

Abstract
Cigarette smoke (CS) is the major cause of lung cancer and contributes to the development of other malignancies. Attempts have been made to construct reduced toxicity cigarettes, presumed to have diminished genotoxic potential. One such product on the market is the tobacco and nicotine free (T&N-free) cigarette type made from lettuce and herbal extracts. We have recently developed a sensitive assay of the genotoxicity of CS based on cytometric analysis of induction of the DNA damage response (DDR) in normal human pulmonary endothelial or A549 pulmonary adenocarcinoma cells. In the present study, we observed that exposure of A549 cells to CS from T&N-free cigarettes induced a smoke-dose dependent DDR as evidenced by phosphorylation (activation) of the Ataxia telangiectasia mutated (ATM) protein kinase and of the histone H2AX (γH2AX). the extent of DDR induced by T&N-free smoke was distinctly greater than that induced by comparable doses of CS from reference cigarettes (2R4F) containing tobacco and nicotine. The pattern of DDR induced by T&N-free smoke was similar to that of 2R4F cigarettes in terms of the cell cycle phase specificity and involvement of reactive oxygen species (ROS). The data also imply that similar to 2R4F exposure of cells to T&N-free smoke leads to formation of double-strand DNA breaks (DSBs) resulting from collapse of replication forks upon collision with the primary ssDNA lesions induced by smoke. Since DSBs are potentially carcinogenic our data indicate that smoking tobacco and nicotine-free cigarettes is at least as hazardous as smoking cigarettes containing tobacco and nicotine.
Keywords: ATM activation, histone H2AX phosphorylation, gammaH2AX, DNA double-strand breaks, DNA replication, laser scanning cytometry, cell cycle
Introduction
Cigarette smoke (CS) is the primary cause of pulmonary cancer and it also contributes to the development of malignancies in other organs (reviewed in refs. 1-4). Genotoxic agents of diverse chemical structures present in CS appear to be the cause of carcinogenesis.3 Extensive studies have been carried out to: (i) identify the carcinogenic agents in CS; (ii) characterize the types of DNA lesions caused by these agents, and (iii) elucidate the molecular mechanisms by which these lesions lead to cancer development. Although progress has been made along these lines, little has been achieved in terms of development of a less harmful cigarette or prevention of CS-induced cancers. Epidemiological studies indicate that there is essentially no difference in lung cancer risk among long-term smokers of different cigarette types (i.e., “ultralight”, “light” or “full-flavor”), and exposure analyses show no significant quantitative differences in several markers of carcinogen and nicotine uptake in these smokers.5-7 Thus, it is clear that the genotoxicity of this inhaled mixture is relatively constant across different brands of cigarettes. The assessment of potential carcinogenicity of particular brands of cigarettes is complicated due to variability in smoking habits; to compensate for differences in nicotine and tar composition smokers often modify their patterns of smoking intensity such as depth, duration and frequency of inhalation (“puffs”) and air intake through the cigarette filters.5,8
We have recently developed a sensitive and rapid assay of genotoxicity applicable to CS. Specifically, using multiparameter laser scanning cytometry (LSC) we were able to detect the induction of the DNA damage response (DDR) in normal human bronchial epithelial cells and in pulmonary carcinoma A549 cells shortly following their exposure to CS.9-13 The function of the DDR is to halt cell cycle progression thereby preventing transfer of damaged DNA to progeny cells, to engage the DNA damage repair machinery and/or to activate the apoptotic pathway in order to eliminate cells with excessive DNA damage (reviewed in refs. 14-21). The biomarkers of DDR in our assay were activation of ataxia telangiectasia mutated (ATM) protein kinase (ATM-Ser1981P), phosphorylation of histone H2AX on Ser139 (γH2AX), Chk2 protein kinase on Thr68 (Chk2-Thr68P) and tumor suppressor p53 on Ser15 (p53-Ser15P) detected immunocytochemically with the respective phospho-specific antibodies.12,13 The induction of DDR signals the formation of DNA lesions, some of which are potentially carcinogenic DNA double-strand breaks (DSBs), following exposure to CS.9-13 Our data also indicated the involvement of reactive oxygen species (ROS) as a factor that, at least in part, mediated the induction of DDR by CS.10
The present study was designed to test whether tobacco and nicotine free (T&N-free) cigarettes constructed with lettuce in place of tobacco can induce DDR. Towards this end we exposed A549 cells to smoke from T&N-free cigarettes, detected activation of ATM and Ser139 phosphorylation of H2AX immunocytochemically using phospho-specific antibodies, and measured cell immunofluorescence (IF) concurrently with DNA content by laser scanning cytometry (LSC). For comparison, under identical conditions we measured the induction of DDR by smoke from 2R4F reference standard cigarettes containing tobacco and nicotine with a composition and construction typical of “light” type commercial cigarette brands. The data clearly demonstrate that the CS of T&N-free cigarettes induced DDR as evidenced by ATM and H2AX phosphorylation. Moreover, the induction of DDR by T&N-free cigarettes was substantially greater than that generated by similar amounts of CS from 2R4F reference cigarettes.
Results
Consistent with our previous findings, exposure of A549 cells to the smoke from conventional, tobacco-containing 2R4F cigarettes induced γH2AX and ATM-S1981P (Fig. 1). While this effect was not apparent after exposure for 10 min, it was very robust after 20 min. Both the induction of γH2AX as well as ATM-S1981P was much more pronounced in S-than G1- or G2M-phase cells. No significant differences in the cell cycle distribution between the mock- and CS-treated cells were evident after 1 h of incubation, as the DNA frequency histograms of the cells from the respective cultures were essentially identical.
Figure 1.
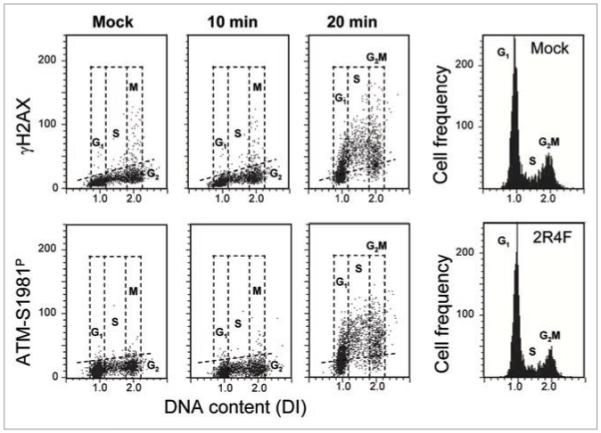
Induction of γH2AX and ATM-S1981P in A549 cells upon their exposure to 2R4F CS. Cells were either mock-treated or exposed for 10 or 20 min to smoke from 2R4F cigarettes and then incubated in culture for 1 h. The bivariate DNA content (DNA index; DI) vs. γH2AX (top) or DNA content vs. ATM-S1981P (bottom) distributions show the expression of γH2AX and ATM-S1981P with respect to the cell cycle phase; cells in G1, S and G2M phases of the cell cycle were identified based on differences on DNA content as shown (see also Materials and Methods). The dashed skewed lines indicate the upper threshold for γH2AX or ATM-S1981P IF for 95% mock-treated cells. The univariate DNA content histograms (right) show no differences in the cell cycle distribution between cells exposed to smoke (20 min) vs. control (mock treated), after a 1 h incubation.
Figure 2 illustrates the effects of exposure of A549 cells to smoke from T&N-free cigarettes. The data show that exposure for 10 or 20 min to whole smoke from these cigarettes led to distinct induction of both γH2AX and ATM-S1981P. The induction was apparent in all phases of the cell cycle. Exposure of cells to T&N-free smoke from cigarettes with four holes (“vents”) made with a 21-gauge needle just above the filter (as suggested by the manufacturer to dilute smoke with ambient air) also led to prominent induction of γH2AX and ATM-S1981P. In this case, induction was more pronounced in S than in G1 or G2M phase cells. Likewise, the vapor phase of T&N-free cigarette smoke induced phosphorylation of both these proteins but appeared to affect all cell cycle phases equally.
Figure 2.
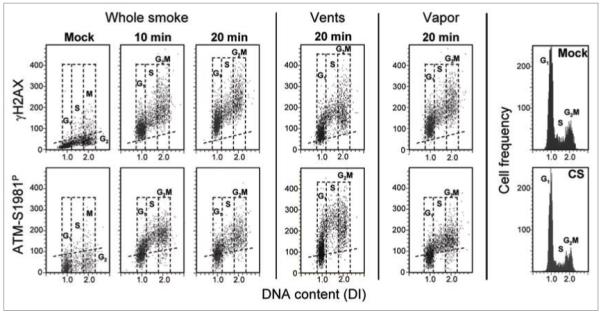
Induction of γH2AX and ATM-S1981P in A549 cells exposed to smoke from the T&N-free cigarettes. The left side three panels (top and bottom) show the effect on A549 cells of exposure to whole smoke from the T&N-free cigarettes, the mid-panels show the effect of smoke from T&N-free cigarettes with vents whose function is to dilute the smoke with air, and the right panels illustrate the effect of vapor phase produced as described in Materials and Methods. The DNA content frequency histograms of the mock- and T&N-free CS (whole smoke, 20 min) treated cells are shown on the right.
The induction of γH2AX by smoke from 2R4F and T&N-free cigarettes in relation to the cell cycle phase is presented in Table 1. After exposure to whole smoke from T&N-free cigarettes, the mean of γH2AX expression of the G1 phase cells was increased 7.6-fold above that of the mock-treated cells compared to 4.51-fold increase in the case of cells exposed to 2R4F smoke. Additionally, the mean of γH2AX expression of G2M cells exposed to whole T&N-free smoke (4.56-fold) was markedly higher than that of G2M cells exposed to 2R4F smoke (2.63-fold). While response of cells to smoke from the T&N-free cigarettes with vents or to the vapor phase of these cigarettes was diminished compared to whole T&N-free smoke, it was within a similar range to that caused by whole 2R4F smoke. Induction of ATM-S1981P was also more pronounced in cells exposed to smoke from T&N-free than from 2R4F cigarettes smoke. In this case, even air-diluted smoke from vented cigarettes or the vapor fraction of T&N-free cigarettes were more effective in inducing H2AX and ATM phosphorylation compared with whole smoke from 2R4F cigarettes. Interestingly, the smoke from vented T&N-free cigarettes diluted with air induced higher levels of ATM-S1981P in S-phase compared to G1 or G2M phase cells.
Table 1.
Induction of γH2AX and ATM-S1981P by smoke from 2R4F and T&N-free cigarettes in relation to the cell cycle phase
| Smoke | G1 | S | G2M |
|---|---|---|---|
| γ H2AX | |||
| 2R4F whole smoke | 4.51 ± 0.65 | 4.14 ± 0.54 | 2.63 ± 0.27 |
| T&N-free whole smoke | 7.60 ± 0.44 | 4.35 ± 0.15 | 4.56 ± 0.19 |
| T&N-free vents | 4.54 ± 0.65 | 3.90 ± 0.02 | 3.18 ± 0.11 |
| T&N-free vapor | 6.04 ± 0.02 | 4.18 ± 0.14 | 4.14 ± 0.60 |
| ATM-S1981P | |||
| 2R4F whole smoke | 2.92 | 3.09 | 2.84 |
| T&N-free whole smoke | 5.55 | 4.83 | 4.32 |
| T&N-free vents | 3.99 | 4.23 | 3.81 |
| T&N-free vapor | 5.14 | 4.71 | 4.09 |
A549 cells were exposed for 20 min either to whole 2R4F smoke or to whole smoke, smoke from cigarettes having air dilution holes (vents) or to the vapor phase (vapor) of T&N-free cigarettes and then incubated for 1 h, as described in Materials and Methods. The mean values of γH2AX (±SE, n = 4) and ATM-S1981P IF intensity (integral) for cells gated in G1, S and G2M phases of the cell cycle are presented as n-fold increase over the mean values of the mock-treated cells in the respective phases of the cell cycle.
The kinetics of induction of γH2AX and ATM-S1981P in A549 cells by exposure to smoke from 2R4F and T&N-free cigarettes is shown in Figure 3. It is evident that while exposure to 2R4F smoke for 5 or 10 min had no detectable effect on either phosphorylation of H2AX or ATM, both these proteins were phosphorylated following 10 min exposure to smoke from T&N-free cigarettes. The level of phosphorylation of both these proteins in cells exposed to T&N-free smoke for 10, 15 or 20 min was markedly higher compared to cells exposed to 2R4F smoke at identical time points.
Figure 3.
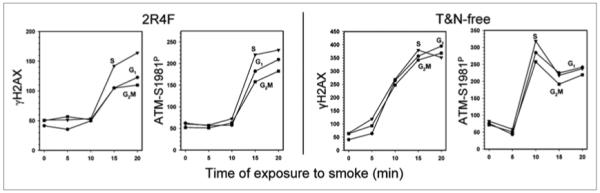
Extent of induction of γH2AX and ATM-S1981P by whole smoke from 2R4F or T&N-free cigarettes in relation to duration of cell exposure to smoke. A549 cells were exposed to smoke for 0–20 min. The plot shows the mean values for γH2AX and ATM-S1981P for G1, S and G2M phase cell populations, in arbitrary units of fluorescence intensity (IF), measured at identical settings of lasers intensity and photomultipliers sensitivity (voltage) for both 2R4F and T&N-free cigarettes. Note higher values of IF for T&N-free compared to 2R4F cigarettes on the respective coordinates.
In the next set of experiments, we explored whether the induction of γH2AX by CS from T&N-free cigarettes is mediated by reactive oxygen species. In our prior studies we have determined that this is the case for smoke from tobacco cigarettes, including 2R4F.10 In the present study A549 cells were exposed to CS from T&N-free cigarettes and to its vapor phase in the absence and presence of the free radical scavenger N-acetyl-L-cysteine (NAC) (Fig. 4). The data clearly indicate that NAC greatly reduced the extent of induction of γH2AX by both whole and vapor phase T&N-free smoke, with the reduction being more pronounced when whole T&N-free smoke was used.
Figure 4.
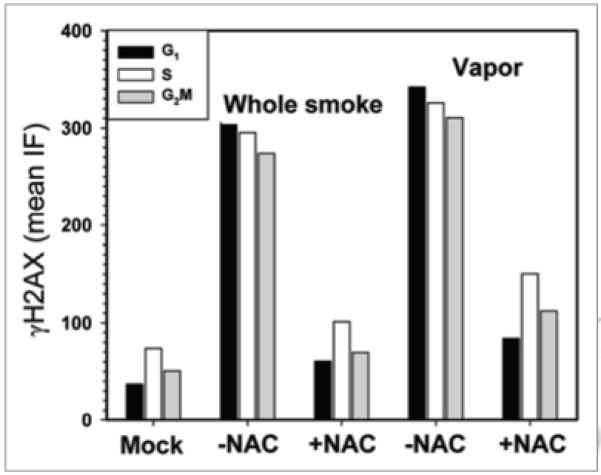
effect of N-acetyl-L-cysteine (NAC) on induction of γH2AX by smoke from T&N-free cigarettes. A549 cells were exposed to whole smoke or to vapor phase from T&N-free cigarettes in the presence or absence of 25 mM NAC and then incubated for 1 h. The bars represent mean values of γH2AX IF (in arbitrary IF units) estimated for populations of G1, S and G2M phase cells, measured under identical LSC laser intensity and photomultiplier settings.
Discussion
Exposure of A549 cells to smoke from either conventional tobacco cigarettes (2R4F) or from T&N-free cigarettes (Bravo) led to activation of ATM (phosphorylation on Ser1981) concurrent with phosphorylation of histone H2AX on Ser139. The induction of γH2AX and ATM-S1981P was dependent on the duration of exposure to smoke, and was seen at shorter times of exposure to CS from T&N-free (5–10 min) compared to 2R4F (15–20 min) cigarettes (Fig. 3). Since the concentration of smoke was constant during the cell’s exposure, the effect of duration of the exposure on the cell’s response reflects the dose of the genotoxic agents taken up by the cell during this time period. Slopes in Figure 3 thus show the smoke-dose dependence of the response. The observed lag in phosphorylation of these proteins during the initial 10 min of exposure to 2R4F or 5 min to T&N-free smoke most likely reflects the time necessary for genotoxic substances in the smoke to diffuse through the thin layer (approximately 1 mm) of D-PBS overlaying the cells in the slide chambers, pass the plasma membrane and cytoplasmic compartment surrounding the nucleus, and finally reach nuclear DNA and initiate DNA damage.13
The degree of phosphorylation of ATM and H2AX induced by the T&N-free smoke was distinctly greater than that induced by the standard tobacco and nicotine-containing 2R4F cigarette. This was reflected by higher intensity of induction of γH2AX and ATM-S1981P over the level of the constitutive phosphorylation of these proteins (Table 1) and by the shorter lag-time in response after exposure to CS (Fig. 3). It should be noted that a large portion of constitutive H2AX phosphorylation and ATM activation seen in the untreated cells reflects an ongoing DNA damage response triggered by oxidative DNA damage caused by endogenous ROS generated during aerobic metabolism.22-26
Since cigarettes and cigarette-like products may differ in both burn rate and “tar” content, the number of cigarettes smoked and the “tar” yield during a 20 min exposure varies from product to product. When smoked using FTC conditions as described in Materials and Methods, a typical 20 min exposure consumed an average of 2.2 and 1.3 cigarettes and yielded 16.0 and 40.4 mg TPM (“tar”) for 2R4F and T&N-free cigarettes, respectively. Thus, the T&N-free cigarettes generated approximately 2.5 times more TPM than 2R4F cigarettes during a typical 20 min exposure.
In the present study we investigated the effect of smoke from T&N-free cigarettes containing air dilution holes (vents). The presence of vents mimics the structure of so-called “light” or “extra-light” cigarettes; their function is to dilute the inhaled smoke with the ambient air entering through the vents and thereby decrease the concentration of the inhaled smoke.8 Since the instructions on the Bravo T&N-free cigarette package advised smokers to “puncture two, three or more pinholes” in the area just above the filter tip (where similar holes are found in conventional cigarettes), thus creating vents to “regulate Bravo’s taste and mildness”, we chose to test these cigarettes with four holes punched using a 21-gauge needle. This resulted in a decreased burn rate such that only one cigarette was required for a 20 min exposure, for which we measured a yield of 15.3 mg “tar”. Thus, on a per-cigarette basis, the T&N-free cigarettes used in this study delivered more “tar” and resulted in more DNA damage than conventional “light”-style cigarettes, even with air dilution holes.
Exposure of cells to smoke from T&N-free cigarettes containing vents exhibited a distinct pattern of cell cycle response. Specifically, we observed a preferential induction of ATM-S1981P in S-phase cells whereas exposure to CS or the vapor phase of smoke affected cells indiscriminately across the cell cycle (Fig. 2, Table 1). This was less apparent in the case of H2AX phosphorylation when the results were expressed as n-fold increase in expression of γH2AX over the mock-treated cells (Table 1). One should keep in mind, however, that because the level of constitutive expression of γH2AX (i.e., in mock-treated cells) is greater in S-phase than in G1 phase cells22-24 the data expressed as n-fold (or percent) increase above the constitutive level may not be a precise reflection of the increase as represented in arbitrary units of IF intensity. The observation that S-phase cells were more sensitive in terms of induction of DNA damage response upon exposure to T&N-free smoke is consistent with our prior data on conventional tobacco cigarettes showing that dilution of smoke with air (decrease of the dose of genotoxic agents) leads to similar change in the response pattern with respect to the cell cycle phases.12,13
The preferential induction of γH2AX and ATM-S1981P in S-phase cells has been observed in response to DNA damage by oxidants,23,26 UV light,27,28 low doses of tobacco smoke,12,13 and to smoke from T&N-free cigarettes containing vents (Fig. 2; Table 1). The primary effects induced by all these genotoxic agents are ssDNA lesions such as pyrimidine dimers, 6-4 (T-C) photoproducts,29 or 8-oxo-7,8-dihydro-2′-deoxyguanosine (oxo8dG) and base ring fragmentation.30,31 Such lesions are induced regardless of the phase of the cell cycle. However, the observed preferential response of the S-phase cells in all probability indicates formation of DNA double-strand breaks as a result of stalling of DNA replication forks upon encountering the ssDNA lesions. Consistent with this, halting DNA replication using the DNA polymerase α inhibitor aphidicolin prevents activation of ATM and H2AX phosphorylation induced by UV or by oxidants.27,28 We have also observed that exposure of cells pretreated for 20 min with 2 μM aphidicolin to T&N-free smoke followed by their incubation for 1 h in the presence of 2 μM aphidicolin decreased the level of induction of γH2AX and ATM-S1981P in S-phase cells by about 40% compared with the sum of the induction of γH2AX and ATM-S1981P in the cells exposed to smoke and aphidicolin alone (data not shown). It should be noted, however, that DNA replication stress induced by aphidicolin alone triggers H2AX and ATM phosphorylation in S-phase cells.27,32
The induction of H2AX and ATM phosphorylation by exposure of cells to T&N-free CS and vapor phase smoke was strongly attenuated in the presence of the ROS scavenger NAC (Fig. 4). This data indicates that, similar to our prior findings on the smoke from standard tobacco and nicotine containing cigarettes,10 the induction of DNA damage by T&N-free smoke, to a large extent, is mediated by ROS, and that they are present in both CS and the vapor component. This is analogous to other genotoxic events such as oxidative burst in phorbol-treated leukocytes33 or upon cell exposure to nitrogen oxide-releasing aspirin,34 when a significant component of oxidative DNA damage was seen predominantly in the S-phase cells.
Assessment of DDR induced by exposure of A549 cells to CS as revealed by phosphorylation of ATM and H2AX provides important information about the genotoxicity of smoke. Concurrent ATM activation and H2AX phosphorylation, particularly when accompanied by the presence of γH2AX foci, provide strong evidence of the induction of DSBs by CS.35-40 Because repair of DSBs is error-prone, such lesions are potentially carcinogenic.
It should be noted that concurrent induction of γH2AX foci and diffusely (uniformly) localized ATM-S1981P can occur in the absence of detectable DNA damage. Such a pseudo-DNA damage response was observed in cells undergoing senescence.41 The authors of that study postulated that cellular senescence led to activation of atypical DNA damage response and that the latter is a marker of general over-activation of senescent cells. However, while less likely, it cannot be excluded that the persistent DNA damage response induced by DNA replication stress in cells arrested in the cell cycle was in actuality the cause of cell senescence.42 Regardless of the mechanism, there was no evidence of cell senescence in the present study after a short 1 h incubation following exposure to smoke.
The fact that cigarette smoking is hazardous, contributing to the development of lung cancer and other malignancies, is indisputable. Yet approximately 23% of adult Americans continue to smoke despite high public awareness of the health risks associated with smoking, placing these individuals at increased risk for developing cancers of the lung as well as other organs. Consequently, there have been ongoing efforts by both conventional tobacco manufacturers and non-conventional companies to produce “safer” cigarettes, with supposedly diminished genotoxic properties. As is evident from our analysis, the T&N-free cigarettes appear to have an even greater propensity to induce potentially carcinogenic DNA lesions compared to conventional, nicotine-containing 2R4F cigarettes.
The methodology to assess the potential carcinogenic properties of tobacco smoke based on cytometric measurement of DNA damage response developed by us43 and presented here provides a useful addition to the battery of genotoxic tests aimed to probe cigarette smoke hazards. Such tests, which can be applied to evaluate the effects of cigarettes and cigarette surrogate products on human health, are needed by regulatory agencies such as the Food and Drug Administration.
Materials and Methods
Cells
Human pulmonary adenocarcinoma A549 cells were purchased from American Type Culture Collection (ATCC #CCL-185, Manassas, VA). The cells were cultured in Ham’s F12K medium with 2 mM L-glutamine adjusted to contain 1.5 g/L sodium bicarbonate (GIBCO/Invitrogen, Carlsbad, CA) and supplemented with 10% fetal bovine serum (GIBCO/Invitrogen). Dual-chambered slides (Nunc Lab-Tek II Fisher Scientific, Pittsburgh, PA) were seeded with 1 ml of 105 cells/ml cell suspension per chamber 48 h before exposure. All incubations were performed at 37°C in a humidified atmosphere of 5% CO2 in air. Cells were grown to 70% confluency, at which time they were treated with smoke.
CS exposure
Slide chamber covers were removed and the slides containing A549 cells were placed in a smoke exposure chamber of 20.6 cm length × 6.7 cm width × 6.3 cm height. The cell culture medium was replaced with 37°C Dulbecco’s PBS (D-PBS) containing calcium and magnesium (GIBCO/Invitrogen) during the smoke exposure. The smoke exposure chamber is designed to deliver smoke uniformly diluted with 5% CO2 in air at a constant flow rate of 500 cm3/min. Whole smoke was generated under Federal Trade Commission (FTC)/International Organization for Standardization (ISO) smoking conditions (35 ± 0.3 cm3 puff of 2 sec duration, one puff every 60 s, with none of the ventilation holes blocked) using a KC 5 Port Smoker (KC Automation, Richmond, VA). The time and distance that the smoke traveled from the end of the cigarette to the exposure chamber was minimized by using the shortest lengths of tubing possible between the parts of the apparatus. Cigarettes were smoked to within 3 mm of the filter tip. Cells were exposed to smoke for time intervals between 5 and 20 min. Mock-exposed (control) cells were treated under identical conditions as cells exposed to CS for 20 min except for the absence of a cigarette in the smoking port. Smoke was generated from the reference cigarette 2R4F (University of Kentucky, Louisville, KY), a “light style” cigarette designed to generate 9.70 mg “tar” and 0.85 mg nicotine, and from T&N-free cigarettes (Bravo cigarettes produced by Safer Smokes Corporation, One University Plaza, Suite 205, Hackensack, NJ). According to the manufacturer T&N-free cigarettes are made “from pure fresh lettuce, processed with enzymes and flavored pleasantly with organic herbal extracts”. They are designed to contain no nicotine but do generate considerable amounts of total particulate matter (TPM, often referred to as “tar”) when smoked. TPM (“tar”) was measured by smoking the cigarettes according to FTC protocol as described above, and collecting the TPM on a pre-weighed 44-mm Cambridge pad glass fiber filter (Performance Systematix Inc., Caledonia, MI). By this analysis, the T&N-free cigarettes contained, on average, 30.9 mg of TPM per cigarette. Using the same method, in our hands, the reference cigarette 2R4F generated 8.9 mg TPM per cigarette. In some experiments, cells were exposed to smoke from a T&N-free cigarette in which 4 air dilution holes (vents) were made just above the filter tip using a 21-gauge needle. T&N-free cigarettes with vents generated 15.3 mg TPM per cigarette. In addition, in some experiments the cells were exposed to the vapor phase of CS from T&N-free cigarettes. This was accomplished by placing a Cambridge pad just after the cigarette port such that the smoke flowed through it before reaching the exposure chamber removing >99% of all particles 0.3 micron or larger from whole smoke. Following smoke-, vapor phase- or mock-treatment the D-PBS was aspirated and replaced with 1 ml per chamber of fresh culture medium at 37°C. The slides were placed in the 37°C, 5% CO2 incubator and incubated for 1 h. At the end of the incubation, medium from each chamber was carefully aspirated and 1 ml of 1% fresh methanol-free formaldehyde in 1x D-PBS was added to each chamber and the cells fixed by gently rocking the slides at room temperature for 15 minutes. Following aspiration of the fixative, the chamber slides were disassembled and the slides submerged in 70% ethanol. The fixed slides were stored at 4°C prior to analysis.
Treatment of cells
N-acetyl cysteine (NAC) was obtained from Sigma Chemical Co., St. Louis, MO. In experiments which included NAC treatment, a 25 mM solution of NAC in D-PBS was prepared and used in place of D-PBS in the slide chambers during the exposure to smoke. The pH of the NAC-containing D-PBS was adjusted to 7.2 with NaOH before use, as described before.10
Immunocytochemical detection of phosphorylated histone H2AX (γH2AX) and activated ATM
After fixation, the cells were washed twice in D-PBS and treated on slides with 0.1% Triton X-100 (Sigma) in D-PBS for 15 min, and then in a 1% (w/v) solution of bovine serum albumin (BSA; Sigma) in D-PBS for 30 min to suppress nonspecific antibody (Ab) binding. The cells were then incubated in a 100 μl volume of 1% BSA containing 1:200 dilution of phospho-specific (Ser139) γH2AX mAb (Biolegend, San Diego, CA) or 1:100 dilution of phosphospecific (Ser1981) ATM mAb (Cell Signaling, Danvers, MA), for 1.5 h at room temperature or overnight at 4°C. The secondary fluorochrome-tagged Abs used were either Alexa Fluor 488 tagged Ab (Invitrogen/Molecular Probes, Carlsbad, CA, at 1:100 dilution) or Alexa Fluor 633 Ab (Invitrogen/Molecular Probes, at 1:100 dilution). Prior to fluorescence measurement on the laser scanning cytometer (LSC), the cells were counterstained with 2.8 μg/ml 4,6-diamidino-2-phenylindole (DAPI; Sigma) in D-PBS for 15 min. Each experiment was performed with an IgG control in which cells were labeled only with secondary antibody, Alexa Fluor 488 goat anti-mouse IgG (H + L) or Alexa Fluor 633 goat anti-rabbit IgG (H + L) without primary antibody incubation to estimate the extent of nonspecific binding of the secondary antibody to the cells. Other details of cell incubations with the primary and secondary Ab were presented before.13,28,44
Measurement of cell fluorescence by LSC
Cellular green or far red IF representing binding of the respective phospho-specific Abs and the blue emission of DAPI stained DNA was measured using an LSC (iCys; CompuCyte, Cambridge, MA) utilizing standard filter settings; fluorescence was excited with 488-nm argon, helium-neon (633 nm) and violet (405 nm) lasers.45,46 The intensities of maximal pixel and integrated fluorescence were measured and recorded for each cell. At least 3,000 cells were measured per sample. Gating analysis was carried out to obtain mean values (±SE) of the respective IF for G1 (DNA Index; DI = 0.9–1.1), S (DI = 1.2–1.8) and G2M (DI = 1.9–2.1) cell populations in each experiment. The standard deviation was estimated based on Poisson distribution of cell populations. Other details are given in figure legends.
Acknowledgements
Supported by NCI grant R01 28 704.
Abbreviations
- ATM
ataxia telangiectasia mutated protein kinase
- CS
cigarette smoke
- DAPI
4,6-diamidino-2-phenylindole
- DDR
DNA damage response
- D-PBS
Dulbecco’s phosphate buffered saline
- DSB
DNA double-strand break
- FTC
federal trade commission
- LSC
laser scanning cytometry
- ISO
international organization for standardization
- NAC
N-acetyl-L-cysteine
- PBS
phosphate buffered saline
- ROS
reactive oxygen species
- T&N-free
tobacco- and nicotine-free
- TPM
total particulate matter (“tar”)
References
- 1.Wynder EL, Hoffmann D. Re: Cigarette smoking and the histopathology of lung cancer. J Natl Cancer Inst. 1998;90:1486–8. doi: 10.1093/jnci/90.19.1486. [DOI] [PubMed] [Google Scholar]
- 2.Vineis P, Alavanja M, Buffler P, Doll R. Tobacco and cancer: recent epidemiological evidence. J Natl Cancer Inst. 2004;96:99–106. doi: 10.1093/jnci/djh014. [DOI] [PubMed] [Google Scholar]
- 3.Hecht SS. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nature Reviews: Cancer. 2003;3:733–44. doi: 10.1038/nrc1190. [DOI] [PubMed] [Google Scholar]
- 4.Kuper H, Boffetta P, Adami HO. Tobacco use and cancer causation: association by tumour type. J Intern Med. 2002;252:206–24. doi: 10.1046/j.1365-2796.2002.01022.x. [DOI] [PubMed] [Google Scholar]
- 5.Hecht SS, Murphy SE, Carmella SG, Li S, Jensen J, Le C, et al. Similar uptake of lung carcinogens by smokers of regular, light and ultralight cigarettes. Cancer Epidemiol Biomarkers Prev. 2005;14:693–8. doi: 10.1158/1055-9965.EPI-04-0542. [DOI] [PubMed] [Google Scholar]
- 6.Godtfredsen NS, Prescott E, Vestbo J, Osler M. Smoking reduction and biomarkers in two longitudinal studies. Addiction. 2006;101:1516–22. doi: 10.1111/j.1360-0443.2006.01542.x. [DOI] [PubMed] [Google Scholar]
- 7.Harris JE, Thun MJ, Mondul AM, Calle EE. Cigarette tar yields in relation to mortality from lung cancer in the cancer prevention study II prospective cohort 1982–8. BMJ. 2004;328:72. doi: 10.1136/bmj.37936.585382.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kozlowski LT, O’Connor RJ. Cigarette filter ventilation is a defective design because of misleading taste, bigger puffs and blocked vents. Tob Control. 2002;11:40–50. doi: 10.1136/tc.11.suppl_1.i40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Albino AP, Huang X, Yang J, Gietl D, Jorgensen E, Traganos F, Darzynkiewicz Z. Induction of histone H2AX phosphorylation in A549 human pulmonary epithelial cells by tobacco smoke and in human bronchial epithelial cells by smoke condensate: A new assay to detect the presence of potential carcinogens in tobacco. Cell Cycle. 2004;3:1062–8. [PubMed] [Google Scholar]
- 10.Albino AP, Huang X, Jorgensen E, Gietl D, Traganos F, Darzynkiewicz Z. Induction of DNA double-strand breaks in A549 and normal human pulmonary epithelial cells by cigarette smoke is mediated by free radicals. Int J Oncol. 2006;28:1491–505. [PubMed] [Google Scholar]
- 11.Tanaka T, Huang X, Jorgensen E, Gietl E, Traganos F, Darzynkiewicz Z, Albino AP. ATM activation accompanies histone H2AX phosphorylation in A549 cells upon exposure to tobacco smoke. BMC Cell Biol. 2007;8:26. doi: 10.1186/1471-2121-8-26. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albino AP, Jorgensen E, Rainey P, Gillman G, Clark TJ, Gietl D, et al. γH2AX: A potential DNA damage response biomarker for assessing toxicological risk of tobacco products. Mutation Res. 2009;678:43–52. doi: 10.1016/j.mrgentox.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao H, Albino AP, Jorgensen E, Traganos F, Darzynkiewicz Z. DNA damage response induced by tobacco smoke in normal human bronchial epithelial and A549 pulmonary adenocarcinoma cells assessed by laser scanning cytometry. Cytometry A. 2009;75:840–7. doi: 10.1002/cyto.a.20778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–86. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 15.Helt CE, Cliby WA, Keng PC, Bambara RA, O’Reilly MA. Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage. J Biol Chem. 2005;280:1186–92. doi: 10.1074/jbc.M410873200. [DOI] [PubMed] [Google Scholar]
- 16.Downs JA, Cote J. Dynamics of chromatin during the repair of DNA double-strand breaks. Cell Cycle. 2005;4:1373–6. doi: 10.4161/cc.4.10.2108. [DOI] [PubMed] [Google Scholar]
- 17.Bakkenist CJ, Kastan MB. Initiating cellular stress responses. Cell. 2004;118:9–17. doi: 10.1016/j.cell.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 18.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 19.Lee J-H, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–4. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 20.Paull TT, Lee JH. The Mre11/Rad50/Nbs1 complex and its role as a DNA-double strand break sensor for ATM. Cell Cycle. 2005;4:737–40. doi: 10.4161/cc.4.6.1715. [DOI] [PubMed] [Google Scholar]
- 21.Stoyanova T, Roy N, Kopanja D, Raychaudhuri P, Bagchi S. DDB2 (Damaged-DNA binding protein 2) in nucleotide excision repair and DNA damage response. Cell Cycle. 2009;8:4067–71. doi: 10.4161/cc.8.24.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants. Cell Cycle. 2006;5:1940–5. doi: 10.4161/cc.5.17.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao H, Tanaka T, Halicka HD, Traganos F, Zarebski M, Dobrucki J, Darzynkiewicz Z. Cytometric assessment of DNA damage by exogenous and endogenous oxidants reports the aging-related processes. Cytometry A. 2007;71:905–14. doi: 10.1002/cyto.a.20469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang X, Tanaka T, Kurose A, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation on Ser-139 in cells untreated by genotoxic agents is cell cycle phase specific and attenuated by scavenging reactive oxygen species. Int J Oncol. 2006;29:495–501. [PubMed] [Google Scholar]
- 25.Tanaka T, Kajstura M, Halicka HD, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation are strongly amplified during mitogenic stimulation of lymphocytes. Cell Prolif. 2007;40:1–13. doi: 10.1111/j.1365-2184.2007.00417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao H, Traganos F, Albino AP, Darzynkiewicz Z. Oxidative stress induces cell cycle-dependent Mre11 recruitment, ATM and Chk2 activation and histone H2AX phosphorylation. Cell Cycle. 2008;7:1490–5. doi: 10.4161/cc.7.10.5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Halicka HD, Huang X, Traganos F, King MA, Dai W, Darzynkiewicz Z. Histone H2AX phosphorylation after cell irradiation with UV-B: Relationship to cell cycle phase and induction of apoptosis. Cell Cycle. 2005;4:339–45. [PubMed] [Google Scholar]
- 28.Zhao H, Traganos F, Darzynkiewicz Z. Kinetics of the UV-induced DNA damage response in relation to cell cycle phase. Correlation with DNA replication. Cytometry A. 2010;77:285–93. doi: 10.1002/cyto.a.20839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinha RP, Häder D-P. UV-induced DNA damage and repair: a review. Photochem Photobiol Sci. 2002;1:225–36. doi: 10.1039/b201230h. [DOI] [PubMed] [Google Scholar]
- 30.Cadet J, Delatour T, Douki T, Gasparutto D, Pouget JP, Ravanat JL, Sauvaigo S. Hydroxyl radicals and DNA base damage. Mutat Res. 1999;424:9–21. doi: 10.1016/s0027-5107(99)00004-4. [DOI] [PubMed] [Google Scholar]
- 31.Marnett LJ. Oxy radicals, lipid peroxidation and DNA damage. Toxicology. 2002;181:219–22. doi: 10.1016/s0300-483x(02)00448-1. [DOI] [PubMed] [Google Scholar]
- 32.Kurose A, Tanaka T, Huang X, Traganos F, Dai W, Darzynkiewicz Z. Effects of hydroxyurea and aphidicolin on phosphorylation of ATM on Ser 1981 and histone H2AX on Ser 139 in relation to cell cycle phase and induction of apoptosis. Cytometry A. 2006;69:212–21. doi: 10.1002/cyto.a.20241. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka T, Halicka HD, Traganos F, Darzynkiewicz Z. Phosphorylation of histone H2AX on Ser 139 and activation of ATM during oxidative burst in phorbol estertreated human leukocytes. Cell Cycle. 2006;5:2671–5. doi: 10.4161/cc.5.22.3472. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka T, Kurose A, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Nitrogen oxide-releasing aspirin induces histone H2AX phosphorylation, ATM activation and apoptosis preferentially in S-phase cells; involvement of reactive oxygen species. Cell Cycle. 2006;5:1669–74. doi: 10.4161/cc.5.15.3100. [DOI] [PubMed] [Google Scholar]
- 35.Abraham RT, Tibbetts RS. Guiding ATM to broken DNA. Science. 2005;308:510–1. doi: 10.1126/science.1112069. [DOI] [PubMed] [Google Scholar]
- 36.Soutoglou E. DNA lesions and DNA damage response: even long lasting relationship needs a “break”. Cell Cycle. 2008;7:3653–8. doi: 10.4161/cc.7.23.7178. [DOI] [PubMed] [Google Scholar]
- 37.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. γH2AX and cancer. Nat Reviews. 2008;8:957–67. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakamura AJ, Rao VA, Pommier Y, Bonner WM. The complexity of phosphorylated H2AX foci formation and DNA repair assembly at DNA double-strand breaks. Cell Cycle. 2010;9:389–97. doi: 10.4161/cc.9.2.10475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mah L-J, El-Osta A, Karagiannis TC. γH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010 doi: 10.1038/leu.2010.6. Epub. [DOI] [PubMed] [Google Scholar]
- 40.Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6:931–42. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- 41.Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009;8:4112–8. doi: 10.4161/cc.8.24.10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Darzynkiewicz Z. When senescence masquerades as DNA damage: is DNA replication stress the culprit? Cell Cycle. 2009;8:3809–15. [PMC free article] [PubMed] [Google Scholar]
- 43.Albino AP, Jorgenson ED, Traganos F, Darzynkiewicz Z, Jin W. Approaches to Identify Less Harmful Tobacco and Tobacco Products. US patent No 7,662,565. 2010 issued.
- 44.Halicka HD, Zhao H, Podhorecka M, Traganos F, Darzynkiewicz Z. Cytometric detection of chromatin relaxation, an early reporter of DNA damage response. Cell Cycle. 2009;8:2233–7. doi: 10.4161/cc.8.14.8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Darzynkiewicz Z, Bedner E, Gorczyca W, Melamed MR. Laser scanning cytometry. A new instrumentation with many applications. Exp Cell Res. 1999;249:1–12. doi: 10.1006/excr.1999.4477. [DOI] [PubMed] [Google Scholar]
- 46.Pozarowski P, Holden E, Darzynkiewicz Z. Laser scanning cytometry: Principles and applications. Meth Molec Biol. 2006;319:165–92. doi: 10.1007/978-1-59259-993-6_8. [DOI] [PMC free article] [PubMed] [Google Scholar]