

Abstract
Fibronectin (FN) purified by gelatin affinity chromatography is unstable and undergoes fragmentation. The cleavage has been ascribed to inherent autolytic protease activities as well as co-purified matrix metalloproteinases (MMP). Understanding the mechanism by which the proteolysis of FN occurs is important, because the FN fragments have biological activities that differ from those of intact FN. Having excluded contributions of other plasma-derived proteases, the present experiments demonstrated that cleavage of FN by MMP-2 to distinct fragments occurred in synergy with inherent FN activities. Limited heat treatment of FN at 56 °C for 30 min inactivated the inherent protease activities sharply reducing autolysis of FN in a manner similar to that seen in the presence of serine proteinase inhibitors. Heat treatment did not alter cell attachment to FN, but significantly increased the susceptibility of FN to enzymatic cleavage by MMP-2. The carboxyl-terminal hemopexin-like domain (PEX) of MMP-2 was shown to possess critical exodomain properties required for the interactions of MMP-2 with FN, and FN was cleaved at a significantly reduced rate by an MMP-2 variant with deletion of PEX. Verifying the specificity of interactions, isolated PEX competed FN cleavage by MMP-2 in a concentration-dependent manner. These results have further elucidated the synergistic contributions of inherent autolytic serine protease-like activities and MMP-2 to fragmentation of FN and provide the rationale and basis for modified preparation and handling of FN used in biological research.
Keywords: Fibronectin, matrix metalloproteinase, MMP, proteolysis, autolysis, cell attachment
1. Introduction
Fibronectin (FN) is an essential extracellular cell adhesion molecule which was first identified as a plasma component with the ability to support cell adhesion (Klebe, 1974). Cloning of the FN gene revealed a primary structure consisting of repeated homologous types I, II, and III modules and additional alternatively spliced modules resulting in the potential for multiple isoforms with unique functions (Kornblihtt et al., 1983; Skorstengaard et al., 1986; White et al., 2008). A number of biological activities, including binding sites for fibrin, heparin, collagen, integrins, and several bacteria have been localized to distinct modules or domains composed of several modules (Hynes, 1985; Yamada, 1989). By virtue of those functions, FN contributes to a range of biological processes, which include but are not limited to embryonic development, coagulation, and wound healing (Pankov and Yamada, 2002).
Significant efforts have been devoted to understanding mechanisms of FN cleavage because FN fragments have been detected in vivo in sites of chronic diseases from lung lavages (Castell et al., 1988), periodontal disease (Talonpoika et al., 1989; Huynh et al., 2002; Stanley et al., 2008), arthritis (Xie et al., 1992), poorly healing diabetic ulcers (Wysocki and Grinnell, 1990; Stanley et al., 2008) and experimental mammary cancer in mice (Xu et al., 2005b).
To define functional domains of FN, investigators applied various enzymes for analytical fragmentation of FN. By those experiments, several FN fragments with distinct biological functions were identified (For review see (Hynes, 1990)). For example, a 30 kDa N-terminal fragment had greater chemotactic activity for monocytes than intact FN and also reduced monocyte attachment to fibrin (Carsons et al., 1985). Surprisingly, cathepsin-generated FN fragments were shown to have proteolytic activities on collagen and FN (Planchenault et al., 1990; Lambert Vidmar et al., 1991a; Lambert Vidmar et al., 1991b).
More recent studies have used protein engineering technologies to express FN segments with highly defined borders and extensions. Such recombinant FN fragments have enabled investigators to very precisely map unique functions to specific residues, modules, and domains within FN, exemplified by the heparin binding residues in module III-13 (Busby et al., 1995), the collagen binding modules (Banyai et al., 1990; Steffensen et al., 2002; Katagiri et al., 2003), apoptotic effects of the heparin binding region (Kapila et al., 1999), and anti-angiogenic and anti-metastatic properties of the carboxylterminal fragment of the first type III module in FN (Anastellin) (Briknarova et al., 2003).
The preponderance of available evidence indicates that members of the matrix metalloproteinase family (MMP) can cleave FN. MMP-2 purified from human rheumatoid synovial cells induced significant FN fragmentation (Okada et al., 1990) and recent proteomics screens analyzing MMP-2 proteolytic activities of fibroblasts identified FN as an important substrate for MMP-2 (Dean and Overall, 2007). However, some investigators detected no cleavage when FN was incubated with MMP-2 (Kraft et al., 2001; Al Hazmi et al., 2007)and, in preliminary experiments, we found substantial variation in the efficiency of FN cleavage by recombinant MMP-2. These differences in the sensitivity of FN as a substrate for MMP-2 pointed to possible effects of catalytic activities inherent to FN (Lambert Vidmar et al., 1991b) or could derive from the activities of MMPs or other enzymes co-purified from plasma with FN (Johansson and Smedsrod, 1986; Pal et al., 2009).
For cleavage of substrates, MMPs typically utilize exosites that bind and position substrates relative to the catalytic site (Overall, 2001; Xu et al., 2007). Collagen binding occurs via the carboxyl-terminal domain in MMP-1 and MMP-3 (Murphy et al., 1992), whereas a distinct collagen binding domain consisting of three in-tandem FN type II-like modules inserted into the catalytic domains of MMP-2 and -9 is essential for binding and cleavage of collagen and elastin in these MMPs (Banyai et al., 1994; Murphy et al., 1994; Steffensen et al., 1995; Shipley et al., 1996). In context of FN cleavage, it was demonstrated that the carboxyl-terminal hemopexin-like domain of MMP-2 (PEX) is essential for binding of FN (Wallon and Overall, 1997). However, it has not yet been established whether the PEX serves as an exosite that is required for cleavage of FN by MMP-2.
On this basis, the present studies investigated the mechanisms by which inherent proteolytic activities and co-purified MMP-2 contribute to the cleavage of FN. Our experiments demonstrated that the inherent protease activities were abrogated by limited heat treatment of FN or by serine protease inhibitors. Importantly, the analyses eliminated the possibility that these activities were from other contaminating plasma proteases. MMP-2 cleaved FN most efficiently in the presence of the inherent FN activities or following limited heat treatment pointing to exposure of MMP-2 susceptible cryptic cleavage sites. After heat inactivating inherent FN proteolytic activities, we demonstrated in a reproducible manner that the carboxylterminal hemopexin-like domain (PEX) exosite is essential for MMP-2 interaction with and cleavage of FN.
2. Results
2.1 FN degradation involves MMP-2 and autolytic activities
In our preliminary efforts to isolate intact FN from human plasma for studies of its proteolysis by MMP-2, it became evident that FN purified by standard affinity purification protocols with gelatin-Sepharose (Engvall and Roushlahti, 1977) underwent self-degradation (not shown). This was consistent with the observation that preparations of FN isolated by this approach contain matrix metalloproteinases MMP-2 and MMP-9 that both have gelatin binding properties (Johansson and Smedsrod, 1986). Our enzymographic analysis of fractions eluted from the gelatin-Sepharose column during purification of FN from human plasma revealed that some of the gelatinolytic activities were eliminated during 1 M NaCl washes designed to eliminate non-specifically binding proteins, but also that the final purified FN fractions contained both MMP-2 and MMP-9 (Fig. 1). Of note, while these enzymatic activities are readily detectable by gelatin substrate enzymography they not are visible by regular SDS-PAGE and have not been reported widely.
Figure 1. MMP-2 and -9 activities co-purified with FN from human plasma by gelatin-Sepharose affinity chromatography.
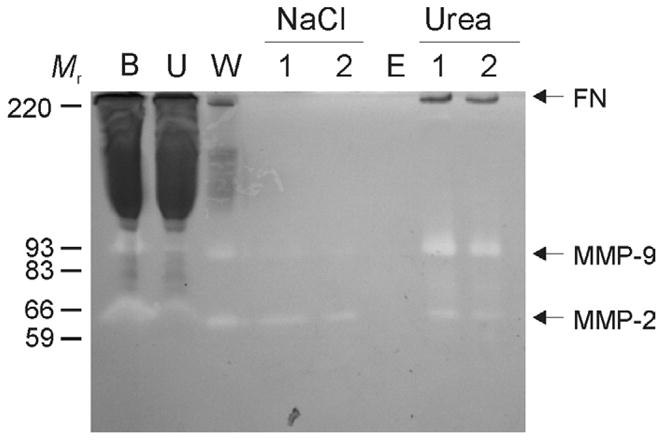
Residual gelatinolytic activities, including MMP-2 and -9, were monitored by gelatin substrate enzymography in column fractions during purification of fibronectin (FN). For analytical purification 500 μl of human plasma (B) was loaded on a gelatin Sepharose (Vt 25 μ1) mini column after sedimentation of insoluble aggregates at 2,500 × g. After collection of unbound proteins (U), the column was thoroughly washed (20 × Vt) with chromatography buffer (W; 50 mM Tris, 0.15 M NaCl, pH7.4) followed by 1M NaCl (N; 1, 2) in chromatography buffer (10 × Vt) to remove non-specifically bound proteins. After equilibration (E), specifically bound proteins were eluted with 4M urea (U; 1, 2). The enzymography analysis showed that plasma-derived MMP-2 and -9 (B) bound gelatin Sepharose and were co-purified with FN. Positions of FN, MMP-2, MMP-9, and kDa of proteins (Mr) are indicated.
To separate contributions of different proteolytic activities to the cleavage of FN, we tested the effects of the MMP inhibitor 1,10-phenanthroline and the broad-spectrum serine proteinase inhibitor 4-(2-aminoethyl) benzenesulfonyl fluoride (AEBSF). Both of these enzyme inhibitors blocked the cleavage of FN compared to control samples for 48 h during incubation at 22 °C pointing to proteolytic contributions of both co-purified gelatin-binding MMPs and serine protease-like activities (Fig. 2A). This result was verified with an additional MMP-specific inhibitor, BB94 (data not shown).
Figure 2. Stability of FN and susceptibility to cleavage by MMP-2 as a function of purification and protease inhibitors.
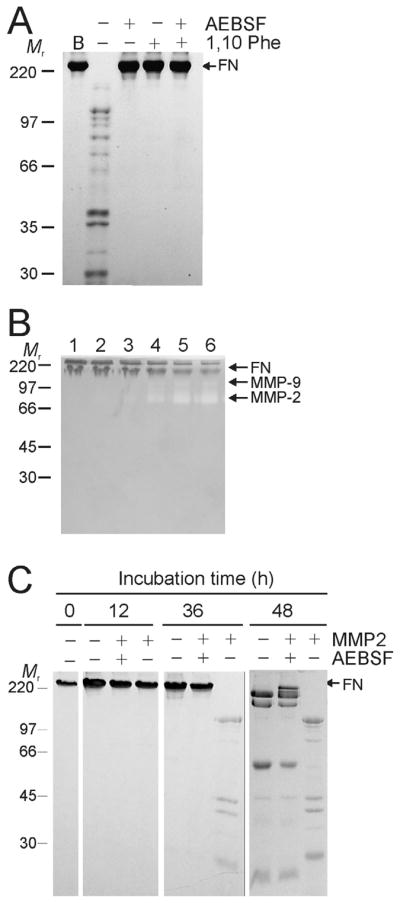
Panel A. The stability of FN in 50 mM Tris, 0.15 M NaCl, pH7.4 following purification from human plasma by gelatin-Sepharose affinity chromatography (B)(See also Fig. 1) was analyzed in the presence or absence of 0.5 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF) serine protease inhibitor and 1.5 mM 1,10 phenanthroline (1,10 Phe) MMP inhibitor. FN cleavage was monitored by 7.5% SDS-PAGE for 48 h at 22°C. FN demonstrated significant fragmentation in the absence of the inhibitors, whereas both inhibitors blocked hydrolysis. Panel B. To eliminate effects of co-purified MMP-2 and -9, urea-eluted FN from the gelatin-Sepharose affinity column was exchanged into 50 mM sodium-phosphate, 150 mM NaCl, pH 7.0 and further purified by gel filtration (HiLoad 16/60 Superdex 200). Eluted fractions in which no gelatinolytic activities were detected by gelatin-zymography (lanes 1, 2) were separated from those that contained MMP-2 and -9 (lanes 3–6), pooled, and used for subsequent analyses. Panel C. The capacity of exogenous recombinant MMP-2 to cleave FN was tested in preparations void of plasma-derived MMPs in the presence or absence of AEBSF over 48 h at 22 °C. Analysis of reactions by 10% SDS-PAGE gels showed significantly greater cleavage of FN by MMP-2 in the absence of AEBSF pointing to synergic actions of MMP-2 and serine protease-like proteolytic activities. Positions of FN and masses (kDa) of protein markers (Mr) are indicated.
To further delineate the specific role of MMP-2 in FN cleavage, FN was subjected to size exclusion chromatography following the initial affinity purification by gelatin-Sepharose. This additional purification step reduced MMP-2 and -9 activities to below detectable levels as assessed by the highly sensitive gelatin zymography (Fig. 2B).
The stability of the highly-purified, gelatinase-free FN samples was then monitored by SDS-PAGE over 48 h at 22 °C in the presence or absence recombinant MMP-2 and AEBSF. Although MMP-2-free FN control samples displayed limited fragmentation over time, addition of exogenous MMP-2 substantially enhanced both the speed and extent of FN cleavage (Fig. 2C). Importantly, MMP-2 had greater activity on FN after 36 and 48 h in absence than in the presence of AEBSF (Fig. 2C). These experiments demonstrated that MMP-2 cleaved FN most efficiently in the presence of serine protease-like activities that may be correspond to those reported to be inherent to FN (Lambert Vidmar et al., 1991b; Bonnefoy and Legrand, 2000). Corroborating the presence of such autolytic FN activities, our analysis of the highly purified preparation of FN by GeLCMS (Lasonder et al., 2002) eliminated the presence of other plasma-derived contaminating proteases in the purified FN preparations.
We then tested the hypothesis that these FN activities expose otherwise cryptic and inaccessible cleavage sites for subsequent hydrolysis by MMP-2. This scenario is known from type I collagen; MMP-2 has low activity on this substrate in its native triple helical conformation, but efficiently hydrolyzes the separated collagen a-chains after heat denaturation or following initial cleavage by MMP-1 which cause unwinding of the collagen helix (Danielsen, 1982; Collier et al., 1988; Tam et al., 2004).
Our experiments demonstrated that pre-treatment of FN at increasing temperatures elevated the susceptibility of FN to cleavage by MMP-2 (Fig. 3A). Heating of FN at 56 °C or greater for 30 min resulted in predictable and increasing FN cleavage by MMP-2 over 24 h. Contrary to preceding experiments (Fig. 2C), no self degradation of FN was detectable for 48 h after heat treatment in the absence of MMP-2 (Fig. 3B). This effect of heat-treatment on the stability of FN mimicked that of AEBSF, suggesting that heating inactivated the inherent serine-protease activities. Thus, heating of FN reduced autolysis of FN and increased the susceptibility to MMP-2.
Figure 3. Heat treatment enhances FN cleavage by MMP-2 and blocks autolysis of FN.
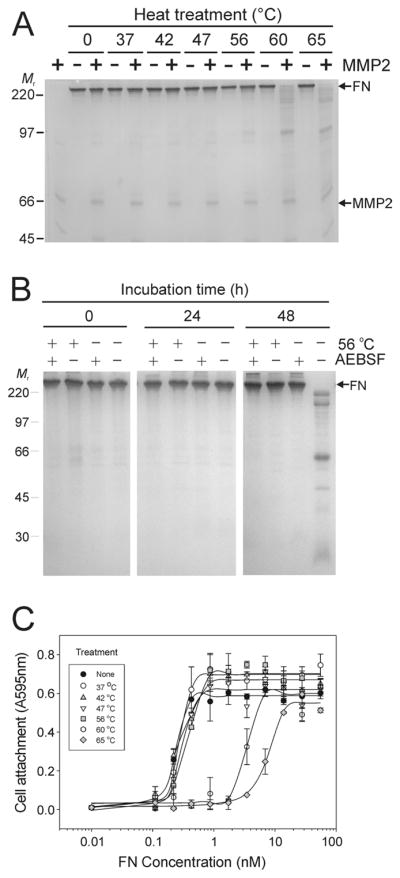
Panel A. Experiments tested the effects of heat treatment on the stability and susceptibility of FN to MMP-2 cleavage. Preparations of FN without detectable plasma MMP-2 or -9 were subjected to limited (30 min) heat treatment between 37 and 65 °C, and then incubated in the presence or absence of exogenous recombinant MMP-2 for 24 h at 22 °C. Analysis by SDS-PAGE showed substantial cleavage of FN samples treated at 56 °C with increasing extent of hydrolysis after treatment at 60 and 65 °C. Panel B. Control FN samples or samples treated for 30 min at 56 °C were subsequently incubated at 22 °C in the presence or absence of AEBSF. Analysis by SDS-PAGE detected only autolysis after 48 h in FN samples that were not heat-treated in the absence of AEBSF, whereas samples containing AEBSF were stable. There were no signs of autolysis of FN after heat-treatment irrespective of the presence or absence of AEBSF implying that heat-treatment inactivated inherent autolytic activities. Panel C. The functional integrity of heat-treated FN was analyzed in cell attachment assays. HT 1080 fibrosarcoma cells in serum-free medium were seeded in 96 microwell tissues culture plates coated with a concentration range of FN samples (70 - 0.01 nM) treated between 37 and 65 °C or control FN. Cell attachment was quantified at 595 nm from re-dissolved crystal violet dye after staining of cells. Cell attachment was comparable to control for FN samples heated up to 56 °C, but substantially reduced when FN was treated at 60 or 65 °C. Positions of FN (FN) and recombinant MMP-2 (MMP2), and kDa of protein standards (Mr) are presented.
Importantly, assays measuring the biological activities of FN revealed that the cell attachment properties of FN were retained after heating for 30 min at 56 °C reflecting functionally folded protein, but were substantially reduced after treatment at or above 60 °C indicating that the molecular structure of FN had been destabilized (Fig. 3C). Therefore, subsequent experiments measuring the MMP-2 proteolytic activities on FN used FN treated at 56 °C.
2.2 Interaction of PEX domain of MMP-2 with Fibronectin
Isolated recombinant hemopexin-like carboxyl-terminal domain of MMP-2 (PEX) binds FN in solid phase assays and by analytical affinity chromatography (Wallon and Overall, 1997). To extend this observation and understand whether exodomain functions of PEX are required for FN cleavage by MMP-2, our experiments first characterized the interactions of native FN with isolated PEX and PEX in the context of the full length MMP-2.
Specifically, we analyzed the interactions of FN with recombinant variants of MMP-2, including full-length MMP-2, a variant of MMP-2 in which the PEX domain was deleted (MMP-2ΔPEX), and isolated recombinant PEX from MMP-2. These protein-protein binding experiments utilized the active site-mutated MMP-2E404A, which retains ligand binding properties, but is proteolytically inactive (Morgunova et al., 1999). Full-length MMP-2E404A bound FN in a concentration-dependent and saturable manner with an apparent Kd of 2 × 10−8 M (Fig 4A). Isolated PEX also bound FN with a Kd of 10−7 M. Of particular interest, MMP-2 with deletion of the PEX domain (MMP-2ΔPEX) interacted in a weak non-saturable manner with FN.
Figure 4. Interaction of FN with the carboxyl-terminal domain of MMP-2.

Panel A. Interactions of FN with the carboxyl-terminal hemopexin-like domain of MMP-2 (PEX) were analyzed. FN was coated in 96 microwell plates (0.5 μg/well), blocked with BSA (10 mg/ml), and reacted with a concentration range (3 × 10−6 – 10−10 M) of biotinylated MMP-2, PEX, or MMP-2ΔPEX in which the PEX is deleted. Protein binding to coated FN was detected with alkaline phosphatase-conjugated streptavidin and PNPP substrate, and quantified at 405 nm. MMP-2 and PEX bound in a concentration-dependent saturable manner to FN. In comparison, MMP-2ΔPEX showed weak and non-saturable binding to this ligand. Panel B. The specific contribution of PEX to the binding was measured in competitive protein-protein binding assays in which biotinylated MMP-2 was added simultaneously with a concentration range of unlabeled MMP-2, PEX or ovalbumin. Like unlabeled MMP-2 (positive control), isolated PEX competitively inhibited the binding of MMP-2 to FN in a concentration-dependent manner pointing to PEX as an essential exosite for MMP-2 interactions with FN.
Additional competitive binding assays were designed to verify that PEX mediated the interaction of full-length MMP-2 with FN. When added simultaneously with MMP-2E404A to surfaces coated with FN, soluble PEX inhibited MMP-2E404A binding to FN in a concentration dependent manner (Fig. 4B). Whereas unlabeled MMP-2E404A as a positive control likewise competed labeled MMP-2E404Abinding to FN, a non-specific ovalbumin control did not alter MMP-2E404Abinding to FN.
These experiments demonstrated that PEX is essential for MMP-2 interactions with FN.
2.3 Contributions of PEX to cleavage of FN by MMP-2
Having established an assay system for analyzing MMP-2 catalytic effects on FN without interference from autolytic FN activities as detailed above, we characterized the contributions of PEX to cleavage of FN by MMP-2. The experiments first compared proteolysis of FN by full-length MMP-2 or MMP-2ΔPEX. In the presence of MMP-2, 50% of the intact FN was reduced to short or non-detectable fragments after 5 h at 22 °C. The degradation increased to 78% after 12 h as assessed by densitometric analysis of SDS-PAGE gels (Fig 5A, B). In comparison, the quantity of intact FN was reduced by only 7% after 5 h and 40% after 12 h in the presence of MMP-2ΔPEX (Fig 5A, B). Moreover, the velocity of FN cleavage was significantly greater for MMP-2 (2.56 RDU × s−1) compared to MMP-2ΔPEX (0.35 RDU ×s−1) and FN without MMP-2 (0.05 RDU × s−1) (Fig. 5B). These results were consistent with the observations from the preceding experiments that implicated PEX as the essential FN-binding exodomain on MMP-2, and showed that FN cleavage is significantly reduced in the absence of the PEX from MMP-2.
Figure 5. Contribution of the carboxyl-terminal domain to cleavage of FN by MMP-2.

The role of the carboxyl-terminal domain (PEX) of MMP-2 to cleavage of FN by the enzyme was evaluated using recombinant wildtype MMP-2, MMP-2ΔPEX which lacks the PEX, and isolated PEX in regular and competitive enzyme activity assays. Panel A. FN (100 μg/ml) after heat-treatment at 56 °C for 30 min were incubated with MMP-2 (MMP-2, +) or MMP-2ΔPEX (MMP-2ΔPEX, +) for 3 - 24 h at 22 °C. Panel B. Equal aliquots of the reactions collected at designated time points were separated by SDS-PAGE. FN cleavage was quantified by densitometric analysis of the intact FN on the gels and plotted for each enzyme as well as control by relative densitometric units (RDU) as a function of time. The rate of FN cleavage (RDU × s−1) determined in the linear range of the assays is presented. Panel C. Assays analyzed the capacity of a concentration range (0 and 0.05 – 3.0 μM) exogenous PEX to inhibit degradation of FITC-labeled FN by MMP-2. Relative fluorescent units (RFU) representing the extent of FN cleavage recorded in the linear range of the assays after 30 min at 22 °C are presented. PEX inhibited FN cleavage by MMP-2 in a concentration dependent manner. Together, these results demonstrated that PEX contributes essential exodomain functions to MMP-2 cleavage of FN. The positions of FN, MMP-2, MMP-2ΔPEX, and kDa of proteins (Mr) are shown.
To confirm the contributions of PEX to the cleavage of FN, subsequent assays used a FN substrate that was labeled with FITC at high density to achieve self-quenching. Kinetic measurements of released fluorophore reflecting proteolysis of FN by MMP-2 enabled us to quantify the competitive effects of a concentration range of PEX on MMP-2 activities on FN. PEX inhibited MMP-2 cleavage of FN in a concentration dependent manner (Fig. 5C) with half-maximal inhibition at a PEX:MMP-2 molar ratio of 1.7:1.
Overall, the present protein binding and enzyme activity experiments have established that cleavage of FN by MMP-2 occurs in concert with autolytic activities that expose MMP-2 sensitive cleavage sites on FN. Serine protease inhibitors and limited heat treatment abolished the self-degradation of FN and increased the sensitivity of FN to cleavage by MMP-2 without disrupting the cell attachment properties. Protein-protein binding studies and competition assays demonstrated that the carboxyl-terminal PEX domain of MMP-2 is an important exosite for MMP-2 interactions with and cleavage of FN.
3. Discussion
In our initial efforts to decipher the mechanism of FN cleavage by MMP-2, experiments yielded significant variability with respect to both the stability of purified FN and the efficiency of FN cleavage by MMP-2. Subsequent experiments designed to optimize the experimental conditions demonstrated that inherent proteolytic activities of FN acted in synergy with MMP-2 activities during fragmentation of the molecule.
Earlier analyses of a range of enzymes, including trypsin, chymotrypsin, plasmin, and cathepsin D showed that these enzymes can cleave FN and generate fragments pending the enzyme-specific cleavage sites that typically are located in the connecting inter-modular segments (Hynes, 1990; Pankov and Yamada, 2002). Of particular interest to the present study, investigators have reported proteolytic activities inherent to FN or FN fragments, including activities that could degrade FN itself (termed fibronectinase)(Lambert Vidmar et al., 1991b), gelatin, laminin, and type IV collagen (Planchenault et al., 1990; Emod et al., 1990; Lambert Vidmar et al., 1991a; Lambert Vidmar et al., 1991b; Unger and Tschesche, 1999; Schnepel and Tschesche, 2000). Further, serine proteinase inhibitors, including phenylmethylsulfonyl fluoride (PMSF), blocked the fibronectinase activity (Planchenault et al., 1990).
The capacity of MMP-2 to cleave FN was established by in vitro assays (Okada et al., 1990; Lee and Lo, 2004) and confirmed in recent proteomics screens for MMP-2 substrates (Dean and Overall, 2007). An interesting observation from our experiments was that samples of FN isolated from human plasma showed fragmentation over time, even when the purification scheme combined the established gelatin-Sepharose affinity chromatography protocol (Engvall and Roushlahti, 1977) with gel filtration to eliminate any co-purified, gelatin-binding MMP-2 and -9. Our analyses of fractions during purification of FN by the highly sensitive GeLCMS confirmed that gel filtration eliminated MMP-2 and -9 and no other plasma-derived contaminating proteases were detected. These observations enabled us to conclude that the non-MMP-induced FN-degradation, which was inhibited by serine proteinase inhibitors PMSF and AEBSF, was caused by autolytic FN activities.
Experiments in which we then added exogenous recombinant MMP-2 to MMP-free FN preparations confirmed that MMP-2 cleaves FN (Okada et al., 1990; Lee and Lo, 2004). The hydrolysis of FN by MMP-2 proceeded significantly faster in the absence than in the presence of AEBSF, and control samples containing AEBSF but no exogenous MMP-2 displayed very limited self degradation. These new results indicate that the autolytic activities expose FN sites that are sensitive to MMP-2 cleavage. Therefore, eliminating MMPs from FN preparations may in itself extend the shelf-life, but does not completely prevent FN fragmentation in the absence of proteinase inhibitors (Pal et al., 2009).
To understand whether the initial autolytic cleavage of FN might emulate conformational disruption, we heat-treated FN over a range of temperatures from 37 – 65 °C prior to analytical cleavage by MMP-2. Heating of FN at 56 °C for 30 min eliminated autolysis but did not abrogate the important cell attachment properties (Klebe, 1974). Moreover, heat treatment significantly enhanced the susceptibility of FN to hydrolysis by MMP-2. Of importance, the increased sensitivity to MMP-2 occurred prior to the maximal thermal denaturation which occurs with a midpoint between 62 and 64 °C (Ingham et al., 1984) below the point at which heating induces FN fragmentation at 70 °C (Miekka, 1983). That heating of FN to 56 °C enhanced the resistance to autolysis in a manner similar to that resulting from addition of AEBSF indicates that both treatments inactivate inherent proteolytic activities. Therefore, following purification, the integrity of FN may be best retained in the presence of an inhibitor of serine protease activities even when chromatography protocols are used which eliminate MMPs from the preparation.
Our understanding of the precise mechanism underlying the inherent catalytic activities of FN is developing. Recent studies have added support for the notion of distinct catalytic activities and catalytic sites in FN. First, an active type IV collagenase of the segment of FN, which is defined by modules I6 – I9 and includes the two type II modules in the collagen binding region, was expressed as recombinant protein (Schnepel and Tschesche, 2000). Recently, zinc-binding properties in the N-terminal part of FN were found to be important for collagen binding (Graille et al., 2010). Moreover, there is a putative catalytic site HEXXH in module I8, which is homologous with the zinc-binding consensus sequence in the active site in members of the metzincin superfamily of enzymes. A recombinant protein consisting of a 77 kDa N-terminal segment of FN and including 10 residues of module III1 had catalytic activities that were inhibited with inhibitors of zinc-dependent metalloproteinases and by mutations in the HEXXH sequence that reduced zinc-binding (Houard et al., 2005). Thus, evidence for distinct structural elements and functions of an FN-protease is increasingly convincing.
Important functional roles have been attributed to exosites in the MMPs (Overall et al., 2000; Overall and Lopez-Otin, 2002) exemplified by the requirement for the collagen binding domain in MMPs 2 and -9, and the carboxyl terminal domain (PEX) in MMP-1 and -3, respectively, to achieve collagen binding and cleavage (Murphy et al., 1994; Steffensen et al., 1995; Xu et al., 2004). In MMP-2, PEX has several key functional properties, including TIMP-2 binding by latent MMP-2 during its activation by MT1-MMP (Strongin et al., 1993; Strongin et al., 1995). However, in spite of the numerous critical biological properties of FN, no exosites in MMP-2 have previously been defined for cleavage of FN.
Extending the observation that PEX in MMP-2 interacts specifically with FN (Wallon and Overall, 1997), we tested whether PEX was required for FN cleavage by MMP-2. Our protein-protein binding experiments determined that full-length MMP-2 bound to FN in a specific and concentration dependent manner, whereas MMP-2 without the PEX domain (MMP-2ΔPEX) had greatly reduced and non-saturable binding to FN. The PEX – FN binding occurs by charge based interactions that require 0.3 M NaCl for disruption pointing to a biologically pertinent interaction (Wallon and Overall, 1997). Our experiments showed that binding of full-length MMP-2 to FN exceeded that of isolated PEX, reflected by apparent Kds of 2 × 10−8 and 10−7 M, respectively. This is consistent with reduced Kds measured for substrate interactions with other isolated recombinant modules and domains of MMP-2 and MMP-9 (Xu et al., 2005a). Likewise, studies of collagen binding domains in FN showed that the two type II modules in FN require at least the flanking type I6 and I7 modules to achieve collagen binding (Steffensen et al., 2002) and that even greater affinity may be achieved by integrating the flanking modules I8 and I9 in the construct (Katagiri et al., 2003). To further verify the contribution of PEX to binding of MMP-2 to FN and eliminate the possibility that the loss of binding for MMP-2ΔPEX resulted from structural perturbations due to the deletion of the 26 kDa PEX, we confirmed that soluble PEX competed interactions of MMP-2 with FN in competitive protein-protein binding assays. Of note, all binding experiments utilized MMP-2 in which a mutation of the active site E to A resulted in a catalytically inactive enzyme without affecting exosite functions.
PEX was also essential for MMP-2 catalytic activities on FN. Our enzyme activity assays demonstrated that deletion of PEX from MMP-2 produced an enzyme variant with very low activity on FN. The rate of cleavage was reduced by nearly 8-fold. Moreover, soluble PEX inhibited cleavage of FN by MMP-2 in a concentration dependent manner.
Overall, the initial methodological experiments demonstrated that MMP-2 cleavage of FN occurs in synergy with inherent autolytic activities of FN that are blocked by inhibitors of serine protease inhibitors or inactivated by limited heat treatment at 56 °C. Heat treatment also increased the susceptibility of FN to MMP-2 cleavage without causing FN fragmentation or disrupting the biological activities of FN reflected by unaltered cell attachment properties. The present protein binding and enzymatic assays have now for the first time established that the carboxyl-terminal domain of MMP-2 (PEX) is the key exosite for interactions of MMP-2 with FN and for cleavage of FN by MMP-2
4. Experimental procedures
4.1 Purification of fibronectin
FN was purified from human plasma by gelatin-Sepharose affinity chromatography according to the established procedures (Engvall and Roushlahti, 1977). Briefly, particulate matter was sedimented by centrifugation of the plasma at 2,500 × g for 20 minutes, which was then diluted 3 fold in chromatography buffer (50 mM Tris, 0.15 M NaCl, pH 7.4) and loaded on a gelatin-Sepharose affinity column. After extensive washes, non-specifically–bound proteins were removed with buffer containing 1 M NaCl, and bound FN was eluted with 4 M urea in chromatography buffer. To separate FN from the gelatin binding MMP-2 and MMP-9, samples concentrated to ~3 mg/ml were further purified by gel filtration using a HiLoad 16/60 Superdex 200 prep grade column (Amersham-Pharmacia, Piscataway, NJ) with 50 mM Tris, 150 mM NaCl, pH 7.4 as chromatography buffer. Fractions were analyzed for purity by SDS-PAGE, and the identity of the final product was verified by Western blotting using anti-FN antibodies. Elimination of MMP-2 and -9 and other gelatinolytic activities were verified by gelatin enzymography (Overall and Limeback, 1988; Steffensen et al., 1995). Purified FN was quantified by the BCA assay (Pierce, Rockford, IL), frozen slowly, and stored at −80°C.
4.2 Detection of contaminating plasma proteases
4.2.1 Enzymography
The presence of gelatinolytic activities in FN fractions was monitored during the purification by enzymography using 10% SDS-PAGE minislab gels co-polymerized with 150 μg/ml type I gelatin (BioRad, Hercules, CA). At each step of the purification process, representative samples were separated under non-reducing conditions. Gels were incubated, processed, and analyzed for residual gelatinolytic activities as detailed previously (Overall and Limeback, 1988; Steffensen et al., 1995).
4.2.2 Gel Liquid Chromatography Mass Spectrometry
The presence of other contaminating plasma-derived proteases was analyzed by gel liquid chromatography mass spectrometry (GeLCMS)(Lasonder et al., 2002) at the UTHSCSA Institutional Mass Spectrometry core facility. Protein samples were collected following gelatin Sepharose affinity chromatography and after subsequent gel filtration (see above). After separation by SDS-PAGE, the protein containing regions of the gels were cut into small pieces (1 – 2 mm each) and digested with trypsin (modified; Promega, Madison, WI). Digests were analyzed by capillary high pressure liquid chromatography - electrospray ionization tandem mass spectrometry (HPLC-ESI-MS/MS) using an LTQ linear ion trap mass spectrometer (Thermo Fisher, Milford, MA). Survey scans were acquired followed by data dependent collision-induced dissociation (CID) spectra of the seven most intense ions. Uninterpreted CID spectra were searched against databases by means of Mascot (Matrix Science, Boston, MA) and results were cross correlated using X! Tandem. Protein probabilities were determined by Scaffold (Proteome software, Portland, OR) and relative abundances of identified proteins were assessed through spectrum counting (Zybailov et al., 2005).
4.3 Expression and purification of recombinant proteins
Recombinant MMP-2 and MMP-2E404A proteins were expressed and purified as described previously (Steffensen et al., 1995; Xu et al., 2004; Xu et al., 2005a). The gene sequence encoding PEX from MMP-2 was amplified by PCR from our previously reported MMP-2 construct (Xu et al., 2004) with primers adding NheI and Hind III sites for directional primer, 5′-ctagctagcGGGGCCTCTCCTGACATTG-3′; reverse primer, 5′-gggaagcttTCAGCAGCCTAGCCAGTCG-3′, lower case letters indicate restriction sites). To construct MMP-2 with deleted PEX domain (MMP-2ΔPEX), the coding gene sequence was amplified by PCR from our previously MMP-2 construct (Xu et al., 2004) (forward primer, 5′-ccgctcgaGTACAACTTCTTCCCTCGCAAG-3′; reverse primer, 5′-cggaattcTCACCCATAGAGCTCCTGAATGC-3′). Primers added XhoI and EcoRI restriction sites and a stop codon) allowing for ligation into the pRSETA expression vector (Invitrogen, San Diego, CA, U.S.A.), which expresses proteins with a His × 6 tag. The construct the expression vector for MMP-2ΔPEXE404A, which has intact ligand-binding properties but is catalytically inactive (Morgunova et al., 1999), the active site Glu of MMP-2ΔPEX was mutated to an alanine by overlap-extension PCR as previously described for generating MMP-2E404A (Xu et al., 2005a). All new expression constructs and mutations were verified by double-stranded DNA sequencing.
The recombinant proteins were expressed in E. coli Le392 or E. coli BL21(DE3) as inclusion bodies that required denaturation with 8 M urea, 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole pH 8.0. The first round of purification of denatured recombinant proteins was by nickel affinity chromatography (Amersham Pharmacia Biotech, Piscataway, NJ) under denaturing conditions. After extensive washes with 8 M urea, 50 mM Na2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0, to remove non-specifically bound bacterial proteins, the recombinant protein was eluted with 150 mM imidazole in the same chromatography buffer. Proteins were refolded by dialysis against 0.5 mM reduced L-glutathione/0.5 mM oxidized L-glutathione in 50 mM NaH2PO4, pH 8.0, and then exchanged into 50 mM NaH2PO4, 300 mM NaCl, pH 8.0, or PBS pH 7.4. The refolded recombinant proteins were further purified by a second round of nickel affinity chromatography under native conditions in chromatography buffer without urea, and with elution at 60 – 80 mM imidazole. After final dialysis with 50 mM Tris-HCl, pH 7.4, all proteins were flash frozen in liquid nitrogen and stored at −80 °C until activity assays.
4.4 Heat treatment of fibronectin
To test the effects of heat treatment on the stability and susceptibility to cleavage by MMP-2, aliquots of purified FN at a concentration of 600 μg/ml in 50 mM Tris, 150 mM NaCl, pH 7.4, were incubated at 37, 42, 47, 56, 60, or 65 °C for 30 min. After heating, FN samples were kept on ice until further analyses.
4.5 Cell attachment to heat – treated fibronectin
We used human fibrosarcoma cell line HT1080 to test cell attachment on heat-treated FN. HT1080 cells were maintained in DMEM (Sigma St. Louis, MO) supplemented with 10% newborn calf serum (NCS), 100 units/ml penicillin, and 100 μg/ml streptomycin. The experiments were carried out as previously described (Stanley et al., 2008) {Stanley, 2008 5763/id}. Briefly, 96-microwell plates were coated with a concentration range of heat-treated FN (25 to 0.025 μg/ml and 0 μg/ml) in 50 μl PBS for 18 hours at 4 °C. A total of 4 × 104 cells were added in serum-free DMEM media and incubated for 90 min at 37 °C. Attached cells were fixed with 4% formaldehyde in PBS and stained with 0.1% crystal violet in 200 mM boric acid, pH 6.0, after which cell-bound stain was dissolved with 10% acetic acid and quantified at 595 nm in an Opsys MR microplate reader (Dynex Technologies, Chantilly, VA). Cell attachment to non-coated wells served to adjust for non-specific attachment. Experiments were performed in duplicate.
4.6 Interactions of the PEX domain of MMP-2 with fibronectin
Interactions of FN with MMP-2 were tested by plate binding assays. Recombinant MMP-2E404A, PEX, and MMP-2 PEXE404A were biotinylated as described previously (Xu et al., 2005a). FN was coated in 96-well plates at 0.5 μg/well in 0.1 M NaHCO3/Na2CO3, pH 9.6, at 4 °C overnight, followed by incubation with 10 mg/ml BSA for 1 h at 22 °C to block non-specific binding sites. Then, serially diluted biotinylated MMP-2E404A, PEX, or MMP-2ΔPEXE404A at a concentration range from 3 to 0.004 μM in 50mM Tris–HCl, pH 7.4, was added to the FN coated wells and incubated for 1 h at 22 °C. After washes with 50 mM Tris-HCl, 0.05% (v/v) Tween 20, pH 7.4 to remove unbound protein, bound biotinylated proteins were detected by alkaline phosphatase (AP)-conjugated streptavidin (Pierce, Rockport, IL) diluted 1:10,000 and 1 mg/ml PNPP substrate (Sigma, St. Louis, MO) at 405nm in an Opsys MR plate reader (Dynex, Chantilly, VA). Apparent Kds were calculated from binding curves using a four-parameter, non-linear curve-fitting algorithm (Sigma Plot, SPSS Corp., Chicago, IL, U.S.A.).
To determine whether recombinant PEX could block the binding of MMP-2 to FN, binding of 10 nM biotinylated MMP-2E404A to coated FN (0.5 μg/well) was competed with a concentration range (4 - 0.008 μM) of unlabeled PEX in competitive protein binding assays. Bound biotinylated MMP-2E404A was detected with AP-conjugated streptavidin and PNPP as detailed above, whereas non-labeled competing proteins remained undetected. If effective, the competing PEX would reduce the binding of the biotinylated protein. The reduction in binding of biotinylated MMP-2E404A in the presence of competing proteins was expressed in per cent of the binding of biotinylated MMP-2E404A binding in the absence of competing protein. Competition with non-labeled MMP-2E404A served as positive control and ovalbumin served as negative control. All experiments were performed in duplicate and repeated at least twice.
4.7 Measurements of FN cleavage
4.7.1 FN cleavage by recombinant MMP-2 and MMP-2ΔPEX by SDS-PAGE
Heat-treated or control samples of purified FN diluted to 100 μg/ml in collagenase assay buffer (50 mM Tris-HCl, 200 mM NaCl, 5 mM CaCl2, 1 μM ZnCl2, 0.05% Brij35, pH 7.0) were incubated with recombinant MMP-2 or MMP-2ΔPEX at 0.75 nM for 0 – 48 h at 22 °C without protease inhibitors or in the presence of 0.5 mM 4-(2-aminoethyl benzenesulfonyl fluoride (AEBSF) serine protease inhibitor or 1.5 mM 1,10-phenanthroline MMP inhibitor. Aliquots were collected at designated time points (see results), immediately mixed with 4 × SDS-PAGE sample loading buffer (2 M urea, 2% SDS, 10 mg/ml DTT in 0.125 M Tris- HCl, pH 6.8) to stop the cleavage, and stored at -20 °C until use. The cleavage of FN by MMP-2 and MMP-2ΔPEX were analyzed by 10% SDS-PAGE. Coomassie Brilliant Blue R-250 stained gels were digitized and the intensity of the protein band corresponding to full-length intact FN was quantified by the Kodak 1D Image Analysis Software (Rochester, NY).
4.7.2 Contribution of MMP-2 PEX to FN cleavage by fluorescence assays
In competitive enzyme activity assays, rMMP-2 enzymatic activities on FITC-labeled FN substrate were analyzed in the presence of a concentration range of PEX. FN substrate was heavily labeled with FITC at 22 °C for 1 h following which remaining reactive sites were blocked with glycine at 4 °C for 2 h. Unbound FITC was removed by gel filtration with Sephadex G-10 (Sigma, St. Louis, MO) monitoring the protein concentration at 280 nm and FITC with λex at 495 nm and λem at 515nm. FITC-labeled FN (4 μg/well) and recombinant MMP-2 (0.15 μM) were added to enzyme assay buffer (50 mM Tris–HCl, 150 mM NaCl, 5 mM CaCl2, pH 7.6) either alone or simultaneously with a concentration range 1.5 – 0.05 μM and 0 μM of PEX. Substrate cleavage represented by fluorescent signal was monitored at 22 °C with λex at 495 nm and λem at 515 nm with a SpectraMAX Gemini XS plate reader (Molecular Devices, Sunnyvale, CA). The enzyme activity was expressed in RFU and presented as means of duplicate measurements from at least two separate experiments.
Acknowledgments
We greatly appreciate the constructive recommendations to the experiments by Dr. Robert J. Klebe, Department of Cellular and Structural Biology, University of Texas Health Science Center at San, Antonio, San Antonio, Texas, and Dr Susan Weintraub at the UTHSCSA Mass Spectrometry Laboratory for guidance in sample analyses and data interpretation. Supported by grants DE017139, DE016312, DE 014236, and DE018135 from the National Institutes of Health, Bethesda, MD.
Abbreviations are
- FN
fibronectin
- MMP-2
matrix metalloproteinase-2
- PEX
hemopexin-like domain of MMP-2
- MMP-2ΔPEX
MMP-2 with deleted PEX
- MMP-2E404A
catalytically inactive ligand binding MMP-2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al Hazmi N, Thomas GJ, Speight PM, Whawell SA. The 120 kDa cell-binding fragment of fibronectin upregulates migration of alphavbeta6-expressing cells by increasing matrix metalloproteinase-2 and -9 secretion. Eur J Oral Sci. 2007;115:454–458. doi: 10.1111/j.1600-0722.2007.00481.x. [DOI] [PubMed] [Google Scholar]
- Banyai L, Tordai H, Patthy L. The gelatin-binding site of human 72 kDa type IV collagenase (gelatinase A) Biochem J. 1994;298:403–407. doi: 10.1042/bj2980403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banyai L, Trexler M, Koncz S, Gyenes M, Sipos G, Patthy L. The collagen-binding site of type-II units of bovine seminal fluid protein PDC-109 and fibronectin. Eur J Biochem. 1990;193:801–806. doi: 10.1111/j.1432-1033.1990.tb19403.x. [DOI] [PubMed] [Google Scholar]
- Bonnefoy A, Legrand C. Proteolysis of subendothelial adhesive glycoproteins (fibronectin, thrombospondin, and von Willebrand factor) by plasmin, leukocyte cathepsin G, and elastase. Thromb Res. 2000;98:323–332. doi: 10.1016/s0049-3848(99)00242-x. [DOI] [PubMed] [Google Scholar]
- Briknarova K, Akerman ME, Hoyt DW, Ruoslahti E, Ely KR. Anastellin, an FN3 fragment with fibronectin polymerization activity, resembles amyloid fibril precursors. J Mol Biol. 2003;332:205–215. doi: 10.1016/s0022-2836(03)00890-8. [DOI] [PubMed] [Google Scholar]
- Busby TF, Argraves WS, Brew SA, Pechik I, Gilliland GL, Ingham KC. Heparin binding by fibronectin module III-13 involves six discontinuous basic residues brought together to form a cationic cradle. J Biol Chem. 1995;270:18558–18562. doi: 10.1074/jbc.270.31.18558. [DOI] [PubMed] [Google Scholar]
- Carsons S, Lavietes BB, Diamond HS, Kinney SG. The immunoreactivity, ligand, and cell binding characteristics of rheumatoid synovial fluid fibronectin. Arthritis Rheum. 1985;28:601–612. doi: 10.1002/art.1780280602. [DOI] [PubMed] [Google Scholar]
- Castell JV, Guillen MI, Marco V, Menendez R, Nauffal D, Gomez-Lechon MJ. Presence of fibronectin fragments in bronchoalveolar lavages: An immunoblotting study. Biochem Soc Trans. 1988;16:378–379. [Google Scholar]
- Collier IE, Wilhelm SM, Eisen AZ, Marmer BL, Grant GA, Seltzer JL, Kronberger A, He C, Bauer EA, Goldberg GI. H-ras oncogene-transformed human bronchial epithelial cells (TBE-1) secrete a single metalloprotease capable of degrading basement membrane collagen. J Biol Chem. 1988;263:6579–6587. [PubMed] [Google Scholar]
- Danielsen CC. Difference in thermal stability of type-I and type-II collagen from rat skin. Biochem J. 1982;203:323–326. doi: 10.1042/bj2030323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean RA, Overall CM. Proteomics discovery of metalloproteinase substrates in the cellular context by iTRAQ labeling reveals a diverse MMP-2 substrate degradome. Mol Cell Proteomics. 2007;6:611–623. doi: 10.1074/mcp.M600341-MCP200. [DOI] [PubMed] [Google Scholar]
- Emod I, Lafaye P, Planchenault T, Lambert Vidmar S, Imhoff JM, Keil-Dlouha V. Potential proteolytic activity of fibronectin: Fibronectin laminase and its substrate specificity. Biol Chem. 1990;371:129–135. doi: 10.1515/bchm3.1990.371.1.129. [DOI] [PubMed] [Google Scholar]
- Engvall E, Roushlahti E. Binding of soluble form of fibroblast surface protein, fibronectin, to collagen. Int J Cancer. 1977;20:1–5. doi: 10.1002/ijc.2910200102. [DOI] [PubMed] [Google Scholar]
- Graille M, Pagano M, Rose T, Ravaux MR, van Tilbeurgh H. Zinc induces structural reorganization of gelatin binding domain from human fibronectin and affects collagen binding. Structure. 2010;18:710–718. doi: 10.1016/j.str.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Houard X, Germain S, Gervais M, Michaud A, van den BF, Foidart JM, Noel A, Monnot C, Corvol P. Migration-stimulating factor displays HEXXH-dependent catalytic activity important for promoting tumor cell migration. Int J Cancer. 2005;116:378–384. doi: 10.1002/ijc.21053. [DOI] [PubMed] [Google Scholar]
- Huynh QH, Wang SH, Tafolla E, Gansky SA, Kapila S, Armitage GC, Kapila YL. Specific fibronectin fragments as markers of periodontal disease status. J Periodontol. 2002;73:1101–1110. doi: 10.1902/jop.2002.73.10.1101. [DOI] [PubMed] [Google Scholar]
- Hynes R. Molecular biology of fibronectin. Ann Rev Cell Biol. 1985;1:67–90. doi: 10.1146/annurev.cb.01.110185.000435. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Fibronectins. Springer-Verlag; New York: 1990. Structures of fibronectins. [Google Scholar]; Springer Series in Molecular Biology. :113–175. [Google Scholar]
- Ingham KC, Brew SA, Broekelmann TJ, McDonald JA. Thermal stability of human plasma fibronectin and its constituent domains. J Biol Chem. 1984;259:11901–11907. [PubMed] [Google Scholar]
- Johansson S, Smedsrod B. Identification of a plasma gelatinase in preparations of fibronectin. J Biol Chem. 1986;261:4363–4366. [PubMed] [Google Scholar]
- Kapila YL, Wang S, Johnson PW. Mutations in the heparin binding domain of fibronectin in cooperation with the V region induce decreases in pp125FAK levels plus proteoglycan-mediated apoptosis via caspases. J Biol Chem. 1999;274:30906–30913. doi: 10.1074/jbc.274.43.30906. [DOI] [PubMed] [Google Scholar]
- Katagiri Y, Brew SA, Ingham KC. All six modules of the gelatin-binding domain of fibronectin are required for full affinity. Journal of Biological Chemistry. 2003;278:11897–11902. doi: 10.1074/jbc.M212512200. [DOI] [PubMed] [Google Scholar]
- Klebe RJ. Isolation of a collagen-dependent cell attachment factor. Nature. 1974;250:248–251. doi: 10.1038/250248a0. [DOI] [PubMed] [Google Scholar]
- Kornblihtt AR, Vibe-Pedersen K, Baralle FE. Isolation and characterization of cDNA clones for human and bovine fibronectins. Proc Natl Acad Sci, USA. 1983;80:3218–3222. doi: 10.1073/pnas.80.11.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft PJ, Haynes-Johnson DE, Patel L, Lenhart JA, Zivin RA, Palmer SS. Fluorescence polarization assay and SDS-PAGE confirms matrilysin degrades fibronectin and collagen IV whereas gelatinase A degrades collagen IV but not fibronectin. Connect Tissue Res. 2001;42:149–163. doi: 10.3109/03008200109014256. [DOI] [PubMed] [Google Scholar]
- Lambert Vidmar S, Lottspeich F, Emod I, Imhoff JM, Keil-Dlouha V. Collagen-binding domain of human plasma fibronectin contains a latent type-IV collagenase. Eur J Biochem. 1991a;201:79–84. doi: 10.1111/j.1432-1033.1991.tb16258.x. [DOI] [PubMed] [Google Scholar]
- Lambert Vidmar S, Lottspeich F, Emod I, Planchenault T, Keil-Dlouha V. Latent fibronectin-degrading serine proteinase activity in N-terminal heparin-binding domain of human plasma fibronectin. Eur J Biochem. 1991b;201:71–77. doi: 10.1111/j.1432-1033.1991.tb16257.x. [DOI] [PubMed] [Google Scholar]
- Lasonder E, Ishihama Y, Andersen JS, Vermunt AM, Pain A, Sauerwein RW, Eling WM, Hall N, Waters AP, Stunnenberg HG, Mann M. Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature. 2002;419:537–542. doi: 10.1038/nature01111. [DOI] [PubMed] [Google Scholar]
- Lee SR, Lo EH. Induction of caspase-mediated cell death by matrix metalloproteinases in cerebral endothelial cells after hypoxia-reoxygenation. J Cereb Blood Flow Metab. 2004;24:720–727. doi: 10.1097/01.WCB.0000122747.72175.47. [DOI] [PubMed] [Google Scholar]
- Miekka SI. Heat-induced fragmentation of human plasma fibronectin. Biochim Biophys Acta. 1983;748:374–380. doi: 10.1016/0167-4838(83)90182-6. [DOI] [PubMed] [Google Scholar]
- Morgunova E, Tuuttila A, Bergmann U, Isupov M, Lindquist Y, Schneider G, Tryggvason K. Structure of human pro-matrix metalloproteinase-2: Activation mechanism revealed. Science. 1999;284:1667–1670. doi: 10.1126/science.284.5420.1667. [DOI] [PubMed] [Google Scholar]
- Murphy G, Allan JA, Willenbrock F, Cockett MI, O’Commell JP, Docherty AJP. The role of the C-terminal domain in collagenase and stromelysin specificity. J Biol Chem. 1992;267:9612–9618. [PubMed] [Google Scholar]
- Murphy G, Nguyen Q, Cockett MI, Atkinson SJ, Allan JA, Knight CG, Willenbrock F, Docherty AJP. Assessment of the role of the fibronectin-like domain of gelatinase A by analysis of a deletion mutant. J Biol Chem. 1994;269:6632–6636. [PubMed] [Google Scholar]
- Okada Y, Morodomi T, Enghild JJ, Suzuki K, Yasui A, Nakanishi I, Salvesen G, Nagase H. Matrix metalloproteinase 2 from human rheumatoid synovial fibroblasts. Purification and activation of the precursor and enzymic properties. Eur J Biochem. 1990;194:721–730. doi: 10.1111/j.1432-1033.1990.tb19462.x. [DOI] [PubMed] [Google Scholar]
- Overall CM. Matrix metalloprotinase substrate binding domains, modules, and exosites. Overview and experimental strategies. In: Clark IM, editor. Matrix metalloproteinase protocols. Humana Press; Totowa, N.J: 2001. [PubMed] [Google Scholar]; Methods in molecular biology. 1:79–120. [PubMed] [Google Scholar]
- Overall CM, Limeback H. Identification and characterization of enamel proteinases isolated from developing enamel. Biochem J. 1988;256:965–972. doi: 10.1042/bj2560965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2:657–672. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- Overall CM, Wallon UM, Steffensen B, De Clerk Y, Tschesche H, Abbey RS. Substrate and TIMP interactions with human gelatinase A recombinant COOH-terminal hemopexin-like and fibronectin type II-like domains: Both the N- and C-domains of TIMP-2 bind the C-domain of gelatinase A. In: Edwards D, et al., editors. Inhibitors of Metalloproteinases in Development and Disease. Gordon & Breach; Amsterdam, Holland: 2000. pp. 57–69. [Google Scholar]
- Pal S, Chen Z, Xu X, Mikhailova M, Steffensen B. Co-purified gelatinases alter the stability and biological activities of human plasma fibronectin preparations. J Periodontal Res. 2009;45:292–295. doi: 10.1111/j.1600-0765.2009.01241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115:3861–3863. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- Planchenault T, Lambert Vidmar S, Imhoff JM, Blondeau X, Emod I, Lottspeich F, Keil-Dlouha V. Potential proteolytic activity of human plasma fibronectin: Fibronectin gelatinase. Biol Chem Hoppe Seyler. 1990;371:117–128. doi: 10.1515/bchm3.1990.371.1.117. [DOI] [PubMed] [Google Scholar]
- Schnepel J, Tschesche H. The proteolytic activity of the recombinant cryptic human fibronectin type IV collagenase from E. coli expression. J Protein Chem. 2000;19:685–692. doi: 10.1023/a:1007104420017. [DOI] [PubMed] [Google Scholar]
- Shipley JM, Doyle GAR, Fliszar CJ, Ye QZ, Johnson LL, Shapiro SD, Welgus HG, Senior RM. The structural basis for the elastolytic activity of the 92-kDa and 72-kDa gelatinases. Role of the fibronectin type II-like repeats. J Biol Chem. 1996;271:4335–4341. doi: 10.1074/jbc.271.8.4335. [DOI] [PubMed] [Google Scholar]
- Skorstengaard K, Jensen MS, Sahl P, Petersen TE, Magnusson S. Complete primary structure of bovine plasma fibronectin. Eur J Biochem. 1986;161:441–453. doi: 10.1111/j.1432-1033.1986.tb10464.x. [DOI] [PubMed] [Google Scholar]
- Stanley C, Wang Y, Pal S, Klebe RJ, Harkless LB, Xu X, Steffensen B. Fibronectin-fragmentation is a feature of both periodontal disease sites and diabetic foot and leg wounds and modifies cell behavior. J Periodontol. 2008;79:861–875. doi: 10.1902/jop.2008.070492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen B, Wallon UM, Overall CM. Extracellular matrix binding properties of recombinant fibronectin type II-like modules of human 72-kDa gelatinase/type IV collagenase. High affinity binding to native type I collagen but not native type IV collagen. J Biol Chem. 1995;270:11555–11566. doi: 10.1074/jbc.270.19.11555. [DOI] [PubMed] [Google Scholar]
- Steffensen B, Xu X, Martin P, Zardeneta G. Human fibronectin and MMP-2 collagen binding domains compete for collagen binding sites and modify cellular activation of MMP-2. Matrix Biol. 2002;21:399–414. doi: 10.1016/s0945-053x(02)00032-x. [DOI] [PubMed] [Google Scholar]
- Strongin AY, Collier IE, Bannikov G, Marmer BL, Grant GA, Goldberg GI. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem. 1995;270:5331–5338. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- Strongin AY, Marmer BL, Grant GA, Goldberg GI. Plasma membrane-dependent activation of the 72-kDa type IV collagenase is prevented by complex formation with TIMP-2. J Biol Chem. 1993;268:14033–14039. [PubMed] [Google Scholar]
- Talonpoika J, Heino J, Larjava H, Hakkinen L, Paunio K. Gingival crevicular fluid fibronectin degradation in periodontal health and disease. Scand J Dent Res. 1989;97:415–421. doi: 10.1111/j.1600-0722.1989.tb01455.x. [DOI] [PubMed] [Google Scholar]
- Tam EM, Moore TR, Butler GS, Overall CM. Characterization of the distinct collagen binding, helicase and cleavage mechanisms of matrix metalloproteinase 2 and 14 (gelatinase A and MT1-MMP): the differential roles of the MMP hemopexin c domains and the MMP-2 fibronectin type II modules in collagen triple helicase activities. J Biol Chem. 2004;279:43336–43344. doi: 10.1074/jbc.M407186200. [DOI] [PubMed] [Google Scholar]
- Unger J, Tschesche H. The proteolytic activity and cleavage specificity of fibronectin-gelatinase and fibronectin-lamininase. J Protein Chem. 1999;18:403–411. doi: 10.1023/a:1020684508212. [DOI] [PubMed] [Google Scholar]
- Wallon UM, Overall CM. The COOH-terminal hemopexin-like domain of human gelatinase A (MMP-2) requires Ca2+ for fibronectin and heparin binding: Binding properties of recombinant gelatinase A C-domain to extracellular matrix and basement membrane components. J Biol Chem. 1997;272:7473–7481. doi: 10.1074/jbc.272.11.7473. [DOI] [PubMed] [Google Scholar]
- White ES, Baralle FE, Muro AF. New insights into form and function of fibronectin splice variants. J Pathol. 2008;216:1–14. doi: 10.1002/path.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki AB, Grinnell F. Fibronectin profiles in normal and chronic wound fluid. Lab Invest. 1990;63:825–831. [PubMed] [Google Scholar]
- Xie DL, Meyers R, Homandberg GA. Fibronectin fragments in osteoarthritic synovial fluid. J Rheumatol. 1992;19:1448–1452. [PubMed] [Google Scholar]
- Xu X, Chen Z, Wang Y, Bonewald L, Steffensen B. Inhibition of MMP-2 gelatinolysis by targeting exodomain-substrate interactions. Biochem J. 2007;406:147–155. doi: 10.1042/BJ20070591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Chen Z, Wang Y, Yamada Y, Steffensen B. Functional basis for the overlap in ligand interactions and substrate specificities of matrix metalloproteinases-9 and -2 (MMP-9 and -2) Biochem J. 2005a;392:127–134. doi: 10.1042/BJ20050650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Wang Y, Chen Z, Sternlicht MD, Hidalgo M, Steffensen B. MMP-2 contributes to cancer cell migration on collagen. Cancer Res. 2005b;65:130–136. [PubMed] [Google Scholar]
- Xu X, Wang Y, Lauer-Fields JL, Fields GB, Steffensen B. Contributions of the MMP-2 collagen binding domain to gelatin cleavage. Substrate binding via the collagen binding domain is required for MMP-2 degradation of gelatin but not short peptides. Matrix Biol. 2004;23:171–181. doi: 10.1016/j.matbio.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Yamada KM. Fibronectins: structure, functions and receptors. Curr Opin Cell Biol. 1989;1:956–963. doi: 10.1016/0955-0674(89)90065-3. [DOI] [PubMed] [Google Scholar]
- Zybailov B, Coleman MK, Florens L, Washburn MP. Correlation of relative abundance ratios derived from peptide ion chromatograms and spectrum counting for quantitative proteomic analysis using stable isotope labeling. Anal Chem. 2005;77:6218–6224. doi: 10.1021/ac050846r. [DOI] [PubMed] [Google Scholar]