

Abstract
Bacterioferritin (BFR) from Escherichia coli is a member of the ferritin family of iron storage proteins and has the capacity to store very large amounts of iron as an Fe3+ mineral inside its central cavity. The ability of organisms to tap into their cellular stores in times of iron deprivation requires that iron must be released from ferritin mineral stores. Currently, relatively little is known about the mechanisms by which this occurs, particularly in prokaryotic ferritins. Here we show that the bis-Met-coordinated heme groups of E. coli BFR, which are not found in other members of the ferritin family, play an important role in iron release from the BFR iron biomineral: kinetic iron release experiments revealed that the transfer of electrons into the internal cavity is the rate-limiting step of the release reaction and that the rate and extent of iron release were significantly increased in the presence of heme. Despite previous reports that a high affinity Fe2+ chelator is required for iron release, we show that a large proportion of BFR core iron is released in the absence of such a chelator and further that chelators are not passive participants in iron release reactions. Finally, we show that the catalytic ferroxidase center, which is central to the mechanism of mineralization, is not involved in iron release; thus, core mineralization and release processes utilize distinct pathways.
Keywords: Heme, Iron Metabolism, Metalloproteins, Metals, Protein Metal Ion Interaction, Ferritin, Iron Release
Introduction
The insolubility and potential toxicity of the essential metal iron led to the evolution of the ferritin family of proteins, which function to maintain cellular stores of iron in a non-toxic form (1, 2). Ferritins are generally composed of 24 α-helical subunits that pack to form a highly symmetric dodecahedron protein coat with a hollow center in which large amounts of iron, in the form of a ferric oxy-hydroxide mineral, can be stored (3–5).
To satisfy the cellular requirements for iron, ferritins must exhibit flexibility of iron flux, depositing it within their central cavity in times of excess and releasing it back to the cellular machinery in times of environmental deficit (6–8). The mechanism by which ferritins generate their iron mineral cargo has been the focus of much attention (3–5). Central to this process is an intrasubunit di-iron site called the ferroxidase center, which is found only in H-chain and H-chain-like subunits. Another type of subunit, the L-chain, found in animal ferritins and isostructural with the H-chain in terms of intersubunit contacts, does not contain the ferroxidase center. The mechanism of mineral formation depends on the type of ferritin. In eukaryotic ferritins, the ferroxidase center operates as a gated iron pore, facilitating the transfer of oxidized iron into the central cavity. Some prokaryotic ferritins also exhibit this type of mechanism, whereas others, including the bacterioferritin (BFR) from Escherichia coli and the ferritin-like protein (Ftn) from Pyrococcus furiosus, exhibit a quite different mechanism, in which the ferroxidase center functions as a true catalytic center, cycling between diferrous and bridged diferric forms as Fe2+ ions are oxidized within the central cavity (see Ref. 9 and references therein).
In general, much less is known about the mechanism(s) of iron release. It was shown that E. coli cells carrying iron-loaded FtnA (a bacterial ferritin) have an iron-restricted growth advantage over those lacking FtnA (6), indicating that iron stored within ferritins can be mobilized for cellular anabolic processes. However, the mechanism by which this occurs is not clear. One possibility involves proteolytic degradation of the ferritin protein shell, and in animal ferritins, this occurs in the lysosome (10). However, this represents a highly uncontrolled release process, and it is likely that in most cases iron release is carefully controlled to avoid the toxic effects of iron. It has been shown that, in general, this requires a supply of electrons to reduce mineral Fe3+ to Fe2+, together with a chelator to accept the iron upon reduction. The ability of a wide range of reductants (including dithionite, thiols, ascorbate, superoxide, dihydroflavins, and diphenols) to drive the release of ferritins has been reported (11–17). Despite these general principles, there is a lack of consensus about the means by which electrons are transferred to the ferric mineral. For example, it was demonstrated that reductants too large to traverse the protein coat (e.g. flavoproteins and ferredoxins) are efficient in promoting core reduction (18), leading to the conclusion that electron transfer across the protein coat must occur. On the other hand, whereas dihydroflavins are efficient in promoting iron release, it was also shown that immobilized versions of the same flavins are not, implying that entry into the cavity is a necessary step (15). It has also been shown that reduction of the ferric mineral core results in an Fe2+ core, without significant iron release (18–20). Thus, in addition to a reductant, a strong Fe2+ chelator is also apparently required to effect iron release.
In BFRs, an important consideration for the mechanism of iron release is the presence of 12 intersubunit heme groups, which are not present in other types of ferritins (21, 22) and which are not required for the formation of the mineral (23), indicating that the heme serves another function. Interestingly, heme-deficient variants of BFR have been isolated containing significantly more iron than the wild-type protein produced under identical conditions (23), suggesting that these variants are deficient in iron release and that heme groups may be involved in iron release. More direct evidence appeared to come from in vitro kinetic studies (24), although subsequently the same authors reported that the P. aeruginosa BFR used in this work may have been damaged in some way (25).
Although the mechanism of iron core mineralization has been characterized in detail for E. coli BFR, the mobilization of iron from the protein's mineral core has not been studied. An understanding of this process would provide insight into how the uptake and release processes within a single BFR may be balanced to provide flexible and dynamic iron management within a cell. Here we show that the rate at which iron is mobilized from the iron mineral core of E. coli BFR is dependent on heme but not on the ferroxidase center, demonstrating that uptake and release processes are distinct. Furthermore, the data demonstrate that electron transfer into the mineral core is the rate-determining step of the release process. Importantly, we show that iron release occurs readily in the absence of a high affinity Fe2+ chelator, requiring only a reductant.
EXPERIMENTAL PROCEDURES
BFR Expression and Purification
BFR and BFR variants M52H (heme-free (23)), E127Q (ferroxidase center variant (26)), and H46A/D50A (inner surface site (27)) were overexpressed and purified as described previously (28). Apo-BFR was generated by treatment with sodium dithionite and bipyridyl as described previously (29) and verified by iron analysis (30). Protein concentration was measured (before and after iron loading) using the bicinchoninic acid method with bovine serum albumin as a standard (31) or spectrophotometrically using an absorptivity of 33,000 m−1 cm−1 at 280 nm for the apoprotein (32). Heme contents were determined through the heme Soret absorbance intensity, using ϵ418 nm = 107,000 m−1 cm−1 (33), and were found to be ∼1.0 for each BFR 24-mer.
BFR containing a higher heme load (∼5 hemes/24-mer) was produced as above but with the following modifications. bfr expression was induced with a low level of isopropyl β-d-1-thiogalactopyranoside (∼2 μm), followed by the addition of aminolevulinic acid (1 mm) and ammonium ferric citrate (200 μm), and grown for a further ∼20 h. We subsequently noted that the omission of aminolevulinic acid had little effect on the heme loading.
Preparation of Mineralized BFRs
Iron cores were generated by the aerobic addition (by Hamilton syringe) of ferrous ammonium sulfate (freshly prepared in deoxygenated ultrapure water with concentrated hydrochloric acid (25 μl/50 ml) to prevent autoxidation) to 0.5 μm BFR (wild type and variants) in 100 mm Mes (pH 6.5) at 25 °C, in additions of 400 Fe2+/24-mer up to a loading of 1200 Fe3+/24-mer. The mineralization process was followed by measuring A340 nm changes (PerkinElmer λ35 spectrophotometer). Fe2+ oxidation was allowed to go to completion and centrifuged (10,950 × g) for 5 min to remove any Fe3+ precipitate not associated with the protein, prior to the subsequent addition. Iron contents were determined by iron analysis (30).
Iron Mobilization and Reduction Assays
Stock solutions of ∼50 mm sodium dithionite were prepared and quantified anaerobically by measuring A320 nm (ϵ320 nm = 8000 m−1 cm−1) (11). Unless otherwise stated, all subsequent manipulations were performed under strictly anaerobic conditions in an anaerobic cabinet (Faircrest) in which O2 was maintained at <3.0 ppm. All buffers were sparged with nitrogen gas for a minimum of 1 h prior to transfer into the anaerobic chamber. A mixed buffer system (referred to as MBS: potassium acetate, Mes, Tris, Mops (all at 10 mm), 200 mm NaCl, pH 6 or 7) was used, and iron release was monitored using the Fe2+ chelators ferrozine (3-[2-pyridyl]-5,6-diphenyl-1,2,4-triazine-4,4′-disulfonic acid; [Fe(II)(ferrozine)3]4− logβ3 = 15.4 (34)) or 2,2′-bipyridyl ([Fe(II)(2,2′-bipyridyl)3]2+ logβ3 = 16.4–17.6 (35)). Absorbance at 562 nm ([Fe(II)(ferrozine)3]4− ϵ562 nm = 28,000 m−1 cm−1) or 523 nm ([Fe(II)(2,2′-bipyridyl)3]2+ ϵ523 nm = 8430 m−1 cm−1) was used to quantify the released Fe2+ as a function of time. In most cases, the chelator was present at 1 mm and BFR was present at 0.05 μm (containing 1200 Fe3+), and the reaction was initiated by the addition of sodium dithionite by a microsyringe (Hamilton). For iron release experiments with flavin mononucleotide (FMN), generation of the reduced dihydroflavin form (FMNH2) was carried out using stoichiometric or excess concentrations of sodium dithionite (36). Assays in MBS were performed at pH 7.0 and 15 °C, with chelator, dithionite (100 μm), and variable FMN (0–100 μm). Iron release experiments in the absence of a chelator were carried out using the same reductants as above. Released (or weakly protein-associated) iron was determined at increasing time points following reduction by the addition of ferrozine, and the instantaneous increase in absorbance at 562 nm was measured.
Absorbance versus time plots were fitted to an exponential function (single or double, as appropriate) using Origin 8 (Microcal). Where a double exponential function was required, the initial, more rapid phase was found to be the major phase in terms of amplitude, and the apparent rate constant associated with this phase was used for rate constant plots. Alternatively, data were subjected to an initial rate analysis, from which values in μm/min were calculated using the ferrozine or bipyridyl extinction coefficient. Initial rate/rate constant data are presented as the average of replicates (n ≥ 3), and the error is the S.D. Where data from spectroscopic measurements are shown, these are representative of (n = 3) replicates. Iron release in the absence of a chelator was also measured by incubating BFR (0.1 μm, 1200 irons) with dithionite (400 μm) at pH 7, followed by passage down an anaerobic Sephadex G25 gel filtration column (PD-10, GE Healthcare). Fractions eluting from the column were collected and assayed for protein (by SDS-PAGE/densitometry) and iron (as above), which indicated >90% recovery. An equivalent experiment was performed in the presence of ferrozine (1 mm).
BFR core reduction was measured directly through absorbance changes at 400 nm following the addition of sodium dithionite to BFR (0.25 μm) loaded with 1200 irons at 25 °C. Data were fitted to a double exponential function as described above, and the extent of reduction was calculated from ϵ400 nm = 870 m−1 cm−1/iron, determined from absorbance data from BFR samples containing accurately determined levels of core iron. All absorbance measurements were recorded using a PerkinElmer Life Sciences λ35 or λ800 UV-visible spectrophotometer.
RESULTS
Chelator-detected Iron Release from BFR
The release of iron from BFR was investigated using a commonly employed chelator-based assay, in which it was assumed that the chelator does not interfere in the release reaction (i.e. formation of the Fe2+-chelator complex is not the rate-determining step of the assay reaction; see Reactions 1–3).
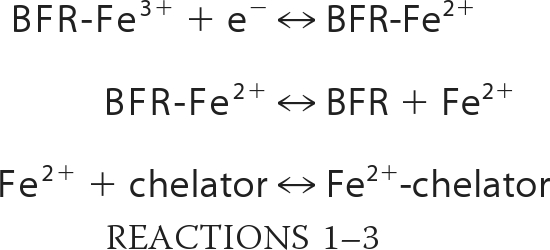 |
Control reactions, in which the high affinity Fe2+ chelator ferrozine (see supplemental Fig. S1A) was present (at 1 mm), demonstrated that Fe2+ was not available for complexation in iron-loaded BFR samples in the absence of a reductant, as evidenced by the complete lack of absorbance increase at 562 nm, which characterizes the [Fe(ferrozine)3]4− complex. Variable additions of the strong reductant sodium dithionite in the range 25–500 μm to BFR containing 1200 Fe3+/protein at pH 7 (Fig. 1A) and pH 6 (supplemental Fig. S2A) led to increases in absorption over time, indicative of the formation of Fe2+-chelator complex. The kinetic profiles varied as a function of the reductant concentration. At 100 μm and below, the profiles fitted well to a double exponential function, with the majority of the amplitude associated with the first phase. As reductant concentration increased, the amplitude of the second phase increased, and, above 100 μm reductant, essentially equal amplitudes were associated with the two phases. The data indicated, therefore, that there are two phases of iron release: a more rapid phase and a slower phase, with the slower phase only occurring to a significant extent at large excesses of reductant. This behavior is similar to that previously reported for Azotobacter vinelandii BFR, for which reductant saturation kinetics were also observed, and under excess dithionite, the majority of iron release was associated with the first exponential phase (11). Plots of apparent rate constants as a function of dithionite concentration are shown in Fig. 1B. The data indicated that the apparent rate constant for the iron release reaction (judged through the availability of Fe2+ to form a colored chelator complex) is pH-dependent, being significantly higher at lower pH. This was also clear from an initial rates analysis (see supplemental Fig. S2B). The data (particularly at pH 6) also showed that the rate constant was essentially linear with reductant at low reductant concentration but subsequently leveled off at higher reductant concentrations.
FIGURE 1.

Iron release from E. coli BFR followed by a dithionite-ferrozine assay. A, plots of ΔA562 nm as a function of time following the addition of varying concentrations (as indicated) of sodium dithionite to BFR (0.05 μm) containing ∼1200 irons/protein in MBS, 1 mm ferrozine, pH 7, at 25 °C. B, apparent rate constants, obtained from fitting the release data in A (and from similar experiments) as described under “Experimental Procedures,” plotted as a function of dithionite concentration. The left ordinate corresponds to data at pH 6 (see supplemental Fig. S2), and the right ordinate corresponds to data at pH 7. C, plots of the concentration of iron detected as the [Fe(II)(ferrozine)3]4− complex at the end point of each release reaction as a function of dithionite concentration. Data in B and C and in other figures are presented as averages (n ≥ 3) ± S.D. (error bars).
To test whether the iron loading of BFR affected the release properties, samples containing an average of 400 Fe3+ ions/protein were also investigated. Very similar behavior to that observed for the 1200 Fe3+ samples was observed, with the initial rate being first order with respect to protein (data not shown).
To test whether the nature of the chelator affects the iron release characteristics, 2,2′-bipyridyl was also used. The two chelators have different electrostatic properties; ferrozine carries two negative charges, whereas bipyridyl is neutral (see supplemental Fig. S1B). The effect of varying the concentration of each chelator in the range 200 μm to 2 mm was investigated. For both ferrozine and bipyridyl, a relatively small variation in the initial rate of Fe2+-chelator complex formation was observed over the range 0.2–2 mm (supplemental Fig. S1, C–F). The data demonstrated that although some differences were observed, there was no great variation in release kinetics upon varying the chelator concentration under any one set of conditions, in general agreement with the assumption that binding of Fe2+ by the chelator is not the rate-limiting step of the release process. However, the fact that some variation was observed indicated that the chelator is not entirely innocent in the release assay. This conclusion was reinforced when comparing the two chelators. At pH 7, the plots for ferrozine and bipyridyl were similar; at pH 6, however, rates of complex formation were significantly greater for ferrozine compared with bipyridyl (supplemental Fig. S1, C and E, circles), demonstrating a clear effect of the nature of the chelator. This also indicates that the origin of the pH effect on iron release observed above (Fig. 1B) is associated with the chelator rather than with the iron release mechanism per se.
Reductant-Protein Association or Electron Transfer Is the Rate-limiting Step in Iron Release from BFR
Although an effective reductant of BFR core iron, dithionite alone has no physiological significance. GSH is a low molecular weight thiol that is highly abundant in the cytoplasm of many bacteria, including E. coli, in which it plays an important role in maintaining the reducing environment of the cytoplasm (37). Thiol compounds have been used previously to stimulate iron release from ferritins, so we tested the iron release activity of GSH (see supplemental Fig. S3). Under the conditions used, very little Fe2+ was released (∼1% of the total iron after 30 min), demonstrating that GSH is not able to function as an effective reductant for iron release from BFR.
Flavins are commonly found as redox cofactors within the cell cytoplasm and have been previously utilized in studies of iron release from ferritins, as a coupled system involving a reductant, such as NADH or dithionite, together with FMN (15, 36, 38–40). Under the conditions used here, NADH alone was not able to reduce core iron, and even when combined with FMN (in the absence of an NADH:flavin oxidoreductase), the rate at which NADH reduced FMN was too slow to provide a useful system for core reduction (data not shown). However, dithionite provided essentially instantaneous and quantitative reduction of FMN to FMNH2 (see supplemental Fig. S4), so iron release from BFR was investigated using a dithionite/FMN reduction system. Initial experiments indicated that the rate of release was significantly greater than observed with dithionite alone, so to enable measurement of Fe2+-chelator formation by conventional spectrophotometry, experiments with variable concentrations of FMN (with fixed dithionite) were conducted at 15 °C. The data revealed a significant increase in the rate of iron release in solutions containing FMN compared with the dithionite-only control (see Fig. 2A). Data at both pH 6 and 7 fitted well to a double exponential function, although compared with the dithionite data, the second, slower phase accounted for a more significant proportion of the released iron. These data are similar to those reported for flavin-mediated iron release from horse spleen ferritin (12). Plots of the apparent rate constant associated with the initial, rapid phase as a function of FMN concentration are shown in Fig. 2B. As for dithionite alone, the apparent rate constant exhibited a significant pH dependence. Also, the apparent rate constant was linear with FMN at low concentration, before leveling out at higher concentration at pH 6. The observed increase in the rate of iron release upon changing the reducing system from dithionite alone to dithionite/FMN indicated that the rate-determining step in the release reaction is the transfer of electrons into the iron storage cavity. This is associated with either the rate at which the reductant and protein form a complex or the rate of electron transfer itself.
FIGURE 2.

Iron release from E. coli BFR followed by a dithionite/FMN-ferrozine assay. A, plots of ΔA562 nm as a function of time following the addition of varying concentrations of FMN (as indicated) together with a fixed concentration of sodium dithionite (100 μm) to BFR (0.05 μm) containing ∼1200 irons/protein in MBS, 1 mm ferrozine, pH 7, at 15 °C. B, apparent rate constants at pH 6 (circles) and pH 7 (squares), obtained from fitting the data in A (and similar experiments), are plotted as a function of FMN concentration. C, the concentration of iron detected as the [Fe(II)(ferrozine)3]4− complex at the end point of each release reaction as a function of FMN concentration. Error bars, S.D.
To investigate the possibility that reduced FMN might form a stable complex with BFR that facilitates electron transfer, reduced FMN (100 μm) was added to apo-BFR and BFR containing 1200 irons/protein (0.5 μm), and the resulting solutions were passed down an anaerobic gel filtration column. UV-visible absorbance spectra of eluted BFR were recorded. In both cases, the spectra were indistinguishable from control samples from which FMN was omitted (not shown). Because oxidized FMN gives rise to a more intense spectrum, particularly in the visible region, BFR samples were subsequently exposed to air, and spectra were rerecorded after a few min. No changes in the spectra were observed. Control experiments showed that stoichiometrically reduced FMN was reoxidized within 3 min of exposure to air, giving large absorbance increases in the 350–500 nm range. Slower running fractions from the gel filtration column rapidly developed yellow color upon air exposure. Taken together, the data indicated that reduced FMN does not co-elute with BFR, and we conclude that the effect of FMN on iron release does not result from the formation of a stable complex between FMN and BFR.
Heme Enhances the Rate and Extent of Iron Release from BFR
BFRs are unique among the ferritin family in that they are hemoproteins. The 12 heme groups of BFR are located at an intersubunit site, coordinated by Met52 from each of the two subunits (22, 41) (see Fig. 3A). We have previously demonstrated that the heme groups do not play an important role in the formation of the iron core (23). Therefore, we investigated whether they are important for iron release. To obtain heme-free BFR, a variant of BFR in which the heme-coordinating Met52 residue was substituted with a His residue was used. Although His is commonly found to coordinate heme, in this case, it is not able to do so, and the protein was isolated in a heme-free state (23). Our standard conditions for the overproduction of E. coli BFR lead to a protein containing only ∼1 heme/24-mer (26, 42). Therefore, to obtain BFR containing higher levels of heme, growth conditions were altered, as described under “Experimental Procedures,” to increase heme incorporation into the overproduced protein. Five hemes per 24-mer was the maximum loading we were able to reproducibly achieve. Additional heme cannot be titrated into native 24-mer BFR because the heme binding site is not accessible from the outside of the protein (22, 25).
FIGURE 3.

The effect of heme on iron release from E. coli BFR followed by a dithionite-ferrozine assay. A, ribbon diagram of a BFR subunit dimer generated from Protein Data Bank entry 1BCF (22) using PyMOL (57). A heme molecule (in a stick representation) is bound at the monomer-monomer interface, coordinated by Met52 from each subunit. The dinuclear ferroxidase center, outer, and inner protein surfaces are also indicated. B, plots of ΔA562 nm as a function of time following the addition of 200 μm sodium dithionite to either wild-type BFR containing 5 hemes/24-mer or to heme-free M52H BFR. Both BFRs (0.05 μm) contained ∼1200 irons/protein and were in MBS, 1 mm ferrozine, pH 7. The release reaction was conducted at 25 °C. C, apparent rate constants, obtained from fitting the data in A (and similar experiments in which dithionite concentration was varied), plotted as a function of dithionite concentration. D, the concentration of iron detected as the [Fe(II)(ferrozine)3]4− complex at the end point of each release reaction as a function of dithionite concentration. Error bars, S.D.
Iron release from proteins containing 1200 Fe3+ ions was measured at pH 7 using the dithionite-chelator and the FMN/dithionite-chelator assays (see Figs. 3 and 4). Data were fitted as described above, and the apparent rate constant corresponding to the initial release event was plotted as a function of reductant concentration. For the dithionite-chelator assay, an increase in the rate constant was observed in the presence of heme, with rate constants ∼2-fold greater (at both pH 7 (Fig. 3C) and pH 6 (supplemental Fig. S5)). For the FMN/dithionite-chelator assay (Fig. 4), the heme effect was significantly greater, with rate constants increased 4-fold in the heme-loaded compared with heme-free BFR samples.
FIGURE 4.

The effect of heme on iron release from E. coli BFR followed by a dithionite/FMN-ferrozine assay. A, plots of ΔA562 nm as a function of time following the addition of 100 μm sodium dithionite and 75 μm FMN to either wild-type BFR containing 5 hemes/24-mer or to heme-free M52H BFR. Both BFRs (0.05 μm) contained ∼1200 irons/protein in MBS, 1 mm ferrozine, pH 7. The release reaction was conducted at 15 °C. B, apparent rate constants, obtained from fitting the data in A (and similar experiments in which FMN concentration was varied), plotted as a function of FMN concentration. C, the concentration of iron detected as the [Fe(II)(ferrozine)3]4− complex at the end point of each release reaction as a function of FMN concentration. Error bars, S.D.
In each of the iron release assays described here, the amount of iron released could be calculated from the end points of the kinetic profiles. In each case, plots of iron released as a function of reductant concentration were pH-independent. In the dithionite-chelator assays at high concentrations of reductant, 45–61% of the iron originally stored within BFR was subsequently detected as the chelator complex (see Fig. 1C), whereas for the FMN/dithionite-chelator assay, only 17–25% of iron was released (see Fig. 2C). Importantly, the extent to which iron was released was also dependent on heme. For the dithionite-chelator assay, approximately twice as much iron was released to chelator in the presence of heme compared with that in the heme-free BFR variant (Fig. 3C). An enhancement for the heme-containing BFR was also observed for the FMN/dithionite-chelator assays (Fig. 4C), although the extent of this was less. Overall, the data demonstrated that the presence of heme increases both the rate and extent of iron release.
Iron Release Occurs in the Absence of a High Affinity Chelator
The ferritin iron release literature indicates that a chelator is required in order to achieve significant iron release following reduction of core Fe3+ to Fe2+ (18–20). To test this for E. coli BFR, we sought to determine to what extent iron release could occur in the absence of a high affinity chelator. Dithionite was added to a solution of BFR containing 1200 irons at pH 7 (time 0), and then chelator (ferrozine) was added at increasing time points. The instantaneous increase in absorbance at 562 nm, due to the formation of the [Fe(II)(ferrozine)3]4− complex, was measured and plotted as a function of the increasing time between addition of reductant and the addition of the chelator, thus determining the concentration of Fe2+ in solution or weakly associated with the protein (and therefore immediately available to the chelator) (see Fig. 5). The data indicate that Fe2+ was not entirely retained within the BFR cavity following reduction of the Fe3+ core; in fact, the extent of iron release was significant, corresponding to up to ∼50% of the iron initially present. The kinetic data fitted well to a single exponential function, with an apparent rate constant of k = 0.23 ± 0.02 min−1, indicating that release of iron occurred in a single phase. Under similar conditions in the presence of the chelator, two phases of release were observed, although one of these was relatively minor.
FIGURE 5.

Iron reduction and release from BFR in the absence of a high affinity Fe2+ chelator. A, plot of ΔA562 nm as a function of time following the addition of sodium dithionite (200 μm) to BFR (0.1 μm) containing ∼1200 irons/protein in MBS, pH 7, at 25 °C. Ferrozine (1 mm) was added at the indicated time following the addition of reductant, and the instantaneous observed jump in A562 nm was taken to represent immediately available, released Fe2+. A fit of the data to a single exponential function is drawn in. B, plots of relative protein and iron concentrations following gel filtration separation of BFR and Fe2+ after mineral core reduction by dithionite (400 μm). BFR (0.1 μm) containing ∼1200 irons/protein in MBS, pH 7. C, as in B, but reduction was performed in the presence of ferrozine (1 mm). D, plots of ΔA400 nm as a function of time following the addition of sodium dithionite (200 μm) to BFR (0.25 μm) containing ∼1200 irons/protein in MBS at 25 °C and pH 6 (circles) and pH 7 (squares). Note that full reduction of the heme present in the sample would have resulted in a decrease in A400 nm of <0.005; therefore, the vast majority of the observed absorbance decrease resulted from the reduction of the Fe3+ mineral.
Similar experiments were conducted using FMN/dithionite as reductant (see Fig. 6). Again, significant release of iron was observed, although, as above, this was less than observed for dithionite alone, with ∼20% of the iron initially present being released for complexation. Kinetic data at pH 7 (and 15 °C) and with 1 heme/24-mer fitted well to a single exponential, giving a rate constant of 0.26 min−1. Data at pH 6 gave an essentially identical apparent rate constant, consistent with the conclusion that the rate of iron release is pH-independent and that pH effects described above resulted from the effect of a chelator. We note that with both reductant systems, the rate constant measured in the absence of chelator was lower than that obtained under similar conditions when chelator was present throughout the assay, indicating an effect of chelator on the rate of iron release.
FIGURE 6.

The influence of heme and pH on iron release from BFR in the absence of a high affinity Fe2+ chelator. A and B, plots of ΔA562 nm as a function of time following the addition of sodium dithionite (100 μm) and FMN (100 μm) to wild-type BFR containing 1 or 5 hemes/24-mer or to heme-free M52H BFR, as indicated. All BFRs (0.05 μm) contained ∼1200 irons/protein in MBS at 15 °C and pH 6 (A) and pH 7 (B). Ferrozine (1 mm) was added at the indicated time point after the addition of reductant, and the immediate jump in A562 nm was taken to represent released Fe2+. Fits of the data to a single exponential function are drawn in. C, bar graph plots of rate constants for iron release (obtained from the fits) and percentage of total Fe3+ released as a function of heme content and pH, as indicated. Error bars, S.D.
FMN/dithionite assays were also carried out with BFR containing variable heme. A significant difference was again observed between heme-free and BFR containing five hemes; at pH 7, the rate constant increased 3–4-fold, and the extent of iron release was ∼2-fold greater in the presence of heme, confirming the importance of heme in both the extent and rate of iron release.
The above experiments demonstrate clearly that a large proportion of BFR-stored iron becomes available for chelation following reduction to Fe2+. To test further whether the Fe2+ ions are actually released into solution in the absence of an obvious candidate chelator, BFR containing 1200 Fe3+/protein molecule was reduced with a ∼3-fold excess (over iron) of dithionite (over a period of 1 h) under anaerobic conditions and subsequently passed down an anaerobic gel filtration column, and fractions were collected and analyzed for protein and iron. A plot of relative iron and protein concentrations as a function of elution fraction (Fig. 5B) demonstrated that the iron and protein peaks were resolved. The eluting iron and protein bands coincided with those observed in control experiments for Fe2+ in solution and apo-BFR, respectively (not shown), and we conclude that a significant proportion of Fe2+ was released from BFR into solution following core reduction. To ensure that none of the components of the mixed buffer system employed was acting as a chelator, the experiment was repeated using poorly chelating Hepes buffer at pH 7. The data (not shown) were essentially identical. The experiment was also repeated in the presence of a high affinity Fe2+ chelator (ferrozine) (Fig. 5C). Surprisingly, the extent of separation was less in the presence of chelator, suggesting that the Fe2+-chelator complex may be associated with the protein, a conclusion supported by a Fe2+-ferrozine control experiment, which showed that this species eluted with a band maximum at 7–8 ml (compared with 6 ml in the presence of BFR; Fig. 5C).
Measurement of core Fe3+ Reduction
The ferric oxy-hydroxide iron core has a characteristic broad absorbance in the UV-visible region of the spectrum, resulting from O-Fe3+ charge transfer transitions. Reduction of the iron core to Fe2+ leads to the loss of such absorbance and thus can be used to monitor the reduction of the core independently of iron release. Although FMN has significant absorbance at 400 nm, dithionite does not, so the latter was used for core reduction studies. The addition of dithionite to BFR containing 1 heme/24-mer and 1200 Fe3+/protein resulted in a significant decrease in absorbance with the reaction complete after ∼25 min (Fig. 5D). The extent of reduction, calculated using ϵ400 nm = 870 m−1 cm−1/iron, was found to correspond to ∼70% of the Fe3+ originally present. Data fitted well to a double exponential function, with a rapid but low amplitude initial phase, followed by a main, slower phase. The main phase corresponded to an apparent rate constant of k = 0.18 ± 0.02 min−1, which is similar to that measured for iron release (Figs. 1 and 5), consistent with the transfer of electrons to the cavity being the rate-limited step of BFR iron release. The initial rapid phase is likely to be due to a small proportional of Fe3+ that is somehow distinct from the bulk of the iron. For example, this could correspond to Fe3+ within the cavity that is not part of the core mineral or not located within the cavity, most likely including iron at the ferroxidase centers. Similar measurements at pH 6 (Fig. 5D) resulted in a lower overall change in absorbance (extent of reduction) but were otherwise very similar, again demonstrating that mineral reduction is not pH-dependent. Further experiments at variable concentrations of dithionite (supplemental Fig. S6), indicated that the extent of reduction reached 100% at higher dithionite concentrations and that the rate constant was independent of dithionite concentration, consistent with electron transfer from reductant to the core being the rate-limiting step in the reduction reaction.
The Ferroxidase Center and Inner Surface Sites of BFR Are Not Involved in Iron Release
The ferroxidase center of BFR, located at the heart of each subunit, plays a key role in the formation of the iron core (26, 27, 43). Two Fe2+ ions bind at each center and, in the presence of oxygen (or hydrogen peroxide), become rapidly oxidized, generating a stable, bridged di-Fe3+ form. Extensive structural and mechanistic studies of E. coli BFR have led to a model in which the center acts as a true catalytic center, continually cycling between oxidized and reduced states, with electrons from the oxidation of Fe2+ in the central cavity being routed to the center, where they are used to reduce oxygen or hydrogen peroxide (26, 27). Recently, an Fe2+-binding site on the inner surface of the cavity, located 10 Å directly below the ferroxidase center, was structurally characterized and shown to play an important role in core formation but not in turnover of the ferroxidase center, consistent with the site being important for the transfer of electrons from the cavity to the ferroxidase center (27). Given the key roles these sites play in core formation, it was of interest to determine whether they play a role in iron core release. Certain residues at the ferroxidase center are critical for core formation such that, in their absence, it is not possible to generate a substantial core. Therefore, we selected a variant of the ferroxidase center, E127Q, that exhibited significantly reduced mineralization activity but nevertheless was able to generate a substantial iron core (26). Likewise, the inner surface site variant H46A/D50A also exhibited a significantly reduced mineralization activity but remained capable of generating a sufficient mineral core to allow iron release studies (27). H46A/D50A and E127Q variant BFRs were loaded with 1200 irons/protein, and iron release was monitored using the dithionite/chelator assay described above. The iron release behavior of both variants (see supplemental Fig. S7) was essentially identical to that of wild type. Therefore, we conclude that neither the ferroxidase center nor the inner surface site plays a key role in iron release under the conditions tested.
DISCUSSION
Reductant and Chelator Dependence of BFR Iron Release Kinetics
We began our in vitro analysis of the iron release properties of E. coli BFR by employing the type of assays that have been used frequently before, involving a two-component reductant-chelator system, in which dithionite or dithionite/FMN was used as reductant and ferrozine or bipyridyl as chelator. The very different rates observed with different reductants (GSH, dithionite, and FMNH2) indicated that the transfer of electrons from reductant to core Fe3+ was the rate-limiting step in the release reaction. With dithionite, a clear first order (linear) dependence was observed at low reductant concentration, which became zero order (independent) of reductant at higher concentration. The data were characteristic of saturation kinetics, indicating that available binding sites for dithionite were saturated at higher concentrations and that the overall reaction became rate-limited by the subsequent electron transfer from reductant to iron.
Dihhydroflavins, such as FMNH2, have been commonly used for ferritin iron release studies (15, 36, 38–40), and it has been proposed that they could play a physiological role in iron release. With FMNH2, a significantly enhanced rate of iron release was observed. Saturation kinetics were not routinely observed with FMNH2, suggesting that binding to BFR was highly transient in nature. Consistent with this was our inability to detect a flavin-BFR complex, and we note that FMN also does not form a stable complex with horse spleen ferritin (15).
In these reductant-chelator assays, the chelator was assumed to act only as a reporter of iron release. The data reported here show that this is not always the case. At pH 6, assays conducted with ferrozine as chelator resulted in significantly greater rates of release than at pH 7, suggesting that iron mobilization was pH-dependent. Iron release involves the dissolution of a ferric oxy-hydroxide mineral, resulting in Fe2+, in a reaction that requires protons (this can be considered to be the reverse of the mineralization reaction, which liberates protons and is enhanced at high pH (5)). Therefore, an enhanced rate at lower pH would be consistent with the mineral dissolution being rate-limiting. However, no pH dependence was observed when bipyridyl was used as chelator, indicating that the pH dependence was an artifact of the chelator assay. Chelator effects on measuring Fe3+ reduction have been noted previously (44). Therefore, we redesigned the release assay so that chelator was not present until the point at which we wished to observe the concentration of immediately available Fe2+. These experiments demonstrated that the iron release reaction rate is pH-independent and that the rate of iron release was dependent on the reductant. Furthermore, the reduction of Fe3+ to Fe2+, measured through the loss of intensity due to the ferric oxy-hydroxide mineral, was found to occur with a rate constant very similar to that of iron release and was also pH-independent. Together, these data demonstrated that the reduction of the mineral core, rather than release of iron, is the rate-limiting step.
Given the above, it is important to consider why the ferrozine release assay data exhibited a pH dependence. The literature on the ability of small molecules to penetrate the coat of ferritin protein is not clear (45), but it is possible that chelators may be able to access the BFR cavity and in doing so may influence the rate at which iron exits the protein. However, if the reduction step is rate-limiting, why should this affect the overall rate of release? One possibility is that the rate of electron transfer into the core may be influenced by binding of chelators at the core surface, which would increase the apparent reduction potential of the Fe3+ ions. In the case of ferrozine, it appears that, at pH 6, it is much better able to access the cavity than at pH 7, consistent with a protonation event. Because the pKa of ferrozine is ∼3.2 (46), this must be associated with the protein, because the chelator will be essentially completely deprotonated at pH 6–7. Even at pH 7, where there was essentially no difference between rates measured through ferrozine and bipyridyl, the release rate was higher than those measured in the absence of chelator (Figs. 5 and 6), demonstrating that the chelator does indeed have some influence on the kinetics of release, and we conclude that it is not possible to directly compare iron release rates measured with different reductants and chelators. Of course, the cell cytoplasm contains many small molecules that could potentially access the BFR cavity and bind Fe2+ and therefore influence the kinetics of release.
The Role of Heme in Iron Release from BFR
The comparison of data for heme-free and heme-containing BFR demonstrates unequivocally that the heme groups play an important role in both the kinetics and thermodynamics of iron release. A significant enhancement in the rate was observed in the presence of heme, indicating that electron transfer occurs through the protein; direct electron transfer at the core surface would not be expected to be influenced by heme. The rate enhancement was larger with FMNH2 as the electron source compared with dithionite, suggesting that dithionite may be able to deliver electrons both via the heme in the protein coat and directly at the core surface. The mechanism by which reductants deliver electrons to core Fe3+ is somewhat uncertain from previous reports. For example, Watt et al. (18) showed that large, low potential reductants (flavoproteins and ferredoxins) react anaerobically with both mammalian ferritin and BFR to generate Fe2+ in the ferritin cavity, leading to the conclusion that reduction can occur without direct interaction of the redox reagent at the mineral core surface. On the other hand, studies of core reduction with dihydroflavins showed that, in solution, these reductants could promote the rapid release of Fe2+, but immobilized versions could not, leading to the conclusion that the flavin must need to pass through the protein shell prior to reduction (15). Here, although our data do not directly address the question of whether the reductant accesses the protein cavity, it is clear that electron delivery by FMNH2 (at least in part) occurs through the protein to a greater extent than for dithionite, as judged from the heme dependence. In other ferritins, which do not contain heme, there must be other mechanisms that facilitate the transfer of electrons to the mineral core. The data presented here are consistent with previous studies of A. vinelandii and P. aeruginosa BFRs, from which it was concluded that heme reduction occurred during core reduction (11, 47). However, neither of these previous studies demonstrated the importance of heme for the rate or extent of iron release.
The extent of iron release observed here for any single addition of reductant to BFR was never more than ∼50% of the original loading and was significantly greater for dithionite than for FMNH2. Again, the literature on the extent of iron release from ferritins is somewhat variable. For example, studies of mammalian ferritin with a variety of reductants, including plant-derived o-diphenols, ascorbate, cysteine, and GSH, resulted in ≤20% iron release (16, 48). On the other hand, studies of release of iron from horse spleen and heart ferritin with dihydroflavins resulted in quantitative reduction and release (15). Other studies of pea seed ferritin using ascorbate as reductant resulted in release of 30–65% of the original iron (depending on pH) (13) but very little release from horse spleen ferritin under similar conditions. The data presented here for E. coli BFR showed that the extent of release was dependent on heme (Figs. 3 and 4) (e.g. increasing from ∼25 to ∼50% of the original loading with dithionite as reductant).
The Importance of a Chelator for Iron Release
Previously, it had been shown that reduction of ferritin and BFR in the absence of a high affinity chelator generated a stable Fe2+ core (18–20). The data presented here demonstrated that significant amounts of Fe2+ are released following reduction and that this can be separated from the protein by gel filtration or can be detected as the fraction of Fe2+ that is immediately available to a chelator added at some point after the initiation of reduction. It is likely that some Fe2+ remains weakly associated with the protein, but this is clearly not protected from being readily chelated or dissociated during gel filtration. We note that Watt et al. (18) also reported that reduced iron was readily available for chelation, but in their study, there was no evidence of release of Fe2+ from the protein during gel filtration. Overall, the results are not very different and may simply reflect different experimental conditions.
Here, the extent of iron release was ≤50% of the original iron, so a significant proportion of the iron remained associated with the protein. Studies of core reduction (Fig. 5D and supplemental Fig. S6) showed that the extent of reduction varied between 70 and 100%, depending on reductant concentration. Therefore, we conclude that upon core reduction, the generated Fe2+ does not remain entirely associated with the protein; rather, up to ∼50% is released. In these relatively complex mixtures, it is difficult to rule out that one of the solution components might be able to complex released iron; for example, it has been proposed that the oxidation products of dithionite, sulfite and sulfate, can act as an iron ligand (12). However, such species do not have very high affinities for Fe2+, and it is clear that a high affinity chelator is not required. We note that evidence for Fe2+ release in the absence of a high affinity chelator has been reported previously for plant ferritin (49).
The Mechanism of Iron Release from the BFR Mineral
The data reported here provide a clear picture of the key mechanistic steps involved in BFR iron release. The rate-determining step of the process is the delivery of electrons from the outside of the protein to the internal cavity. Heme plays an important role in catalyzing this electron transfer step. Even a small amount of heme was found to have a significant effect on the rate and extent of release. Electrons arriving at the mineral result in the reduction of Fe3+ to Fe2+. Although a significant proportion of Fe2+ remains within the cavity, the relatively low affinity of Fe2+ for ligands in the cavity leads to the mobilization of Fe2+, via protein coat channels, to the external surface, where it is available for chelation. This movement of Fe2+ toward the external surface occurs even when a high affinity chelator is absent.
Iron release from BFR occurs independently of the ferroxidase center, so iron core mineralization and release involve different catalytic/cofactor centers; the ferroxidase center is essential for mineralization but appears not to be involved in iron release, and the heme group is dispensable for mineralization but plays an important role in iron release. This reflects that the reactions involved do not proceed in both directions: for mineralization, a high potential oxidant (O2 or H2O2) is required to accept electrons from Fe2+ in the cavity; for release, a low potential reductant is required to supply electrons for reduction of Fe3+.
The nature of the reductant in vivo is not yet known; certainly, a dihydroflavin is a possibility. In this context, we note that, in the plant pathogen Erwinia chrysanthemi, iron cycling by BFR is intimately associated with the Suf iron-sulfur assembly pathway (50). Furthermore, these studies showed that SufC (an ATPase) is important for mediating iron release from BFR. Interestingly, it was recently demonstrated that a SufBC2D complex contains a flavin adenine dinucleotide (FADH2), leading to the proposal that this may be involved in the reductive mobilization of iron for incorporation into iron-sulfur clusters (51, 52).
The Regulation of Iron Release
Our observations raise important questions about how iron release is regulated. For BFR, the data suggest that this must be at the level of electron supply, through the kinetic control of the rate-limiting step of the reaction. An important observation in this context is that GSH, an abundant cytoplasmic reductant, was unable to stimulate significant iron release from E. coli BFR in vitro. Potentially important in this context is the bacterioferritin-associated ferredoxin (Bfd) (1, 53, 54). Expression of bfd is up-regulated under low iron, leading to the proposal that it is involved in iron release from BFR, and it has been shown previously that Bfd binds BFR specifically (47, 54). However, it is currently uncertain as to what form of the protein, the [2Fe-2S] or apo-form, is important in iron release.
Other regulatory mechanisms may also be important for controlling iron release. The protein coat itself could influence the rate of electron delivery to the cavity, for example by modulating its interaction with reductant. Also, the protein channels that facilitate Fe2+ exit could play an important role. For example, in eukaryotic ferritins, it has been shown that the 3-fold channels can undergo localized “melting” in response to environmental changes (such as low chaotrope concentration, temperature, and specific peptide-ferritin interactions), such that iron release can be considered to be “gated” (40, 55, 56). Whether such mechanisms that could affect the access of reductant to the ferric mineral also exist in BFRs is not clear, but we note that several of the residues important for channel melting in eukaryotic ferritin are conserved. Having established the principles of iron release from BFR, we are now in a position to address issues of how iron release is regulated.
Supplementary Material
Acknowledgment
We thank Nick Cull for technical assistance with protein purification.
Footnotes
This work was supported by a grant from the United Kingdom Biotechnology and Biological Sciences Research Council Grants BB/D001943/1 (to N. E. L. B. and G. R. M.) and BB/D002435/1 (to S. C. A.) and a JIF grant from the Wellcome Trust.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S7.
REFERENCES
- 1. Andrews S. C. (1998) Adv. Microb. Physiol. 40, 281–351 [DOI] [PubMed] [Google Scholar]
- 2. Harrison P. M., Arosio P. (1996) Biochim. Biophys. Acta 1275, 161–203 [DOI] [PubMed] [Google Scholar]
- 3. Theil E. C., Matzapetakis M., Liu X. F. (2006) J. Biol. Inorg. Chem. 11, 803–810 [DOI] [PubMed] [Google Scholar]
- 4. Chasteen N. D. (1998) Ferritin. Uptake, storage, and release of iron. in Metal Ions Biol. Syst. 35, 479–514 [PubMed] [Google Scholar]
- 5. Lewin A., Moore G. R., Le Brun N. E. (2005) Dalton Trans. 3597–3610 [DOI] [PubMed] [Google Scholar]
- 6. Abdul-Tehrani H., Hudson A. J., Chang Y. S., Timms A. R., Hawkins C., Williams J. M., Harrison P. M., Guest J. R., Andrews S. C. (1999) J. Bacteriol. 181, 1415–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fu X., Deng J., Yang H., Masuda T., Goto F., Yoshihara T., Zhao G. H. (2010) Biochem. J. 427, 313–321 [DOI] [PubMed] [Google Scholar]
- 8. Marchetti A., Parker M. S., Moccia L. P., Lin E. O., Arrieta A. L., Ribalet F., Murphy M. E., Maldonado M. T., Armbrust E. V. (2009) Nature 457, 467–470 [DOI] [PubMed] [Google Scholar]
- 9. Le Brun N. E., Crow A., Murphy M. E., Mauk A. G., Moore G. R. (2010) Biochim. Biophys. Acta 1800, 732–744 [DOI] [PubMed] [Google Scholar]
- 10. Radisky D. C., Kaplan J. (1998) Biochem. J. 336, 201–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Richards T. D., Pitts K. R., Watt G. D. (1996) J. Inorg. Biochem. 61, 1–13 [DOI] [PubMed] [Google Scholar]
- 12. Funk F., Lenders J. P., Crichton R. R., Schneider W. (1985) Eur. J. Biochem. 152, 167–172 [DOI] [PubMed] [Google Scholar]
- 13. Laulhere J. P., Briat J. F. (1993) Biochem. J. 290, 693–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Biemond P., Swaak A. J., van Eijk H. G., Koster J. F. (1988) Free Rad. Biol. Med. 4, 185–198 [DOI] [PubMed] [Google Scholar]
- 15. Jones T., Spencer R., Walsh C. (1978) Biochemistry 17, 4011–4017 [DOI] [PubMed] [Google Scholar]
- 16. Boyer R. F., Clark H. M., LaRoche A. P. (1988) J. Inorg. Biochem. 32, 171–181 [DOI] [PubMed] [Google Scholar]
- 17. Takagi H., Shi D., Ha Y., Allewell N. M., Theil E. C. (1998) J. Biol. Chem. 273, 18685–18688 [DOI] [PubMed] [Google Scholar]
- 18. Watt G. D., Jacobs D., Frankel R. B. (1988) Proc. Natl. Acad. Sci. U.S.A. 85, 7457–7461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Watt G. D., Frankel R. B., Papaefthymiou G. C. (1985) Proc. Natl. Acad. Sci. U.S.A. 82, 3640–3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Watt G. D., Frankel R. B., Jacobs D., Huang H., Papaefthymiou G. C. (1992) Biochemistry 31, 5672–5679 [DOI] [PubMed] [Google Scholar]
- 21. Stiefel E. I., Watt G. D. (1979) Nature 279, 81–83 [DOI] [PubMed] [Google Scholar]
- 22. Frolow F., Kalb A. J., Yariv J. (1994) Nat. Struct. Biol. 1, 453–460 [DOI] [PubMed] [Google Scholar]
- 23. Andrews S. C., Le Brun N. E., Barynin V., Thomson A. J., Moore G. R., Guest J. R., Harrison P. M. (1995) J. Biol. Chem. 270, 23268–23274 [DOI] [PubMed] [Google Scholar]
- 24. Moore G. R., Kadir F. H., al-Massad F. (1992) J. Inorg. Biochem. 47, 175–181 [DOI] [PubMed] [Google Scholar]
- 25. Moore G. R., Kadir F. H., al-Massad F. K., Le Brun N. E., Thomson A. J., Greenwood C., Keen J. N., Findlay J. B. (1994) Biochem. J. 304, 493–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baaghil S., Lewin A., Moore G. R., Le Brun N. E. (2003) Biochemistry 42, 14047–14056 [DOI] [PubMed] [Google Scholar]
- 27. Crow A., Lawson T. L., Lewin A., Moore G. R., Le Brun N. E. (2009) J. Am. Chem. Soc. 131, 6808–6813 [DOI] [PubMed] [Google Scholar]
- 28. Baaghil S., Thomson A. J., Moore G. R., Le Brun N. E. (2002) J. Chem. Soc. Dalton Trans. 811–818 [Google Scholar]
- 29. Bauminger E. R., Harrison P. M., Hechel D., Nowik I., Treffry A. (1991) Biochim. Biophys. Acta 1118, 48–58 [DOI] [PubMed] [Google Scholar]
- 30. Stookey L. L. (1970) Anal. Chem. 42, 779–781 [Google Scholar]
- 31. Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., Klenk D. C. (1985) Anal. Biochem. 150, 76–85 [DOI] [PubMed] [Google Scholar]
- 32. Yang X., Le Brun N. E., Thomson A. J., Moore G. R., Chasteen N. D. (2000) Biochemistry 39, 4915–4923 [DOI] [PubMed] [Google Scholar]
- 33. Cheesman M. R., le Brun N. E., Kadir F. H., Thomson A. J., Moore G. R., Andrews S. C., Guest J. R., Harrison P. M., Smith J. M., Yewdall S. J. (1993) Biochem. J. 292, 47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gibbs C. R. (1976) Anal. Chem. 48, 1197–1201 [Google Scholar]
- 35. Sillen L. G., Martell A. E. (1964) Stability Constants of Metal-Ion Complexes, The Chemical Society, London [Google Scholar]
- 36. Sirivech S., Frieden E., Osaki S. (1974) Biochem. J. 143, 311–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fahey R. C., Brown W. C., Adams W. B., Worsham M. B. (1978) J. Bacteriol. 133, 1126–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jacobs D. L., Watt G. D., Frankel R. B., Papaefthymiou G. C. (1989) Biochemistry 28, 1650–1655 [DOI] [PubMed] [Google Scholar]
- 39. Chasteen N. D., Ritchie I. M., Webb J. (1991) Anal. Biochem. 195, 296–302 [DOI] [PubMed] [Google Scholar]
- 40. Jin W., Takagi H., Pancorbo B., Theil E. C. (2001) Biochemistry 40, 7525–7532 [DOI] [PubMed] [Google Scholar]
- 41. Cheesman M. R., Thomson A. J., Greenwood C., Moore G. R., Kadir F. (1990) Nature 346, 771–773 [DOI] [PubMed] [Google Scholar]
- 42. Lawson T. L., Crow A., Lewin A., Yasmin S., Moore G. R., Le Brun N. E. (2009) Biochemistry 48, 9031–9039 [DOI] [PubMed] [Google Scholar]
- 43. Le Brun N. E., Andrews S. C., Guest J. R., Harrison P. M., Moore G. R., Thomson A. J. (1995) Biochem. J. 312, 385–392 [PMC free article] [PubMed] [Google Scholar]
- 44. Cowart R. E., Singleton F. L., Hind J. S. (1993) Anal. Biochem. 211, 151–155 [DOI] [PubMed] [Google Scholar]
- 45. Yang X., Chasteen N. D. (1996) Biophys. J. 71, 1587–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thompsen J. C., Mottola H. A. (1984) Anal. Chem. 56, 755–757 [Google Scholar]
- 47. Weeratunga S. K., Gee C. E., Lovell S., Zeng Y., Woodin C. L., Rivera M. (2009) Biochemistry 48, 7420–7431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kontoghiorghes G. J. (1986) Biochem. J. 233, 299–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laulhère J. P., Barcelò F., Fontecave M. (1996) Biometals 9, 303–309 [DOI] [PubMed] [Google Scholar]
- 50. Expert D., Boughammoura A., Franza T. (2008) J. Biol. Chem. 283, 36564–36572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wollers S., Layer G., Garcia-Serres R., Signor L., Clemancey M., Latour J. M., Fontecave M., Ollagnier de Choudens S. (2010) J. Biol. Chem. 285, 23331–23341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Saini A., Mapolelo D. T., Chahal H. K., Johnson M. K., Outten F. W. (2010) Biochemistry 49, 9402–9412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Quail M. A., Jordan P., Grogan J. M., Butt J. N., Lutz M., Thomson A. J., Andrews S. C., Guest J. R. (1996) Biochem. Biophys. Res. Commun. 229, 635–642 [DOI] [PubMed] [Google Scholar]
- 54. Garg R. P., Vargo C. J., Cui X., Kurtz D. M., Jr. (1996) Biochemistry 35, 6297–6301 [DOI] [PubMed] [Google Scholar]
- 55. Liu X., Jin W., Theil E. C. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 3653–3658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu X. S., Patterson L. D., Miller M. J., Theil E. C. (2007) J. Biol. Chem. 282, 31821–31825 [DOI] [PubMed] [Google Scholar]
- 57. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.