

Abstract
The RNA silencing pathway constitutes a defence mechanism highly conserved in eukaryotes, especially in plants, where the underlying working principle relies on the repressive action triggered by the intracellular presence of double-stranded RNAs. This immune system performs a post-transcriptional suppression of aberrant mRNAs or viral RNAs by small interfering RNAs (siRNAs) that are directed towards their target in a sequence-specific manner. However, viruses have evolved strategies to escape from silencing surveillance while promoting their own replication. Several viruses encode suppressor proteins that interact with different elements of the RNA silencing pathway and block it. The different suppressors are not phylogenetically nor structurally related and also differ in their mechanism of action. Here, we adopt a model-driven forward-engineering approach to understand the evolution of suppressor proteins and, in particular, why viral suppressors preferentially target some components of the silencing pathway. We analysed three strategies characterized by different design principles: replication in the absence of a suppressor, suppressors targeting the first protein component of the pathway and suppressors targeting the siRNAs. Our results shed light on the question of whether a virus must opt for devoting more time into transcription or into translation and on which would be the optimal step of the silencing pathway to be targeted by suppressors. In addition, we discussed the evolutionary implications of such designing principles.
Keywords: RNA silencing, silencing suppression, systems and synthetic biology, transcription–translation tradeoff, virus evolution, virus–host interaction
1. Introduction
RNA viruses are difficult to control and eliminate because of their rapid evolution. This high evolvability is a consequence of their high mutation rates, large population size and short generation times [1,2] that confer them an astonishing ability to explore genotypic space. Indeed, RNA viruses typically have mutation rates orders of magnitude higher than their DNA hosts [3]. Eukaryotic organisms have developed a sequence-specific mechanism to modulate gene expression based on RNA interference, which was first found in the nematode Caenorhabditis elegans [4] and later on in many other eukaryotes, including plants [5] and mammals [6]. Likewise, this molecular mechanism is able to silence viral or aberrant genes.
The underlying working principle of RNA silencing relies on the repressive action triggered by the intracellular presence of double-stranded RNAs (dsRNAs) [4]. In the case of single-stranded RNA (ssRNA) viruses, dsRNAs are by-products of genome replication mediated by virus-encoded RNA-dependent RNA polymerases (RdRps). During viral genome replication, the dsRNA intermediates become the target of the first component of the silencing pathway, DICER, a type-III RNase that degrades these dsRNAs into units of 21–24 nucleotides called small interfering RNAs (siRNAs; [7,8]). Subsequently, the cellular RNA-induced silencing complex (RISC), which contains the argonaute (AGO) endonuclease [9], loads the antisense siRNAs, resulting in an active form. Using the antisense siRNA as a guide, AGO cleaves the target viral ssRNA [10]. Furthermore, in a secondary cycle of amplification, the host's RNA-dependent RNA polymerase VI (RDR6) uses siRNAs as primers, together with partially degraded ssRNAs, to produce long dsRNAs that serve as new substrates for DICER, a process known as transitivity [11]. siRNAs systemically move from cell-to-cell, immunizing new cells against infection [11,12]. Given the properties of the RNA silencing pathway (specificity and amplification), it represents a sort of innate immune system for plants [13,14].
Not surprisingly, viruses have evolved strategies to actively evade the RNA silencing surveillance while promoting their own replication [15]. Many viruses encode a suppressor protein (viral suppressor of RNA silencing or VSR) that interacts with elements of the silencing pathway blocking it [16–18]. The targets of these VSRs within the RNA silencing pathway are diverse: DICER, the dsRNA, the siRNA, RISC or the systemic signal [15,19,20]. For example, the helper component-protease (HC-Pro) encoded by the Potyvirus works as suppressor by sequestering siRNAs [21–24]. This binding prevents the incorporation of siRNAs into the RISC. Furthermore, by also binding plant endogenous micro-RNAs and controlling the expression of other genes, HC-Pro may interfere the expression of DICER proteins [25,26], reducing the degradation of dsRNAs and, thus, favouring potyvirus replication. Similarly, the Nodavirus B2 suppressor also sequesters siRNAs [15]. The Tombusviridae P19 and Cucumovirus 2b suppressors interfere with the systemic spread of the 24 nucleotide siRNAs produced by DCL3 [27]. Some suppressors act on the RISC, either avoiding the upload of siRNAs into AGO, like the Closterovirus P21 [28], by binding to AGO1 and avoiding its interaction with other proteins required to assemble the RISC, as the coat protein (CP) of Tombusvirus [29], by inhibiting the RISC activity after its maturation, like the Begomovirus AC4 [30], or by targeting AGO for degradation, as it is the case for Polerovirus P0 protein ([31,32] press). It has also been recently shown that the V2 suppressor of Geminivirus competes with SGS3, a key component of the secondary cycle of siRNA amplification, in binding dsRNAs and thus interferes with transitivity [33]. Finally, the CP of some carmoviruses [34] and the P14 of Aureusvirus [35] can also bind long dsRNAs, resulting in the protection of the intermediaries of replication from DICER activity. Accordingly, VSRs have been divided into three families [20]: (i) those enhancing within-cell virus accumulation, (ii) those essential for cell-to-cell movement but dispensable on virus accumulation in single cells, and (iii) those that facilitate virus long-distance movement and/or intensify disease symptoms but are not essential for viral replication and cell-to-cell movement.
The first mathematical models of the RNA silencing pathway focused on aberrant cellular mRNA as triggers of the silencing response [36–38]. More recent models consider viral RNAs as triggers of the response and focused on the spread of viruses in plants [39,40]. However, on the one hand, these studies did not analyse in detail the possible effect that different viral suppressor strategies may have on the outcome of the interaction. On the other hand, although many kinetic models of intracellular growth have been proposed for different viruses, none of them specifically incorporates the silencing response (e.g. [41–47]). In this work, we present the first model that incorporates the interaction of different suppressor proteins with components of the silencing pathway. We perform a dynamical analysis and show the time course of viral RNA accumulation under a wide set of parameter states. We also show phase diagrams for different combinations of parameters and focus our discussion on the behaviour of the system for different viral replication and translation rates in the presence/absence of different suppressor strategies. These analyses allow us to rationalize why different viruses may opt for different strategies in their investment into producing new genomes (i.e. transcription via antigenomic strains) or into producing large amounts of protein from a few initial sense genomes (i.e. translation). Such models are important to unveil defence strategies and design principles of viral systems.
2. The model
We have constructed a mathematical model based on nonlinear differential equations to describe the interplay between the silencing pathway and a positive-sense RNA virus that encodes for a single polyprotein that is processed into mature peptides, as is the case for picorna-like viruses (e.g. poliovirus, hepatitis C virus, foot-and-mouth disease virus and the potyviruses, which are the largest and more important family of plant viruses). The model involves the following molecular species: genomic and antigenomic ssRNA (S+ and S−, respectively), dsRNA (D), antisense siRNA (I), viral proteins (p), virions (V), primed ssRNA (S*) and secondary dsRNA (D*). Three different viral proteins are considered, the non-structural replicase and VSR and the structural CP. Their corresponding relative abundances are p, q and 1 − p − q, respectively. This constraint is biologically relevant for picornaviruses as all proteins are self-processed from a single polyprotein and, thus, their relative abundances remain constant during infection. In addition, the model accounts for several cellular components: the ribosomes (Z), the RDR6 polymerase involved in transitivity (Y), DICER-like proteins (C) and the inactivated and activated RISC (R and R*, respectively). We assume that at the beginning of infection, a single viral ssRNA genome is present, which in our particular model must be genomic. Notice that genomic strands are those that encode for proteins, whereas antigenomic strands are complementary and, for simplification, we will assume are not coding. To accommodate negative-sense RNA viruses into the model, the equations can be straightforwardly modified (i.e. negative strand are encapsidated and cleaved by RISC) changing the initial conditions. For retroviruses or DNA viruses, the model must be conveniently modified.
The model is constructed following a generalized enzyme kinetics scheme where both substrates and enzymes are limited in the medium [48], and there are competitions between different enzymes for the same substrate and different substrates for the same enzyme [49]. This gives a highly coupled formulation. In figure 1, we show the scheme of the RNA silencing pathway, and the kinetic parameters are shown in table 1, with parameter values taken from different sources.
Figure 1.

Schematic of the RNA silencing pathway and its interaction with viral replication. RNA viruses encode for replicase, suppressors of silencing (VSR) and coat proteins. Three types of suppressors are considered in the scheme: suppressors of DICER (I), sequesters of siRNA (II) and suppressors of RISC (III).
Table 1.
Values for the kinetic parameters used in the model. Other non-kinetic model parameters are p = q = 0.4, ω = 0.1, n = 2n* = 10, f = 0.01, σ = 0.1 and k = 10k0 = 30. The amounts of cellular resources are Z = 105, Y = 105, C0 = 104 and R0 = 104 molecules. In the case of a virus encoding a VSR, the corresponding binding constant (ΓC, ΓI or ΓR) takes the value of Γ. The cell volume is assumed ∼ 10−13 l, then 1 nM ∼ 100 molecules.
| parameter | value | value in the literature |
|---|---|---|
| α | 10 h−1 | 1.7 h−1 for HCV [46] |
| μ | 10 h−1 | 10 h−1 for HCV [46] |
| β | 10 h−1 | 10 h−1 [39,40] |
| δ | 10 h−1 | 228 h−1in vitro for E. coli [59] |
| ρ | 1 h−1 | ∼5 h−1 [39,40] |
| υ | 25 h−1 | 25 h−1in vitro for Drosophila melanogaster [60] |
| λ | 0.1 h−1 | ∼100 M−1 h−1 for nucleation [61] |
| γ | 10 h−1 | ∼105 M−1 h−1 for elongation [61] |
| κS | 0.1 h−1 | 0.5 h−1 [39,40] |
| κI | 1 h−1 | 2 h−1 [39,40] |
| κD | 0.01 h−1 | 0.06 h−1 for HCV [46] |
| κP | 0.01 h−1 | 0.01 h−1 [62] |
| KP | 104 molecules | 225 nM in vitro for TBSV [63] |
| KR | 103 molecules | 8 nM in vitro for D. melanogaster [60] |
| KZ | 104 molecules | 260 nM in vitro [64] |
| KC | 105 molecules | 3780 nM [61] |
| KD | 105 molecules | 335 nM in vitro for E. coli [59] |
| KI | 105 molecules | ∼1000 molecules [39,40] |
| Γ | 103 molecules | 10–1000 nM in vitro p19, p21 and HC-Pro [65] |
Viral replication is a process involving multiple reactions aiming to bypass the defence systems of the cell. The RNA replication rates (J), for both polarities, are given by the following set of equations:
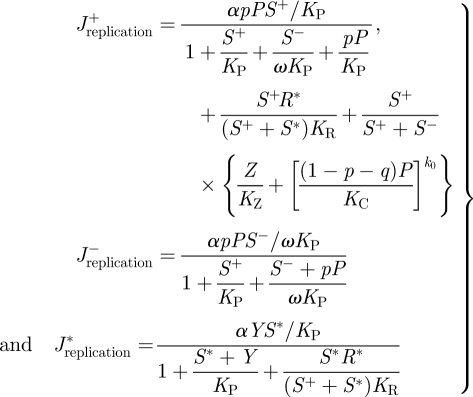 |
2.1 |
where α is the maximum replication rate per molecule of ssRNA, KP, KR, KZ and KC are the binding constants for the replicase, the activated RISC, the ribosomes and the CP, respectively. The affinity of the replicase for the antigenomic strands is incorporated into the model by the parameter ω. If ω = 1, then the RdRp has the same affinity for both strains, whereas ω > 1 would imply a larger affinity for the antigenomic strain. By doing so, we can model replication modes ranging from the geometric (ω = 1) to the stamping machine one (ω ≫ 1) [47].
A molecule of dsRNA can be separated into two ssRNA molecules of complementary polarity at a first-order rate with a constant parameter β
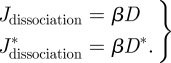 |
2.2 |
In addition, genomic ssRNAs are translated into viral proteins with rate
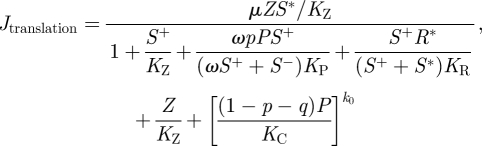 |
2.3 |
where μ is the maximum translation rate per molecule of genomic ssRNA.
The process of RNA silencing is initiated when DICER cleaves dsRNA into siRNAs. The rates describing this process are given by the following set of equations:
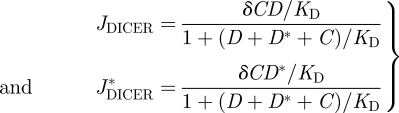 |
2.4 |
where δ and KD are the catalytic and binding constants of DICER, respectively. Afterwards, the RISC is activated by uploading the antisense siRNAs produced by DICER into AGO according to the equation
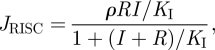 |
2.5 |
where ρ and KI are the catalytic and binding constants of the RISC, respectively. Following the activation of the RISC, it is now capable of directing the cleavage of the viral ssRNA with rate
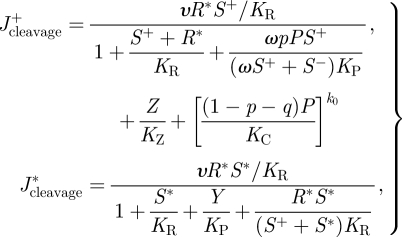 |
2.6 |
where υ is the catalytic constant of RNA cleavage. CPs are pre-assembled with ssRNA to produce immature virions at a rate given by
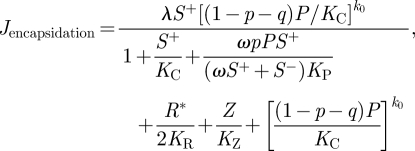 |
2.7 |
where λ is the maximum assembly rate and k0 < k is the number of CP monomers associated to the immature virions, Vimmature. Then, virions are produced at rate
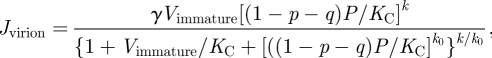 |
2.8 |
where γ is the maximum rate to produce virions, and k is the number of CPs necessary to complete a mature virion. All species are thermodynamically degraded at rates κS (ssRNAs), κD (dsRNAs), κI (siRNAs) and κP (the rest of proteins or protein complexes).
The effect exerted by different VSRs on DICER, RISC and RDR6 can be conveniently modelled by the following three equations, respectively:
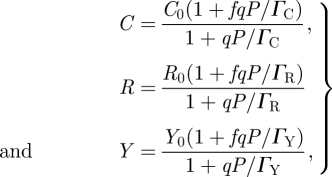 |
2.9 |
where C0, R0 and Y0 are the corresponding amounts of each protein in the cell, which are assumed to be in large excess, and ΓC, ΓR and ΓY are the binding coefficients of the corresponding VSR to their substrate protein, DICER, RISC and RDR6, respectively. The parameter f determines the efficiency at which the suppressor precludes the activity of its target. For example, in the equation for DICER, an f = 0.01 means that even at saturating concentration of the suppressor, 1 per cent of DICER molecules will still be active. To account for the suppression on siRNA, we modify JRISC and introduce a new equation to model the sequestration of siRNAs.
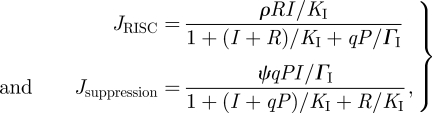 |
2.10 |
where ρ and ψ are, respectively, the rates at which the RISC and the suppressor attach to the siRNA and ΓI, the binding affinity of the suppressor for the siRNAs.
After defining all the relevant rate equations, it is now possible to write a system of coupled differential equations describing the dynamics of the system:
 |
2.11 |
where the stoichiometric parameters n, n* and σ represent, respectively, the number of siRNAs produced per molecule of dsRNA, the number of siRNAs produced in the secondary cycle of amplification and the relative contribution of the secondary siRNA amplification to the degradation of dsRNA relative to the primary siRNAs.
The full model in the Matlab format is available in the electronic supplementary material.
3. Stability analysis
The system (2.11) can be rewritten in a vectorial form as dy/dt = F(y) = ΩJ(y) − Ξy, where Ω is the matrix of stoichiometric coefficients, J(y) the vector of production rates and Ξ a diagonal matrix with the vector of degradation rates. The initial condition for the molecular species involved in the system (y0) depends on the nature of the virus (i.e. the infectious particle containing a genomic or an antigenomic RNA strand). Here, we have considered for our analyses viruses encapsidating genomic RNAs and therefore all the elements in y0 are zero except for S+ = 1. In case of negative-sense RNA viruses, the initial condition would be S− = 1. Accordingly, we construct an initial value problem to obtain the dynamics of the system. The vector of steady states is given by F(y∞) = 0, which serves to calculate the asymptotic behaviour of the system through the eigenvalues of its Jacobian matrix ∇F(y∞). The behaviour can change significantly by modifying pivotal parameters of the system. Thus, the construction of bifurcation diagrams is a useful tool for evaluating the behaviour regimes under different conditions, and also to build up a sensitivity analysis of the parameters of the system.
We show that the trivial solution of the system (i.e. silenced virus) is stable. The Jacobian matrix evaluated at y∞ = 0 is given by
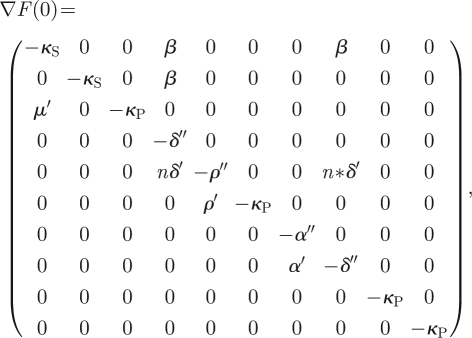 |
where α′ = αY/KP, δ′ = δC0/KD, ρ′ = ρR0/KI, μ′ = μZ/KZ, α″ = α′ + κS, δ″ = δ′ + β + κD and ρ″ = ρ′ + κI. This Jacobian has five negative real eigenvalues (−α″, −ρ″, −δ″, −κS and −κP) that represent an asymptotically stable solution of the system. Three of them have multiplicity greater than one. The system also has a second non-trivial solution in which the virus beats the silencing response and replicates and accumulates in the cell. Although we have verified numerically the existence of this non-trivial solution on the full model, without lost of generality, the stability analysis for this second solution can be done analytically by simplifying the system (2.11) as
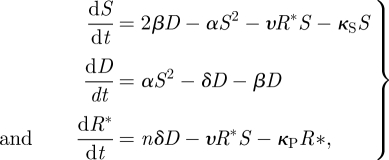 |
3.2 |
where the non-trivial steady state is the solution of S (β − δ)/(β + δ) = κS + nδαυS2/(β + δ) (υS + κP). The characteristic polynomial is −X3 + τX2 − ηX − Δ, where τ = −2αS − υR − κS − β− δ −υS − κP is the trace of the Jacobian matrix, η = (2αS + υR + κS)(β + δ+ υS + κP) + (υS + κP) (β + δ) − 4αβS − υ2RS is the trace of its adjoint matrix and Δ = [4αβS − (2αS + υR + κS) (β + δ)] (υ S + κP) − 2nαδ υS2 + υ2RS(β + δ) its determinant. By applying the Routh–Hurwitz stability criterion, the system will be stable when τ < 0, Δ < 0 and ητ < Δ. Henceforth, by taking the appropriate kinetic parameters that meet these three conditions, the system is characterized by bistability and, therefore, the initial condition is pivotal to determine the outcome of the process.
4. Results
4.1. Virus replication in the absence of a VSR
We have studied the viral replication dynamics by using the mathematical model presented in the previous section. First, we considered the case of RNA viruses that do not encode suppressor proteins. In figure 2, we show several time-course evolutions of the system species (S+, S− and P) for two different sets of initial conditions. When the multiplicity of infection is low (one single viral S+ genome per cell) and for the typical parameter values shown in table 1, we show that the population is extinguished (figure 2a), after a transient where the concentration of P reaches a maximum. The model predicts that in this situation, the amount of antigenomic strains S− produced is meaningless and its dynamics is dominated by the degradation term in the system of equations (2.11).
Figure 2.

Dynamics of viral infection for different initial conditions. (a) The starting condition of the simulation is a single viral genome; this results in the virus being silenced. (b) The starting condition is that 10 viral genomes infect the cell; this high multiplicity of infection results in exponential viral replication after a period of latency of 1 day required to reach a threshold level of RdRps. This successful infection happens even in the absence of a VSR. The parameters values are those shown in table 1.
However, the virus can bypass the silencing mechanism if the multiplicity of infection just increases to S+ = 10 molecules (figure 2b). In this case, after a latency period of about 1 day, viral proteins reach a critical concentration and promote further exponential replication. Analytically, the latency period can be estimated when P reaches ωKP. In all these simulations, the condition S+ > S− holds, in excellent agreement with the observation of an excess of sense siRNAs for positive-sense viral genomes [50]. The effect of further increasing the multiplicity of infection is to reduce the latency period (data not shown).
We performed several sensitivity analyses to study the regions in parameter space in which viral replication occurs (non-trivial solution) or for which viral silencing takes place (trivial solution). We found that the higher the affinity for the negative strand (lower ω), the wider is the parameter space for viral replication (figure 3a). In fact, this can be rationalized because viral RdRps compete with ribosomes and with the activated RISC for genomic strands, whereas they do not compete for antigenomic strands. In addition, high replication rates also allow the virus to escape from the silencing machinery and to minimize the effect of non-specific thermodynamic degradation (figure 3b).
Figure 3.

Phase diagrams identify different viral strategies. (a) The effect of the catalytic constant of DICER cleavage (δ) in the replication rate (α) and the differential affinity of RdRps for positive and negative strands (ω). (b) The relationship between α and the ssRNA degradation rate (κS) for different values of the translation rate (μ). (c) The sensitivity of the binding constants of ribosomes (KZ) and replicases (KP) to α. (d) The effect of KP on μ and α. Rep means viral replication bypassing silencing, and Sil viral extinction by silencing.
One question that arises here is whether a tradeoff between replication and translation exists. Upon uncoating and the strictly necessary first event of translation, a viral genome can be directed either to transcription, and thus increase the concentration of RNA, or to translation, and thus increase the concentration of viral proteins (in this case only replicase and coat). In figure 3c, we analysed such tradeoff by considering the binding affinities to positive strands of replicase (KP) and ribosomes (KZ). We showed that in the absence of a silencing suppressor, silencing is the outcome favoured when translation is more frequent than transcription (KP < KZ). Accordingly, the best strategy for a virus to bypass the RNA silencing response in the absence of a suppressor protein would be to increase the affinity of its RNA to the replicase rather than to optimize its binding affinity to the ribosomes. Likewise, by increasing its transcription efficiency, a virus will produce more copies of its genome up to the point in which the cleavage by DICER would no longer control the accumulation of viral genomes. Figure 3d shows, as expected, that the higher the catalytic constants for transcription and translation, the higher the chances for a successful viral replication.
4.2. Virus replication dynamics in presence of a VSR that acts on DICER
Many, if not all, viruses encode proteins capable of interacting with the cell molecular machinery. The suppression mechanism is often a protein–protein or RNA–protein interaction resulting in a sequestration or blockage of one of the many molecules involved in the silencing pathway that allows the virus to escape from silencing surveillance. Our general model can be used to analyse and study the effect of various suppressors encoded by different viruses. To analyse the effect of a suppressor, we considered the virus replication speed as a characteristic scoring function. This speed can be easily computed as the inverse of the time taken to produce mature virions (TV). In figure 4, we plot 1/TV versus KZ and KP for the case of a VSR operating over DICER. We found that such a suppressor enhances the speed of virus accumulation with respect to a virus without encoding a VSR.
Figure 4.

Virus replication speed (computed as the inverse of the time to form a mature virion, TV) versus the binding constants of ribosomes (KZ) and replicases (KP), with ΓC = 102 molecules. (a) Virus without a VSR. (b) Virus encoding a suppressor that blocks DICER. The benefit associated with carrying such a suppressor is evaluated as the difference between both surfaces and is indicated by the dashed line and the arrow. The other parameters take the values shown in table 1.
To further analyse the suppressor strategy of manipulating DICER, we constructed a phase diagram between the catalytic constant of cleavage by DICER (δ) and the suppressor binding constant (ΓC) (figure 5a). We found that the effect of the suppressor is only significant beyond a threshold level of ΓC (in this case 7000 molecules). In other words, if the affinity of the suppressor is not high enough, it only represents a cost for the virus because it cannot help in its replication. Figure 5b shows the effect that the binding affinity of the suppressor for DICER has on the time necessary for completing a virion as a function of the cellular amounts of DICER (C0). For low amounts of DICER, TV is insensitive to variation in ΓC. In addition, an increase in the number of DICER molecules per cell does not have any effect on TV for suppressors with weak affinity. However, if ΓC increases (moving rightwards in the ordinates axis in figure 5b), then the time to produce virions significantly grows up and becomes infinity (indicating viral silencing) for high amounts of DICER molecules present in the cell at the time of infection.
Figure 5.

(a) Phase diagram to analyse the suppressor effect on DICER between δ and ΓC, with α = 20 h−1 for different values of μ. (b) Time to form one virion (TV) versus the suppressor constant of DICER, with α =μ = 20 h−1 for different values of C0 (in molecules). The other parameters take the values shown in table 1. Rep means viral replication bypassing silencing, and Sil viral extinction by silencing.
4.3. The effect of suppressing downstream steps of the silencing pathway
Next, we sought the effect of VSRs operating downstream in the silencing pathway. Surprisingly, we found that suppressors affecting at other levels of the pathway (e.g. sequestering siRNAs, interfering with RISC or with RDR6) did not enlarge the parameter space in which the virus successfully replicates within a single cell (data not shown). This result suggests that only by suppressing DICER, the first bottleneck to replication imposed by the system, viruses could widen the parameter region, resulting in successful replication. Hence, the question is why other types of VSRs, such as siRNA sequesters, have evolved? Our negative result suggests that the RNA silencing mode of action cannot be rationalized by only looking into a single cell but that a more complex situation in which cell-to-cell effects may contribute should be considered. This leads us to consider the role of the space to analyse such mechanism.
In figure 6a, we plot the relative amount of accumulated siRNAs (normalized by the amount or siRNA produced in the absence of a VSR, I/IΓ→∞), in the presence of two suppression strategies. For illustrative purposes, we have chosen the successful operation over DICER described in the previous section and one based on sequestering siRNAs. By increasing the affinity for the corresponding target molecule (moving rightwards on the ordinate axis) to the maximum value analysed, the strategy based on blocking DICER reduces the concentration of siRNA around two orders of magnitude. However, the strategy based on sequestering siRNAs is far less efficient since at the strongest affinity it only reduces the accumulation of virus-derived siRNAs by one order of magnitude.
Figure 6.

(a) Amount of siRNA (relative to the amount accumulated without a viral suppressor of RNA silencing) versus the suppressor constant (Γ) on DICER or siRNA. (b) Viral RNA dynamics in a cell which has not been immunized by receiving siRNA from neighbouring cells (R*(0) = 0) and in a cell that has received a small input of siRNA from an infected neighbour cell (R*(0) = 10 molecules). The parameters take the values shown in table 1, expect α = 50 h−1.
However, the transfer of siRNAs from infected to neighbouring healthy cells, which allows the peripheral cells to activate the RISC in the absence of viral infection, has the expected effect (figure 6b). In the absence of triggering siRNAs, infection progresses with the time delay already described above. However, if the cell has been already activated, the virus is not able to overcome the cleavage by the RISC and runs to extinction.
5. Discussion
We have presented a deterministic model of the interplay between viral replication and the RNA silencing pathway. For the sake of biological realism, we modelled a particular type of virus, the picorna-like. By doing so, the model pays the cost of reduced generality and the conclusions may not be applicable to viruses with other genomic architectures such as negative-sense RNA, retroviruses or DNA viruses. Although our results have been performed for positive-sense RNA viruses, the model can also be used to study negative-sense viruses with minor changes in some rates and the initial conditions. Readers interested in exploring the interplay between the silencing pathway and any of these viruses must necessarily look at this article as the starting point for developing their own models. Nonetheless, our approximation has allowed us to study and compare different viral suppression strategies. We have shown that the RNA silencing pathway allows a large variety of behaviours, suggesting multiple potential evolutionary trajectories for RNA viruses. Future models will account for different viral genomic organizations and for inherent stochastic effects associated with small numbers of molecules [51]. The model presented here differs from other models of the interaction between virus and the host silencing response [39,40] in which here we have explored the role played by different suppressors of RNA silencing. We have demonstrated and shown in figure 2 that the system has two stable steady states (replication and silencing) and, thus, the initial condition of the system (i.e. the initial amount of ssRNA in the cell) is important to determine its dynamics. Likewise, the higher the initial amount of viral RNA, the higher the zone for exponential viral replication in the parameter space. This suggests that increasing the multiplicity of infection is a possible strategy for virus to escape from the control of RNA silencing.
We have shown that in the presence of an active silencing response, it is to the benefit of the virus to invest into a transcriptional strategy rather than in translation. This may be somehow counterintuitive because one may expect more replication to generate more dsRNA and, therefore, to strength the silencing response and, likewise, more translation to produce more suppressor protein. It can be argued that, after the very initial burst of translation from the infecting genomic sequence resulting in a few viral proteins, the optimal strategy involves synthesizing antigenomic strands and using them as templates for producing a large excess of genomic strands (i.e. using a stamping machine replication strategy) without diverting them into translation. If replication is fast enough, this replicative strategy works even in the absence of a suppressor protein: a positive feedback is established such that the replication overcomes the capacity of the available DICER molecules to keep virus replication under control. Once a significant amount of genomic strands has been produced, then translation may take place. If translation results in a VSR protein, then a synergistic effect between fast transcription and translation appears, resulting in successful viral replication.
Among many possibilities, we have focused on three viral strategies. The first one, consisting of blocking DICER, turns out to be the most efficient promoting viral replication. This result is somehow logical from an optimal design perspective. By hitting the first bottleneck in the pathway, the virus ensures its own replication. Hitting downstream steps would allow DICER to still exert partial control on virus replication. The other three strategies explored, sequestering siRNA, blocking the RISC and disrupting the secondary amplification via RDR6, have been less efficient in promoting intracellular virus accumulation, although they may gain some benefit when looking at cell-to-cell movement. This finding is in good agreement with the observation that Cucumovirus 2b and Tombusvirus P19, which promote systemic and cell-to-cell movements, are not required for intracellular accumulation [15].
Although mathematically convenient, it may be a biological oversimplification to assume that suppressors act at a single stage of the silencing pathway. Evidence exists showing that VSRs may well simultaneously operate at diverse stages of the pathway. For example, the potyviral HC-Pro sequesters siRNAs but also affects the expression of plant genes, including the dcl-like genes encoding for the different DICER proteins in Arabidopsis thaliana [25,26], or by reducing the 3′ methylation of siRNAs, making them sensitive to oligouridilation and subsequent degradation [52,53]. Another example of multiple actions is the Polerovirus P0 that interferes with the silencing pathway at least at two levels: binding siRNAs and avoiding the formation of the activated AGO complex and labelling it for degradation ([31,32] press). Also, a virus may carry more than one VSR, as seems to be the case for some tombusviruses (P19 and CP).
We have also found that in certain regions of parameter space, a virus would be capable of replicating even in the absence of a VSR. The plant subviral pathogens known as viroids do not encode for any protein at all and are still capable of replication in susceptible hosts [54], despite the fact that their RNA molecules are targets of DICER [55]. It has been suggested that viroids may evade silencing because of their highly complex and packed secondary structure [56,57]. Other strategies viruses may use for avoiding silencing consist in replicating within spherules in the endoplasmic reticulum membrane [58], where they remain inaccessible to DICER.
Although we have modelled the effect of VSRs on DICER as a direct protein–protein interaction, VSRs can also interfere with DICER activity by protecting the dsRNA as it is produced as reported for CP [34]. This particular activity would be easily incorporated into our mathematical framework in two simple ways. First, by treating the binding affinity of DICER for dsRNA (KD) as a decreasing function of CP concentration. Second, by defining a new molecular species for the complex dsRNA/CP and writing down the corresponding rates and adding a new differential equation in equation (2.11).
In conclusion, we have shown that from a system design perspective, the best strategy that a virus may take to ensure its replication in the presence of the antiviral response mediated by RNA silencing would be to (i) replicate fast and by producing an excess of genomic strands, (ii) encode for a VSR that interacts with the DICER protein, and (iii) exert some control on the multiplicity of infection, ensuring that multiple genomes infect each cell. Obviously, evolution is not a perfect designer and viruses have acquired suppressor proteins that target at different steps of the silencing pathway. Understanding the exact mechanisms by which these VSRs operate will allow us to develop better models and to increase our ability to predict the outcome of the virus–host interaction. Furthermore, VSRs have clear biotechnological potential as they can be used to maximize the expression of transgenes [15]. Designing optimal suppressors would benefit from the knowledge advanced in this article.
Acknowledgements
This work was supported by the Spanish Ministerio de Ciencia e Innovación grants BFU2009-06993 to S.F.E. and TIN-2006-12860 to A.J, and by FP6-NEST-043340 (BioModularH2), FP7-ICT-043338 (Bactocom), FP7-KBBE-212894 (Tarpol), the Structural Funds of the European Regional Development Fund, the ATIGE-Genopole and the Foundation pour la Recherche Medicale grants (all to A.J.). J.C, G.R. and A.J. also acknowledge the HPC-Europa programme (RII3-CT-2003-506079). G.R. was supported by a graduate fellowship from the Generalitat Valenciana and an EMBO Short-term fellowship. S.F.E. also acknowledges support from the Santa Fe Institute.
References
- 1.Domingo E., Holland J. J. 1997. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51, 151–178 10.1146/annurev.micro.51.1.151 (doi:10.1146/annurev.micro.51.1.151) [DOI] [PubMed] [Google Scholar]
- 2.Elena S. F., Sanjuán R. 2007. Virus evolution: insights from an experimental approach. Annu. Rev. Ecol. Evol. Syst. 38, 27–52 10.1146/annurev.ecolsys.38.091206.095637 (doi:10.1146/annurev.ecolsys.38.091206.095637) [DOI] [Google Scholar]
- 3.Drake J. W., Holland J. J. 1999. Mutation rates among RNA viruses. Proc. Natl Acad. Sci. USA 96, 13 910–13 913 10.1073/pnas.96.24.13910 (doi:10.1073/pnas.96.24.13910) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fire A., Xu S., Mongomery M. K., Kostas S. A., Driver S. E., Mello C. C. 1998. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811 10.1038/35888 (doi:10.1038/35888) [DOI] [PubMed] [Google Scholar]
- 5.Vaucheret H., Beclin C., Fagard M. 2001. Post-transcriptional gene silencing in plants. J. Cell Sci. 14, 3083–3091 [DOI] [PubMed] [Google Scholar]
- 6.Elbashir S. M., Harborth J., Lendeckel W., Yalcin A., Weber K., Tuschl T. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498 10.1038/35078107 (doi:10.1038/35078107) [DOI] [PubMed] [Google Scholar]
- 7.Hamilton A. J., Baulcombe D. C. 1999. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 286, 950–952 10.1126/science.286.5441.950 (doi:10.1126/science.286.5441.950) [DOI] [PubMed] [Google Scholar]
- 8.Vermeulen A., Behlen L., Reynolds A., Wolfson A., Marshall W. S., Karpilow J., Khvorova A. 2005. The contributions of dsRNA structure to DICER specificity and efficiency. RNA 11, 674–682 10.1261/rna.7272305 (doi:10.1261/rna.7272305) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bohmert K., Camus I., Bellini C., Bouchez D., Caboche M., Benning C. 1998. AGO1 defines a novel locus of Arabidopsis controlling leaf development. EMBO J. 17, 170–180 10.1093/emboj/17.1.170 (doi:10.1093/emboj/17.1.170) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rand T., Petersen S., Du F., Wang X. 2005. Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell 123, 621–629 10.1016/j.cell.2005.10.020 (doi:10.1016/j.cell.2005.10.020) [DOI] [PubMed] [Google Scholar]
- 11.Voinnet O., Vain P., Angell S., Baulcombe D. C. 1998. Systemic spread of sequence specific transgene RNA degradation is initiated by localized introduction of ectopic promoter less DNA. Cell 95, 177–187 10.1016/S0092-8674(00)81749-3 (doi:10.1016/S0092-8674(00)81749-3) [DOI] [PubMed] [Google Scholar]
- 12.Himber C., Dunoyer P., Moissiard G., Ritzenthaler C., Voinnet O. 2003. Transitivity-dependent and -independent cell-to-cell movement of RNA silencing. EMBO J. 22, 4523–4533 10.1093/emboj/cdg431 (doi:10.1093/emboj/cdg431) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lecellier C. H., Voinnet O. 2004. RNA silencing: no mercy for viruses? Immunol. Rev. 1998, 285–303 [DOI] [PubMed] [Google Scholar]
- 14.Ding S., Voinnet O. 2007. Antiviral immunity directed by small RNAs. Cell 130, 413–426 10.1016/j.cell.2007.07.039 (doi:10.1016/j.cell.2007.07.039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li F., Ding S. W. 2006. Virus counterdefenses: diverse strategies for evading the RNA-silencing immunity. Annu. Rev. Microbiol. 60, 503–531 10.1146/annurev.micro.60.080805.142205 (doi:10.1146/annurev.micro.60.080805.142205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brigneti G., Voinnet O., Li W. X., Ji L. H., Ding S. W., Baulcombe D. C. 1998. Viral pathogenicity determinants are suppressors of transgene silencing in Nicotiana benthamiana. EMBO J. 17, 6739–6746 10.1093/emboj/17.22.6739 (doi:10.1093/emboj/17.22.6739) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Baulcombe D. 2004. RNA silencing in plants. Nature 431, 356–363 10.1038/nature02874 (doi:10.1038/nature02874) [DOI] [PubMed] [Google Scholar]
- 18.Kasschau K. D., Xie Z., Allen E., Llave C., Chapman E. J., Krizan K. A., Carrington J. C. 2003. P1/HC-Pro, a viral suppressor of RNA silencing, interferes with Arabidopsis development and miRNA function. Dev. Cell 4, 205–217 10.1016/S1534-5807(03)00025-X (doi:10.1016/S1534-5807(03)00025-X) [DOI] [PubMed] [Google Scholar]
- 19.Moissiard G., Voinnet O. 2004. Viral suppression of RNA silencing in plants. Mol. Plant Pathol. 5, 71–82 10.1111/j.1364-3703.2004.00207.x (doi:10.1111/j.1364-3703.2004.00207.x) [DOI] [PubMed] [Google Scholar]
- 20.Díaz-Pendón J. A., Ding S. W. 2008. Direct and indirect roles of viral suppressors of RNA silencing in pathogenesis. Annu. Rev. Phytopathol. 46, 303–326 10.1146/annurev.phyto.46.081407.104746 (doi:10.1146/annurev.phyto.46.081407.104746) [DOI] [PubMed] [Google Scholar]
- 21.Mallory A. C., Reinhart B. J., Bartel D., Vance V. B., Bowman L. H. 2002. A viral suppressor of RNA silencing differentially regulates the accumulation of short interfering RNAs and microRNAs in tobacco. Proc. Natl Acad. Sci. USA 99, 15 228–15 233 10.1073/pnas.232434999 (doi:10.1073/pnas.232434999) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chapman E. J., Prokhnevsky A. I., Gopinath K., Dolja V., Carrington J. C. 2004. Viral RNA silencing suppressors inhibit the microRNA pathway at an intermediate step. Genes Dev. 18, 1179–1186 10.1101/gad.1201204 (doi:10.1101/gad.1201204) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunoyer P., Lecellier C. H., Parizotto E. A., Himber C., Voinnet O. 2004. Probing the microRNA and small interfering RNA pathways with virus-encoded suppressors of RNA silencing. Plant Cell 16, 1235–1250 10.1105/tpc.020719 (doi:10.1105/tpc.020719) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Lakatos L., Szittya G., Silhavy D., Burgyan J. 2004. Molecular mechanism of RNA silencing suppression mediated by p19 protein of tombusviruses. EMBO J. 23, 876–884 10.1038/sj.emboj.7600096 (doi:10.1038/sj.emboj.7600096) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mlotshwa S., et al. 2005. Ectopic DICERLIKE1 expression in P1/HC-Pro Arabidopsis rescues phenotypic anomalies but not defects in microRNA and silencing pathways. Plant Cell 17, 2873–2885 10.1105/tpc.105.036608 (doi:10.1105/tpc.105.036608) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deleris A., Gallego-Bartolome J., Bao J., Kasschau K. D., Carrington J. C., Voinnet O. 2006. Hierarchical action and inhibition of plant DICER-like proteins in antiviral defense. Science 313, 68–71 10.1126/science.1128214 (doi:10.1126/science.1128214) [DOI] [PubMed] [Google Scholar]
- 27.Qi Y., Zhong X., Itaya A., Ding B. 2004. Dissecting RNA silencing in protoplasts uncovers novel effects of viral suppressors on the silencing pathway at the cellular level. Nucleic Acids Res. 32, e179. 10.1093/nar/gnh180 (doi:10.1093/nar/gnh180) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peremyslov V. V., Hagiwara Y., Dolja V. V. 1999. HSP70 homolog functions in cell-to-cell movement of a plant virus. Proc. Natl Acad. Sci. USA 96, 14 771–14 776 10.1073/pnas.96.26.14771 (doi:10.1073/pnas.96.26.14771) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azevedo J., et al. 2010. Argonaute quenching and global changes in Dicer homeostasis caused by a pathogen-encoded GW repeat protein. Genes Develop. 24, 904–915 10.1101/gad.1908710 (doi:10.1101/gad.1908710) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vanitharani R., Chellappan P., Pita J. S., Fauquet C. M. 2004. Differential roles of AC2 and AC4 of Cassava Geminiviruses in mediating synergism and suppression of posttranscriptional gene silencing. J. Virol. 78, 9487–9498 10.1128/JVI.78.17.9487-9498.2004 (doi:10.1128/JVI.78.17.9487-9498.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baumberger N., Tsai C. H., Lie M., Havecker E., Baulcombe D. C. 2007. The polerovirus silencing suppressor P0 targets ARGONAUTE proteins for degradation. Curr. Biol. 17, 1609–1614 10.1016/j.cub.2007.08.039 (doi:10.1016/j.cub.2007.08.039) [DOI] [PubMed] [Google Scholar]
- 32.Csorba T., Lózsa R., Hutvágner G., Burgyán J. 2010. Polerovirus protein P0 prevents the assembly of small RNA containing RISC complexes and leads to degradation of ARGONAUTE1. Plant J. 62, 463–472 10.1111/j.1365-313x.2010.04163.x (doi:10.1111/j.1365-313x.2010.04163.x) [DOI] [PubMed] [Google Scholar]
- 33.Fukunaga R., Doudna J. A. 2009. dsRNA with 5′ overhangs contributes to endogenous and antiviral RNA silencing pathways in plants. EMBO J. 28, 545–555 10.1038/emboj.2009.2 (doi:10.1038/emboj.2009.2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meng C., Chen J., Peng J., Wong S. M. 2006. Host-induced avirulence of Hibiscus chlorotic ringspot virus mutant correlates with reduced gene-silencing suppression activity. J. Gen. Virol. 87, 451–459 10.1099/vir.0.81578-0 (doi:10.1099/vir.0.81578-0) [DOI] [PubMed] [Google Scholar]
- 35.Mérai Z., Keréncyi Z., Molnár A., Barta E., Bisztray G., Havelda Z., Burgyán J., Silhavy D. 2005. Aureusvirus P14 is an efficient RNA silencing suppressor that binds double-stranded RNAs without size specificity. J. Virol. 79, 7217–7226 10.1128/JVI.79.11.7217-7226.2005 (doi:10.1128/JVI.79.11.7217-7226.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bergstrom C. T., McKittrick E., Antia R. 2003. Mathematical models of RNA silencing: unidirectional amplification limits accidental self-directed reactions. Proc. Natl. Acad. Sci. USA 100, 11 511–11 516 10.1073/pnas.1931639100 (doi:10.1073/pnas.1931639100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raab R. M., Stephanopoulos G. 2004. Dynamics of gene silencing by RNA interference. Biotechnol. Bioeng. 88, 121–132 10.1002/bit.20216 (doi:10.1002/bit.20216) [DOI] [PubMed] [Google Scholar]
- 38.Groenenboom M. A. C., Marée A. F. M., Hogeweg P. 2005. The RNA silencing pathway: the bits and pieces that matter. PLoS Comp. Biol. 1, e21. 10.1371/journal.pcbi.0010021 (doi:10.1371/journal.pcbi.0010021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Groenenboom M., Hogeweg P. 2008. The dynamics and efficacy of antiviral RNA silencing: a model study. BMC Syst. Biol. 2, 28. 10.1186/1752-0509-2-28 (doi:10.1186/1752-0509-2-28) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Groenenboom M., Hogeweg P. 2008. RNA silencing can explain chlorotic infection patterns on plant leaves. BMC Syst. Biol. 2, 105. 10.1186/1752-0509-2-105 (doi:10.1186/1752-0509-2-105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Endy D., Kong D., Yin J. 1996. Intracellular kinetics of a growing virus: a genetically structured simulation for bacteriophage T7. Biotechnol. Bioeng. 55, 375–389 (doi:10.1002/(SICI)1097-0290(19970720)55:2<375::AID-BIT15>3.0.CO;2-G) [DOI] [PubMed] [Google Scholar]
- 42.Reddy B., Yin J. 1999. Quantitative intracellular kinetics of HIV type 1. AIDS Res. Hum. Retrovir. 15, 273–283 10.1089/088922299311457 (doi:10.1089/088922299311457) [DOI] [PubMed] [Google Scholar]
- 43.Srivastava R., You L., Summers J., Yinn J. 2002. Stochastic vs. deterministic modeling of intracellular viral kinetics. J. Theor. Biol. 218, 309–321 10.1006/jtbi.2002.3078 (doi:10.1006/jtbi.2002.3078) [DOI] [PubMed] [Google Scholar]
- 44.Sidorenko Y., Reichl U. 2004. Structured model of influenza virus replication in MDCK cells. Biotechnol. Bioeng. 88, 1–14 10.1002/bit.20096 (doi:10.1002/bit.20096) [DOI] [PubMed] [Google Scholar]
- 45.Lim K., Lang V., Lam T., Yin J. 2006. Model-based design of growth-attenuated viruses. PLoS Comput. Biol. 2, e116. 10.1371/journal.pcbi.0020116 (doi:10.1371/journal.pcbi.0020116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dahari H., Ribeiro R. M., Rice C. M., Perelson A. S. 2007. Mathematical modeling of subgenomic hepatitis C virus replication in Huh-7 cells. J. Virol. 81, 750–760 10.1128/JVI.01304-06 (doi:10.1128/JVI.01304-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sardanyés J., Solé R. V., Elena S. F. 2009. Replication mode and landscape topology differentially affect RNA virus mutational load and robustness. J. Virol. 83, 12 579–12 589 10.1128/JVI.00767-09 (doi:10.1128/JVI.00767-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeAngelis D. L., Goldstein R. A., O'Neill R. V. 1975. A model for trophic interaction. Ecology 56, 881–892 10.2307/1936298 (doi:10.2307/1936298) [DOI] [Google Scholar]
- 49.MacRae I. J., Zhou K., Doudna J. A. 2007. Structural determinants of RNA recognition and cleavage by DICER. Nat. Struct. Mol. Biol. 14, 934–940 10.1038/nsmb1293 (doi:10.1038/nsmb1293) [DOI] [PubMed] [Google Scholar]
- 50.Qi X., Bao F. S., Xie Z. 2010. Small RNA deep sequencing reveals role for Arabidopsis thaliana RNA-dependent RNA polymerases in viral siRNA biogenesis. PLoS ONE 4, e4971. 10.1371/journal.pone.0004971 (doi:10.1371/journal.pone.0004971) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gillespie D. T. 1977. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 81, 2340–2361 10.1021/j100540a008 (doi:10.1021/j100540a008) [DOI] [Google Scholar]
- 52.Ebhart H. A., Thi E. P., Wang M. B., Unrau P. J. 2005. Extensive 3′ modification of plant small RNAs is modulated by helper component-proteinase expression. Proc. Natl Acad. Sci. USA 102, 13 398–13 403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mérai Z., Kerényi Z., Kerstész S., Magna M., Lakatos L., Silhavy D. 2006. Double-stranded RNA binding may be a general plant RNA viral strategy to suppress RNA silencing. J. Virol. 80, 5747–5756 10.1128/JVI.01963-05 (doi:10.1128/JVI.01963-05) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Daròs J. A., Elena S. F., Flores R. 2006. Viroids: an Ariadne's thread into the RNA labyrinth. EMBO Rep. 7, 593–598 10.1038/sj.embor.7400706 (doi:10.1038/sj.embor.7400706) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Di Serio F., Gisel A., Navarro B., Delgado S., Martínez de Alba A. E., Donvito G., Flores R. 2009. Deep sequencing of the small RNAs derived from two symptomatic variants of a chloroplastic viroid: implications for their genesis and for pathogenesis. PLoS ONE 4, e7539. 10.1371/journal.pone.0007539 (doi:10.1371/journal.pone.0007539) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang M. B., et al. 2004. On the role of RNA silencing in the pathogenicity and evolution of viroids and viral satellites. Proc. Natl Acad. Sci. USA 101, 3275–3280 10.1073/pnas.0400104101 (doi:10.1073/pnas.0400104101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gómez G., Pallás V. 2007. Mature monomeric forms of Hop stunt viroid resist RNA silencing in transgenic plants. Plant J. 51, 1041–1049 10.1111/j.1365-313X.2007.03203.x (doi:10.1111/j.1365-313X.2007.03203.x) [DOI] [PubMed] [Google Scholar]
- 58.Schwartz M., Chen J., Janda M., Sulivan M., den Boon J., Ahlquist P. 2002. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell 9, 505–514 10.1016/S1097-2765(02)00474-4 (doi:10.1016/S1097-2765(02)00474-4) [DOI] [PubMed] [Google Scholar]
- 59.Sun W., Jun E., Nicholson A. W. 2001. Intrinsic double-stranded-RNA processing activity of Escherichia coli ribonuclease III lacking the dsRNA-binding domain. Biochemistry 40, 14 976–14 984 10.1021/bi011570u (doi:10.1021/bi011570u) [DOI] [PubMed] [Google Scholar]
- 60.Haley B., Zamore P. D. 2004. Kinetic analysis of the RNAi enzyme complex. Nat. Struct. Mol. Biol. 11, 599–606 10.1038/nsmb780 (doi:10.1038/nsmb780) [DOI] [PubMed] [Google Scholar]
- 61.Endres D., Zlotnick A. 2002. Model-based analysis of assembly kinetics for virus capsids or other spherical polymers. Biophys. J. 83, 1217–1230 10.1016/S0006-3495(02)75245-4 (doi:10.1016/S0006-3495(02)75245-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rana T. M. 2007. Illuminating the silence: understanding the structure and function of small RNAs. Mol. Cell. Biol. 8, 23–36 [DOI] [PubMed] [Google Scholar]
- 63.Rajendran K. S., Nagy P. D. 2006. Kinetics and functional studies on interaction between the replicase proteins of Tomato bushy stunt virus: requirement of p33: p92 interaction for replicase assembly. Virology 345, 270–279 10.1016/j.virol.2005.09.038 (doi:10.1016/j.virol.2005.09.038) [DOI] [PubMed] [Google Scholar]
- 64.Scheper G. C., van Kollenburg Dagger B., Hu J., Luo Y., Goss D. J., Proud C. G. 2002. Phosphorylation of eukaryotic initiation factor 4E markedly reduces its affinity for capped mRNA. J. Biol. Chem. 277, 3303–3309 10.1074/jbc.M103607200 (doi:10.1074/jbc.M103607200) [DOI] [PubMed] [Google Scholar]
- 65.Lakatos L., et al. 2006. Small RNA binding is a common strategy to suppress RNA silencing by several viral suppressors. EMBO J. 25, 2768–2780 10.1038/sj.emboj.7601164 (doi:10.1038/sj.emboj.7601164) [DOI] [PMC free article] [PubMed] [Google Scholar]