
Figure 1.
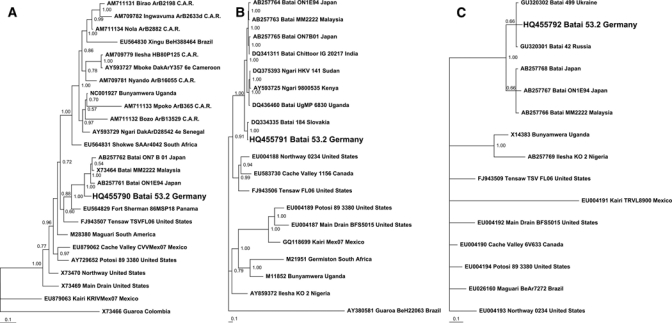
Bayesian phylogenetic tree of selected orthobunyaviruses based on partial S (A; length = 838 nucleotides), M (B; length = 3,152 nucleotides), and L segment (C; length = 200 nucleotides) sequences. Each sequence is identified by GenBank accession number, virus name, strain designation, and strain origin. Phylogenetic analysis was performed using MrBayes 3.0 program.8 Three heated chains and a single cold chain were used in all Markov Chain Monte Carlo (MCMC) analyses, which were run for 1,000,000 generations, sampling one tree every 100 generations. Trees obtained before convergent and stable likelihood values were discarded (i.e., a 2,500 tree burn-in). Four independent runs, each started from different randomly chosen trees, were performed to assess convergence. Posterior probabilities for nodes were assembled from all post–burn-in trees (i.e., 30,004 trees per analysis). Posterior probabilities are shown on each branch. Scale bar indicates number of nucleotide substitutions per site.