different gyrase subunits and domains, the way the DNA and enzyme interact, and help validate many of the major assumptions behind the proposed reaction cycle intermediates.
Towards this goal, we developed a method for purifying large amounts of a DNA-bound gyrase complex spanning a large 137-bp region, centered on a well characterized preferred gyrase binding site (20). The complex is formed and isolated by continuous gel electrophoresis in the absence of ATP, ADP or non-hydrolysable analogues and corresponds to the putative beginning/end of the reaction cycle. The enzymatic state of the complex was confirmed when the addition of ATP or ADP to the preformed complex destabilized it and lead to DNA dissociation. Evidence from DNase I footprinting experiments indicate that the DNA is bound in the active site and wrapped around the CTDs. Stabilization of the complex by cleavage site poisons (25,27–29) also suggests that the DNA is in the appropriate position. AUC sedimentation velocity experiments confirm that the complex is homogeneous in composition and has an acceptable hydrated molecular weight for a single, anisotropically shaped species of unaggregated heterotetramer bound to DNA. Taken as a whole, all the biochemical evidence indicates that the purified complex is stable, homogeneous, with the DNA wrapped around the enzyme, and with the enzyme poised for cleavage.
To obtain a low resolution envelope of the DNA-bound gyrase complex, SAXS experiments with different DNA-bound gyrase complexes were performed. Data processing using GNOM (38) produced RG values consistent with those previously found by small-angle neutron scattering experiments of E. coli gyrase bound to a 172-bp DNA fragment (67). During model calculations (39), it was determined that gyrase does indeed have P2 symmetry at this resolution. All P2 models showing good agreement with the data have a central body with a shorter dimension and four protruding domains, two at each end of the longest dimension. The exact position of these two sets of two domains varies slightly in different models, which resulted in a larger volume for the domains in the final, average model than in individual solutions. Nevertheless, the dimensions of the final envelope are remarkably consistent with the expected dimensions based on structures of crystallographic fragments (12,13,55). Finally, the overall envelope shape seen in each of the full-length gyrase complexes that were prepared and analyzed by SAXS, including samples with and without nalidixic acid, and with two different DNA lengths, was unchanged at this level of resolution. As the hand of the envelope cannot be determined from the SAXS data, a hand was chosen that most easily accommodated positive wrapping around the protein and all models were assigned the same hand.
Different structures of gyrase and the DNA breakage/reunion domain of yeast topoII in complex with DNA were used to build a model for the protein in the DNA-bound gyrase complex consistent with the SAXS envelope. In the SAXS model, the topoII complex structure was placed in the main body of the envelope with the 2-fold axes coinciding. This placement of the yeast fragment resulted in the positioning of the GyrB ATPase domains in the space closest to the DNA-gate, forming the previously seen tadpole shape made by the GyrB ATPase, toprim and tail domains (15), while the GyrA CTDs were placed in the remaining density at the opposite end of the enzyme near the C-gate (Supplementary Movie S1). In each SAXS envelope, the plane formed by the GyrB ATPase domains was rotated 60° relative to the plane formed by the GyrA CTDs, which in turn were rotated 20° from the plane formed by the bent DNA fragment (Figure 3B).
The placement of the CTDs in this distal position was unexpected since models of DNA-bound gyrase (13) corresponding to the conformational state studied place the CTDs almost adjacent to the DNA-gate and near the ATPase domains as in the crystal structure of the topoIV ParC subunit (58). However, hydrodynamic simulations of a model with the CTDs adjacent to the DNA-gate result in an RG of 55 Å, a value significantly smaller than the 70–80 Å RG and RH experimental values obtained for the full-length complexes from SAXS and AUC. Two additional independent observations lend further support to the placement of the CTDs in the distal position. First, in SAXS experiments of a full-length GyrA dimer the CTDs are also observed near the C-gate (14). This position could be accommodated easily by the CTD with the addition of the eight residues missing between the GyrA core and the CTD in the structures, and taking into account the proposed flexibility of the linker helix at the C-terminus of the GyrA core (13,14,16). While these experiments show the CTDs resting against the main body of the protein and not protruding from it, the absence of DNA could be responsible for this tight fit. Second, electron microscopy studies of complexes of gyrase with DNA show remarkable agreement in shape with the SAXS envelope and also show that in some of the particles the DNA strands emerge from what was interpreted as the end distal to the GyrB subunits, which would correspond to the C-gate (68). As the DNA is proposed to be wrapped in these complexes, the micrographs would also suggest that the CTDs can be near or adjacent to the C-gate. The SAXS experiments of gyrase with DNA, GyrA without DNA, and the electron microscopy observations are consistent with the placement of the CTDs near the C-gate.
In the calculated envelopes, the path of the DNA is not directly visible as the modeling assumes beads of uniform density. Nevertheless, other information, such as the placement of the CTDs near the C-gate, places constraints on possible DNA paths. In the final model, the CTDs are located directly ∼65 Å away from the active site. The approximate distance between the edge of the CTD and the eighth base pair from the center of the DNA in the topoII fragment is around 80 Å (70 Å following a direct path that does not avoid the protein core residues), which would correspond to ∼24-bp of straight B DNA. Assuming that two-thirds of the CTD β-pinwheel has to bind DNA in order to redirect the DNA 180°, this would suggest that around 40-bp are bound to the CTD (13) and hence the total coverage of each GyrA monomer would be ∼72-bp of DNA. 144-bp of protection by the heterotetramer is an overestimate since upon modeling of the DNA duplex following a path that goes from the central core and wraps around the downward positioned CTDs only required 138-bp of DNA (Figure 3C, Supplementary Movie S1), a value similar to those found in nuclease protection experiments (20–22). The DNA modeling suggests that in a gyrase complex the DNA would also retain a local bend of ∼90° near the active site although in the opposite direction, towards the C-gate, to the one observed in other type IIA topoisomerase structures (55,57,59). The differences in DNA directionality could be attributed to differences in DNA substrate, including length or nicking, or as a difference between gyrase and other type IIA enzymes.
There have been several related models proposed for the gyrase reaction that involve conformational changes in the protein and the DNA. In the SAXS model, the path of the DNA would remain very similar to that of the most current proposed models (14,15,23). In all proposed models, both ends of the DNA wrap around the CTDs in a right handed manner in order to create a (+) supercoil, but in the SAXS model the DNA travels down the sides of the gyrase molecule towards the CTDs (Figure 3C), while other models have the CTDs and the DNA roughly on the same plane as the active site.
A reaction cycle based on the SAXS model is shown in Figure 4. It has many structural similarities to previous reaction cycle models, but there are important differences as well. In all models, the reaction starts with complex assembly and wrapping of the DNA (Figure 4, steps 1–3). In the SAXS model, the propensity of T-segment capture would be significantly increased by redirection of the DNA towards the N-gate by the CTDs, but the CTDs themselves would not actively drive this capture through a conformational change or movement upon DNA binding, as previous models have suggested. This modified role is supported by the observations that the CTDs do not need to be attached to the N-terminus of GyrA to promote negative supercoiling (28). Although observations of additional global protection of the DNA on the 5′ end and additional local protection of the DNA 3′ of the cleavage site have led to models with either an asymmetric redirection of the wrapped DNA or an asymmetric conformational change of the protein (22,69), the SAXS complex is symmetric at this resolution, making assignment of different sides of the molecule impossible. Therefore, our model shows how capture and passage of either DNA strand of the wrapped segment would introduce (–) supercoils and how wrapping of the DNA has the effect of giving directionality to the supercoiling reaction, providing gyrase with its unique function. While it is unknown what governs which strand is captured by the GyrB ATPase domains, the presence of low-level ATPase activity suggests that the GyrB subunits are continuously searching for a T-segment (70–72). Induced by GyrB subunit dimerization and T-segment capture, the G-segment is then cleaved (Figure 4, step 4) and ATP hydrolysis, strand passage and resealing of the G-segment follows (Figure 4, step 5). Finally, the C-gate is opened, the T-segment exits the protein and the cycle is completed (Figure 4, step 6). Overall, the stages of the catalytic cycle remain similar to previous models, but the shape of the DNA in the complex is different and there would be fewer initial movements of the CTDs which maintain a distal position before and after DNA binding and wrapping.
Structural studies using crystallographic and SAXS methods together with the wealth of biochemical information about gyrase have provided detailed models on the overall mechanism of the enzyme. However, the SAXS models of the complex are the first to depict the conformation of the whole enzyme, and not just of its domains. In particular, the location of the CTDs is crucial for understanding the mechanism of the enzyme. There is currently no data on the possible coordinated intramolecular protein conformational changes that could allow movement of the CTDs, such as the one depicted in steps 4 or 6 of Figure 4 leading to T-segment capture or passage, or on the possibility of multiple conformations of these domains during the catalytic cycle. Nevertheless, current models place them in an upward position. It was shown previously that the CTDs are found in a distal position in the dimerized full-length GyrA subunits (14). The SAXS model of the complex further supports this placement, near the exit gate, at opposite ends from the GyrB subunits, and far from the active site of the enzyme even when the GyrA subunits are in complex with the GyrB subunits and DNA substrate. Confirmation of the model, the path of the DNA and the atomic structure of the subunits in the complex will require methods, such as small-angle neutron scattering, electron microscopy or crystallography, that can resolve the structure of the protein subunits and the DNA unambiguously. Further understanding of the mechanism of gyrase will also require structures in different conformational states along the catalytic cycle. The experiments described here provide a starting point for future higher resolution investigations of the way gyrase interacts with DNA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Use of the APS was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences (DE-AC02-06CH11357). Support from the R.H. Lurie Comprehensive Cancer Center of Northwestern University to the Structural Biology Facility is also acknowledged. This work was supported by the National Institutes of Health (GM51350 to A.M.). Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
ACKNOWLEDGMENTS
We are grateful to S. Halford for the gift of purified EcoRV and J. Ipsaro and R. Rajan for comments and suggestions. We acknowledge staff and instrumentation support from the Keck Biophysics Facility and the Center for Structural Biology at Northwestern University. AUC calculations were supported by NSF Teragrid Grant #MCB070038 (to B. Demeler). Portions of this work were performed at DND-CAT at the APS which is supported by E.I. DuPont de Nemours & Co., The Dow Chemical Company and the State of Illinois.
REFERENCES
- 1.Schoeffler AJ, Berger JM. DNA topoisomerases: harnessing and constraining energy to govern chromosome topology. Q Rev. Biophys. 2008;41:41–101. doi: 10.1017/S003358350800468X. [DOI] [PubMed] [Google Scholar]
- 2.Maxwell A. DNA gyrase as a drug target. Trends Microbiol. 1997;5:102–109. doi: 10.1016/S0966-842X(96)10085-8. [DOI] [PubMed] [Google Scholar]
- 3.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 4.Forterre P, Gribaldo S, Gadelle D, Serre MC. Origin and evolution of DNA topoisomerases. Biochimie. 2007;89:427–446. doi: 10.1016/j.biochi.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 5.Gellert M, Mizuuchi K, O'Dea MH, Nash HA. DNA gyrase: an enzyme that introduces superhelical turns into DNA. Proc. Natl Acad. Sci. USA. 1976;73:3872–3876. doi: 10.1073/pnas.73.11.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baker TA, Sekimizu K, Funnell BE, Kornberg A. Extensive unwinding of the plasmid template during staged enzymatic initiation of DNA replication from the origin of the Escherichia coli chromosome. Cell. 1986;45:53–64. doi: 10.1016/0092-8674(86)90537-4. [DOI] [PubMed] [Google Scholar]
- 7.Liu LF, Wang JC. Supercoiling of the DNA template during transcription. Proc. Natl Acad. Sci. USA. 1987;84:7024–7027. doi: 10.1073/pnas.84.20.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zechiedrich EL, Khodursky AB, Bachellier S, Schneider R, Chen D, Lilley DM, Cozzarelli NR. Roles of topoisomerases in maintaining steady-state DNA supercoiling in Escherichia coli. J. Biol. Chem. 2000;275:8103–8113. doi: 10.1074/jbc.275.11.8103. [DOI] [PubMed] [Google Scholar]
- 9.Brown PO, Cozzarelli NR. A sign inversion mechanism for enzymatic supercoiling of DNA. Science. 1979;206:1081–1083. doi: 10.1126/science.227059. [DOI] [PubMed] [Google Scholar]
- 10.Mizuuchi K, Fisher LM, O'Dea MH, Gellert M. DNA gyrase action involves the introduction of transient double-strand breaks into DNA. Proc. Natl Acad. Sci. USA. 1980;77:1847–1851. doi: 10.1073/pnas.77.4.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 12.Brino L, Urzhumtsev A, Mousli M, Bronner C, Mitschler A, Oudet P, Moras D. Dimerization of Escherichia coli DNA-gyrase B provides a structural mechanism for activating the ATPase catalytic center. J. Biol. Chem. 2000;275:9468–9475. doi: 10.1074/jbc.275.13.9468. [DOI] [PubMed] [Google Scholar]
- 13.Corbett KD, Shultzaberger RK, Berger JM. The C-terminal domain of DNA gyrase A adopts a DNA-bending beta-pinwheel fold. Proc. Natl Acad. Sci. USA. 2004;101:7293–7298. doi: 10.1073/pnas.0401595101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costenaro L, Grossmann JG, Ebel C, Maxwell A. Small-angle X-ray scattering reveals the solution structure of the full-length DNA gyrase a subunit. Structure. 2005;13:287–296. doi: 10.1016/j.str.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 15.Costenaro L, Grossmann JG, Ebel C, Maxwell A. Modular structure of the full-length DNA gyrase B subunit revealed by small-angle X-ray scattering. Structure. 2007;15:329–339. doi: 10.1016/j.str.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 16.Morais Cabral JH, Jackson AP, Smith CV, Shikotra N, Maxwell A, Liddington RC. Crystal structure of the breakage-reunion domain of DNA gyrase. Nature. 1997;388:903–906. doi: 10.1038/42294. [DOI] [PubMed] [Google Scholar]
- 17.Ruthenburg AJ, Graybosch DM, Huetsch JC, Verdine GL. A superhelical spiral in the Escherichia coli DNA gyrase A C-terminal domain imparts unidirectional supercoiling bias. J. Biol. Chem. 2005;280:26177–26184. doi: 10.1074/jbc.M502838200. [DOI] [PubMed] [Google Scholar]
- 18.Wigley DB, Davies GJ, Dodson EJ, Maxwell A, Dodson G. Crystal structure of an N-terminal fragment of the DNA gyrase B protein. Nature. 1991;351:624–629. doi: 10.1038/351624a0. [DOI] [PubMed] [Google Scholar]
- 19.Fu G, Wu J, Liu W, Zhu D, Hu Y, Deng J, Zhang XE, Bi L, Wang DC. Crystal structure of DNA gyrase B' domain sheds lights on the mechanism for T-segment navigation. Nucleic Acids Res. 2009;37:5908–5916. doi: 10.1093/nar/gkp586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher LM, Mizuuchi K, O'Dea MH, Ohmori H, Gellert M. Site-specific interaction of DNA gyrase with DNA. Proc. Natl Acad. Sci. USA. 1981;78:4165–4169. doi: 10.1073/pnas.78.7.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morrison A, Cozzarelli NR. Contacts between DNA gyrase and its binding site on DNA: features of symmetry and asymmetry revealed by protection from nucleases. Proc. Natl Acad. Sci. USA. 1981;78:1416–1420. doi: 10.1073/pnas.78.3.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orphanides G, Maxwell A. Evidence for a conformational change in the DNA gyrase-DNA complex from hydroxyl radical footprinting. Nucleic Acids Res. 1994;22:1567–1575. doi: 10.1093/nar/22.9.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gore J, Bryant Z, Stone MD, Nollmann M, Cozzarelli NR, Bustamante C. Mechanochemical analysis of DNA gyrase using rotor bead tracking. Nature. 2006;439:100–104. doi: 10.1038/nature04319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu LF, Wang JC. Micrococcus luteus DNA gyrase: active components and a model for its supercoiling of DNA. Proc. Natl Acad. Sci. USA. 1978;75:2098–2102. doi: 10.1073/pnas.75.5.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee MP, Sander M, Hsieh T. Nuclease protection by Drosophila DNA topoisomerase II. Enzyme/DNA contacts at the strong topoisomerase II cleavage sites. J. Biol. Chem. 1989;264:21779–21787. [PubMed] [Google Scholar]
- 26.Thomsen B, Bendixen C, Lund K, Andersen AH, Sorensen BS, Westergaard O. Characterization of the interaction between topoisomerase II and DNA by transcriptional footprinting. J. Mol. Biol. 1990;215:237–244. doi: 10.1016/S0022-2836(05)80342-0. [DOI] [PubMed] [Google Scholar]
- 27.Peng H, Marians KJ. The interaction of Escherichia coli topoisomerase IV with DNA. J. Biol. Chem. 1995;270:25286–25290. doi: 10.1074/jbc.270.42.25286. [DOI] [PubMed] [Google Scholar]
- 28.Reece RJ, Maxwell A. The C-terminal domain of the Escherichia coli DNA gyrase A subunit is a DNA-binding protein. Nucleic Acids Res. 1991;19:1399–1405. doi: 10.1093/nar/19.7.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kampranis SC, Maxwell A. Conversion of DNA gyrase into a conventional type II topoisomerase. Proc. Natl Acad. Sci. USA. 1996;93:14416–14421. doi: 10.1073/pnas.93.25.14416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nollmann M, Stone MD, Bryant Z, Gore J, Crisona NJ, Hong SC, Mitelheiser S, Maxwell A, Bustamante C, Cozzarelli NR. Multiple modes of Escherichia coli DNA gyrase activity revealed by force and torque. Nat. Struct. Mol. Biol. 2007;14:264–271. doi: 10.1038/nsmb1213. [DOI] [PubMed] [Google Scholar]
- 31.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 32.Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 2004;375:23–44. doi: 10.1016/s0076-6879(03)75002-2. [DOI] [PubMed] [Google Scholar]
- 33.Shuman S. Analysis of topoisomerase-DNA interactions by electrophoretic mobility shift assay. Methods Mol. Biol. 2001;95:65–74. doi: 10.1385/1-59259-057-8:65. [DOI] [PubMed] [Google Scholar]
- 34.Rau DC, Gellert M, Thoma F, Maxwell A. Structure of the DNA gyrase-DNA complex as revealed by transient electric dichroism. J. Mol. Biol. 1987;193:555–569. doi: 10.1016/0022-2836(87)90266-x. [DOI] [PubMed] [Google Scholar]
- 35.Bates AD, Maxwell A. DNA gyrase can supercoil DNA circles as small as 174 base pairs. Embo J. 1989;8:1861–1866. doi: 10.1002/j.1460-2075.1989.tb03582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Higgins NP, Peebles CL, Sugino A, Cozzarelli NR. Purification of subunits of Escherichia coli DNA gyrase and reconstitution of enzymatic activity. Proc. Natl Acad. Sci. USA. 1978;75:1773–1777. doi: 10.1073/pnas.75.4.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Demeler B, editor. UltraScan: A Comprehensive Data Analysis Software Package for Analytical Ultracentrifugation Experiments. Cardiff, UK: Royal Society of Chemistry; 2005. [Google Scholar]
- 38.Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 1992;25:495–503. [Google Scholar]
- 39.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Cryst. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petoukhov MV, Svergun DI. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wriggers W, Milligan RA, McCammon JA. Situs: a package for docking crystal structures into low-resolution maps from electron microscopy. J. Struct. Biol. 1999;125:185–195. doi: 10.1006/jsbi.1998.4080. [DOI] [PubMed] [Google Scholar]
- 43.Garcia De La Torre J, Huertas ML, Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 2000;78:719–730. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noble CG, Maxwell A. The role of GyrB in the DNA cleavage-religation reaction of DNA gyrase: a proposed two metal-ion mechanism. J. Mol. Biol. 2002;318:361–371. doi: 10.1016/S0022-2836(02)00049-9. [DOI] [PubMed] [Google Scholar]
- 45.Sissi C, Marangon E, Chemello A, Noble CG, Maxwell A, Palumbo M. The effects of metal ions on the structure and stability of the DNA gyrase B protein. J. Mol. Biol. 2005;353:1152–1160. doi: 10.1016/j.jmb.2005.09.043. [DOI] [PubMed] [Google Scholar]
- 46.Sugino A, Peebles CL, Kreuzer KN, Cozzarelli NR. Mechanism of action of nalidixic acid: purification of Escherichia coli nalA gene product and its relationship to DNA gyrase and a novel nicking-closing enzyme. Proc. Natl Acad. Sci. USA. 1977;74:4767–4771. doi: 10.1073/pnas.74.11.4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gellert M, Mizuuchi K, O'Dea MH, Itoh T, Tomizawa JI. Nalidixic acid resistance: a second genetic character involved in DNA gyrase activity. Proc. Natl Acad. Sci. USA. 1977;74:4772–4776. doi: 10.1073/pnas.74.11.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corbett KD, Berger JM. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu. Rev. Biophys. Biomol. Struct. 2004;33:95–118. doi: 10.1146/annurev.biophys.33.110502.140357. [DOI] [PubMed] [Google Scholar]
- 49.Corbett KD, Berger JM. Structural dissection of ATP turnover in the prototypical GHL ATPase TopoVI. Structure. 2005;13:873–882. doi: 10.1016/j.str.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 50.Laponogov I, Sohi MK, Veselkov DA, Pan XS, Sawhney R, Thompson AW, McAuley KE, Fisher LM, Sanderson MR. Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nat. Struct. Mol. Biol. 2009;16:667–9. doi: 10.1038/nsmb.1604. [DOI] [PubMed] [Google Scholar]
- 51.Yoshida H, Bogaki M, Nakamura M, Nakamura S. Quinolone resistance-determining region in the DNA gyrase gyrA gene of Escherichia coli. Antimicrob Agents Chemother. 1990;34:1271–1272. doi: 10.1128/aac.34.6.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshida H, Bogaki M, Nakamura M, Yamanaka LM, Nakamura S. Quinolone resistance-determining region in the DNA gyrase gyrB gene of Escherichia coli. Antimicrob Agents Chemother. 1991;35:1647–1650. doi: 10.1128/aac.35.8.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berger JM, Gamblin SJ, Harrison SC, Wang JC. Structure and mechanism of DNA topoisomerase II. Nature. 1996;379:225–232. doi: 10.1038/379225a0. [DOI] [PubMed] [Google Scholar]
- 54.Classen S, Olland S, Berger JM. Structure of the topoisomerase II ATPase region and its mechanism of inhibition by the chemotherapeutic agent ICRF-187. Proc. Natl Acad. Sci. USA. 2003;100:10629–10634. doi: 10.1073/pnas.1832879100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dong KC, Berger JM. Structural basis for gate-DNA recognition and bending by type IIA topoisomerases. Nature. 2007;450:1201–1205. doi: 10.1038/nature06396. [DOI] [PubMed] [Google Scholar]
- 56.Graille M, Cladiere L, Durand D, Lecointe F, Gadelle D, Quevillon-Cheruel S, Vachette P, Forterre P, van Tilbeurgh H. Crystal structure of an intact type II DNA topoisomerase: insights into DNA transfer mechanisms. Structure. 2008;16:360–370. doi: 10.1016/j.str.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 57.Schmidt BH, Burgin AB, Deweese JE, Osheroff N, Berger JM. A novel and unified two-metal mechanism for DNA cleavage by type II and IA topoisomerases. Nature. 465:641–644. doi: 10.1038/nature08974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corbett KD, Schoeffler AJ, Thomsen ND, Berger JM. The structural basis for substrate specificity in DNA topoisomerase IV. J. Mol. Biol. 2005;351:545–561. doi: 10.1016/j.jmb.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 59.Laponogov I, Pan XS, Veselkov DA, McAuley KE, Fisher LM, Sanderson MR. Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS ONE. 5:e11338. doi: 10.1371/journal.pone.0011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 61.Petoukhov MV, Konarev PV, Kikhney AG, Svergun DI. ATSAS 2.1 – towards automated and web-supported small-angle scattering data analysis. J. Appl. Cryst. 2007;40(Suppl.):s223–s228. [Google Scholar]
- 62.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst. 2003;36:1277–1282. [Google Scholar]
- 63.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat. Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lipfert J, Doniach S. Small-angle X-ray scattering from RNA, proteins, and protein complexes. Annu. Rev. Biophys. Biomol. Struct. 2007;36:307–327. doi: 10.1146/annurev.biophys.36.040306.132655. [DOI] [PubMed] [Google Scholar]
- 65.Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J. Appl. Cryst. 2001;34:33–41. [Google Scholar]
- 66.Nollmann M, Crisona NJ, Arimondo PB. Thirty years of Escherichia coli DNA gyrase: from in vivo function to single-molecule mechanism. Biochimie. 2007;89:490–499. doi: 10.1016/j.biochi.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 67.Krueger S, Zaccai G, Wlodawer A, Langowski J, O'Dea M, Maxwell A, Gellert M. Neutron and light-scattering studies of DNA gyrase and its complex with DNA. J. Mol. Biol. 1990;211:211–220. doi: 10.1016/0022-2836(90)90021-D. [DOI] [PubMed] [Google Scholar]
- 68.Kirchhausen T, Wang JC, Harrison SC. DNA gyrase and its complexes with DNA: direct observation by electron microscopy. Cell. 1985;41:933–943. doi: 10.1016/s0092-8674(85)80074-x. [DOI] [PubMed] [Google Scholar]
- 69.Heddle JG, Mitelheiser S, Maxwell A, Thomson NH. Nucleotide binding to DNA gyrase causes loss of DNA wrap. J. Mol. Biol. 2004;337:597–610. doi: 10.1016/j.jmb.2004.01.049. [DOI] [PubMed] [Google Scholar]
- 70.Maxwell A, Gellert M. The DNA dependence of the ATPase activity of DNA gyrase. J. Biol. Chem. 1984;259:14472–14480. [PubMed] [Google Scholar]
- 71.Mizuuchi K, O'Dea MH, Gellert M. DNA gyrase: subunit structure and ATPase activity of the purified enzyme. Proc. Natl Acad. Sci. USA. 1978;75:5960–5963. doi: 10.1073/pnas.75.12.5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Staudenbauer WL, Orr E. DNA gyrase: affinity chromatography on novobiocin-Sepharose and catalytic properties. Nucleic Acids Res. 1981;9:3589–3603. doi: 10.1093/nar/9.15.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.