

Abstract
Background
Chemoresistance is the major factor limiting long-term treatment success in patients with epithelial ovarian cancers. Most cytotoxic drugs kill cells through apoptosis; therefore defective execution of apoptotic pathways results in a drug resistant phenotype in many tumor types.
Methods
A panel of 24 ovarian cancer cell lines was screened for expression and function of the apoptosome components Apaf-1 and caspase-9. Expression levels were analyzed by immunohistochemistry and immunoblotting; Apaf-1 function was determined by assessing the ability of endogenous Apaf-1 to cleave caspase-9 in the presence or absence of cytochrome c. The effect of the histone deacetylase inhibitor (HDACi) trichostatin A (TSA) on Apaf-1 expression and function was evaluated.
Results
We report here that the resistance of ovarian cancer cells to the pro-apoptotic effects of chemotherapy is due in part to deficient Apaf-1 activity. Although Apaf-1 is expressed in most ovarian cancers, the functional activity is impaired as Apaf-1 has a diminished ability to recruit and activate caspase-9. Treatment of ovarian cancer cells with TSA results in restoration of Apaf-1 function independent of alterations in Apaf-1 expression. Furthermore, treating chemoresistant cells with sublethal doses of TSA restores Apaf-1 function and sensitizes cells to cisplatin induced apoptosis.
Conclusions
Targeting intrinsic pathway defects for therapeutic intervention may result in sensitizing tumors to standard chemotherapy or triggering apoptosis in the absence of other apoptotic signals. The identification of drugs that can use Apaf-1 when it is present, yet can overcome its functional inactivation, may be an important clinical advance.
Keywords: ovarian cancer, ovarian carcinoma, apoptosome, Apaf-1, HDACi, chemoresistance, TSA
Background
Epithelial ovarian carcinoma is the leading cause of death among patients with gynecological cancers in the United States. It is the fifth most frequent cause of cancer death in women, and more than 15,000 women die of this disease annually. Although the majority of tumors initially respond to chemotherapy, most patients succumb to chemoresistant, recurrent disease. Despite improved treatment success for many malignancies during the recent era in clinical oncology, the 5-year survival for patients with advanced stage ovarian cancer remains less than 50% (1).
Chemoresistance is the principal factor limiting long-term treatment success in ovarian carcinoma patients. Chemoresistance has been associated with the failure of chemotherapy drugs to induce apoptosis. Chemotherapeutic agents, such as cisplatin induce apoptosis by engaging multiple signaling pathways that converge on the mitochondria to cause release of cytochrome c. In the presence of dATP/ATP, cytochrome c binds to Apaf-1, which then oligomerizes and binds procaspase-9. The cytochrome c, Apaf-1, caspase-9 complex (apoptosome) enables enzymatic self-activation of caspase-9 that subsequently activates pro-caspase-3, ultimately resulting in apoptotic cell death (2).
Previously, we demonstrated that the majority of primary ovarian tumors and chemoresistant ovarian carcinoma cell lines express detectable levels of the three proteins that constitute the apoptosome, but appear to have deficient apoptosome activity. Moreover, apoptosome dysfunction was associated with a chemoresistance in ovarian carcinoma in vitro, leading us to hypothesize that it contributes to the chemoresistant phenotype associated with ovarian carcinoma clinically (3).
In this study, we demonstrate that dysfunctional apoptosome activation in ovarian carcinoma is due to deficient Apaf-1 function. Although endogenous Apaf-1 expression levels are not predictive of apoptotic response to chemotherapy, exogenous Apaf-1 restored apoptosome function in in vitro assays. Formation of the apoptosome in chemoresistant ovarian carcinoma cells is impaired by diminished binding between Apaf-1 and pro-caspase-9. Of potential therapeutic importance, we show that treating ovarian carcinoma cells with a histone deacetylase inhibitor (HDACi), trichostatin A (TSA) increases both the expression and activity of Apaf-1. HDACi can affect gene expression as well as the functional properties of a variety of non-histone proteins by regulating the balance of acetylated protein residues (4). TSA treatment was found to sensitize chemoresistant ovarian carcinoma cells to cisplatin, independently triggered apoptosis, and resulted in enhanced binding between Apaf-1 and caspase-9. Furthermore, we found that TSA treatment resulted in increased Apaf-1 activity independent of alterations in Apaf-1 expression. Together, these results identify Apaf-1 dysfunction as a specific cause of chemoresistance in ovarian carcinoma, and provide initial evidence that the pharmacodynamic response to TSA specifically overcomes this mechanism of chemoresistance.
Materials and Methods
Chemicals and Reagents
Trichostatin A (5) was obtained from Sigma-Aldrich Chemical Co (St. Louis, MO). Cisplatin was obtained from Ben Venue Labs, Inc. (Bedford,OH).
Cell lines and tumor samples
Normal ovarian surface epithelium (OSE) cells were harvested from fresh normal human ovarian surgical specimens and cultured in medium (M199:MCDB105 (1:1) with 10% FBS). Wild type murine embryonic fibroblasts (MEF) and MEF Apaf-1 −/− cells were a generous gift from Dr. M. Soengas (University of Michigan). The remaining ovarian carcinoma cell lines were obtained from Dr. K. Cho (University of Michigan).
Tissue microarrays (TMAs) were constructed using 302 cores from 86 patients with epithelial ovarian carcinoma and 25 cores from benign ovarian samples. Tumors were histopathologically classified according to the International Federation of Gynecology and Obstetrics (FIGO) criteria. The histology of tumors in the ovarian carcinoma microarray included papillary serous (52%), endometriod (9%), clear cell (9%), undifferentiated (3%) and mixed histology (27%). Clinicopathologic and demographic data was collected from medical records under an IRB-approved protocol (IRBMED # 2004-0814).
All hematoxylin and eosin stained slides of the tumors were reviewed, and areas of tumor were identified. 2-3 tissue cores per tumor were taken from spatially separate areas in a single donor block from each case using a tissue microarrayer (Chemicon Advanced Tissue Arrayer). Cores were arrayed into a recipient block at predetermined coordinates. The H&E stained sections from donor and recipient paraffin blocks were used to confirm the area of tumor from which cores were retrieved.
For immunohistochemistry, 5 μM thick sections were cut from the respective arrays, de-paraffinized, and dehydrated. Immunohistochemical staining for Apaf-1 was performed using a streptavidin peroxidase procedure. Antigen bound primary antibody was detected using standard avidin-biotin immunoperoxidase complex (Dako).
Cytoplasmic expression of Apaf-1 in epithelial ovarian carcinoma cells was determined quantitatively by evaluating the proportion of positive tumor cells over total tumor cells, independent of intensity as previously described (6, 7). Positivity was scored as follows: negative (score = 1, no visible staining); weak (score = 2, < 25% of cells staining); moderate (score = 3, 25-75% of cells staining); and strong (score = 4, >75% of cells staining). All scoring was performed by a single observer. The association of Apaf-1 with response to platinum based chemotherapy (interval between completion of adjuvant chemotherapy and date of first recurrence) was evaluated using two-sample t tests. All analyses were carried out using SAS (Version 9.1, The SAS Institute, NC, USA).
Flow cytometry analysis
Cells were stained with propidium iodide, and percentage of apoptotic cells was determined as described previously (8). Analysis was performed using Lysis II software on a FACScan flow cytometer (Beckton Dickinson, Mountain View, CA).
Immunoblotting and Co-immunoprecipitation
For the preparation of S-100 cytosolic lysates, cells were lysed in Buffer A isotonic lysis buffer as previously described (3). After incubation with secondary antibody, the reaction was developed by enhanced chemiluminescence using the Luminol Reagent kit, (Santa Cruz, Santa Cruz, CA) and exposed to film (Denville Scientific, Metuchen, NJ). Coimmunoprecipitation of endogenous pro-caspase-9 was performed using S100 cytosolic lysates. Monoclonal caspase 9 and Apaf-1 antibodies were obtained from Stressgen (Victoria, Canada) and Travigen (Gaithersburg, MD) respectively.
In Vitro Caspase-9 and Caspase-3 Assay
Cytosolic extracts were prepared from cell lines as described above. The in vitro caspase-9 and caspase-3 assays were performed as described previously (3). Reactions were stopped with 5 × SDS loading buffer, and proteins were resolved on polyacrylamide/SDS gels. Gels were dried and exposed for autoradiography.
Results
Apaf-1 expression levels are not predictive of apoptosome function
The development of resistance to chemotherapy induced apoptosis limits our ability to treat patients with advanced or recurrent ovarian carcinoma. Apoptotic stimuli such as cisplatin stimulates mitochondrial release of cytochrome c, which then complexes with the cytosolic factor Apaf-1. Apaf-1 then binds and activates caspases -9 and -3, ultimately leading to apoptotic cell death. We hypothesized that expression levels of Apaf-1 would be predictive of clinical response to cisplatin in ovarian carcinoma patients. We found that Apaf-1 expression was high in the majority of ovarian tumors analyzed by immunohistochemistry (87 tumors, representative data shown in Figure 1A); however, expression levels did not correlate with response to cisplatin based chemotherapy clinically (data not shown). All patients underwent staging surgery followed by adjuvant cisplatin based chemotherapy. Response to cisplatin was determined by measuring the interval between completion of adjuvant chemotherapy and date of first recurrence (disease free interval). Recurrence was defined as a rise in Ca-125 and/or measurable disease prompting re-initiation of chemotherapy. Patients were placed into two categories: platinum sensitive (disease free interval > 6 months) or platinum resistant (disease free interval < 6 months). Apaf-1 expression levels were uniformly high (3-4+) among platinum sensitive and platinum resistant patients.
Figure 1. Apaf-1 expression is not predictive of response to cisplatin.
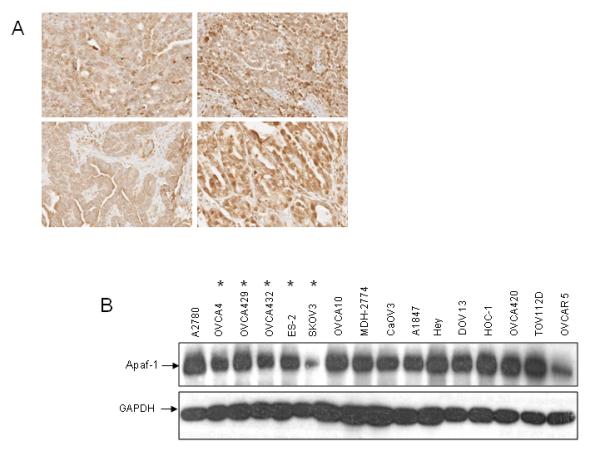
A. Tissue microarrays containing cores from 87 epithelial ovarian tumors were analyzed for Apaf-1 expression with immunohistochemistry. Representative samples shown. B. Immunoblot analysis of cytosolic Apaf-1 expression in ovarian cancer cell lines. The level of Apaf-1 in a panel of ovarian carcinoma cell lines was determined by immunoblotting.
Chemoresistant cell lines are indicated *. C. Representative ovarian carcinoma cell lines were treated with DMSO or cisplatin (5 μg/ml) for 24 or 48 hours. The percent of apoptotic cells was determined by propidium iodide staining and subG0 analysis. Values are expressed as mean±SD (n= 3). D. A. Cytosolic extracts from chemosensitive (A2780) and chemoresistant (OVCA 4, OVCA 429, OVCA 432, ES-2, and SKOV3) cell lines were incubated for 30 minutes in the presence or absence of cytochrome c and dATP. Extracts were fractionated on a Superdex 200 HR column. Equal amounts (50 μl) of each fraction were resolved by SDS PAGE and Apaf-1 was detected by immunoblotting. The elution profiles and sizes of selected standards are indicated.
Next, we screened a panel of ovarian carcinoma cell lines for Apaf-1 expression by immunoblotting (Figure 1B). With the exception of two cell lines (SKOV3 and OVCAR 5), all cell lines had similar expression levels of Apaf-1. Six representative cell lines were incubated with cisplatin, and apoptotic response was assessed (Figure 1 C). Consistent with the data in human tumor samples, expression levels of Apaf-1 did not correlate with response to cisplatin in vitro.
Because total expression levels of Apaf-1 were not predictive of apoptotic response to cisplatin, we hypothesized that Apaf-1 function was altered in the chemoresistant ovarian carcinoma cells. We have previously demonstrated that Apaf-1 multimerization in vitro in response to cytochrome c/dATP was equivalent between chemoresistant SKOV3 and chemosensitive A2780 cells (3). We re-evaluated Apaf-1 multimerization in 4 additional chemoresistant cell lines (OVCA4, OVCA429, OVCA432, ES2) by separating Apaf-1 monomers and oligomers following cytochrome c/dATP activation using gel filtration chromatography. The results shown in Figure 1D demonstrate that in all chemoresistant cells, Apaf-1 formed an oligomer in a cytochrome c/dATP dependent manner, similar to the chemosensitive cells (A2780). As expected, in the absence of cytochrome c/dATP, Apaf-1 eluted in monomeric form in all cell lines (Figure 1D and data not shown).
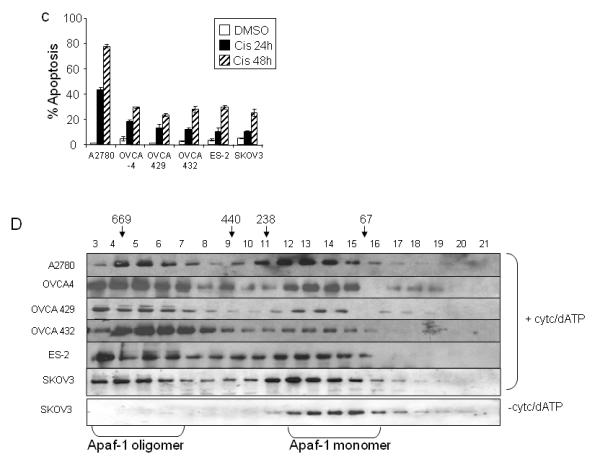
Apaf-1-caspase-9 binding is abnormal in cells with dysfunctional apoptosome activity
While Apaf-1 expression levels and ability to form oligomers is similar in chemoresistant and chemosensitive ovarian cancer cell lines, it is not known whether Apaf-1-caspase-9 binding also occurs with equal efficiency in these cell lines. Cytosolic extracts from chemosensitive (A2780) and chemoresistant (OVCA4, OVCA429, OVCA432, ES-2, SKOV3) cells were activated with cytochrome c/dATP, and binding interaction between caspase-9 and Apaf-1 was determined. After immunoprecipitating caspase-9 from cytosolic extracts, the resulting immune complexes were resolved and blotted to detect co-immunoprecipated Apaf-1. All cell lines expressed similar endogenous levels of Apaf-1 with the exception of SKOV3 (Figure 1B). Compared to the chemosensitive A2780 cell line, reduced levels of Apaf-1 were co-immunprecipitated with caspase-9 in each of the chemoresistant cell line extracts (Figure 2A). Although expression levels of immunoprecipitated pro-caspase-9 are similar across all cell lines, there are reduced levels of activated, or cleaved caspase-9 in the chemoresistant cells as compared to A2780. The absence of a reliable antibody to immnoprecipitate Apaf-1 precluded our ability to test Apaf-1/caspase-9 binding in the reverse direction. Nevertheless, these results suggest that in chemoresistant ovarian carcinoma cell lines, binding of Apaf-1 to pro-caspase-9 is impaired.
Figure 2. Apaf-1-caspase-9 binding is abnormal in cells with dysfunctional apoptosome activity.
A. Cytosolic extracts from cell lines with functional (A2780) and dysfunctional (OVCA 4, OVCA 429, OVCA 432, ES-2, and SKOV3) apoptosome activity were incubated in the presence or absence of cytochrome c and dATP. Lysates were immunoprecipitated with an anti-caspase-9 antibody, and immunoblotting was performed for Apaf-1 and caspase-9. The anti-caspase 9 antibody is also immunoreactive for the pro-caspase 9 cleavage products, p37/35. B. Cytosolic lysates from ovarian cancer cell lines and normal ovarian epithelial cells (OSE) were incubated with in vitro translated 35S-pro-caspase-9 in the presence or absence of cytochrome c and dATP as indicated. After 30 minutes of incubation, 35S-pro-caspase-9 and its cleavage products p37/35, were separated by SDS-PAGE. Gels were dried and exposed for autoradiography.
To further evaluate Apaf-1 function, we prepared cytosolic extracts from 16 ovarian cancer cell lines and tested the ability of endogenous Apaf-1 to activate procaspase-9 in response to cytochrome c in vitro (Figure 2B). Apaf-1 function was assayed by determining the cleavage of in vitro translated, [35S] labeled pro-caspase-9 (Figure 2B) and pro-caspase-3 (data not shown) into the proteolytic fragments p37/35 and p19/17 respectively, in the presence of cytochrome c and dATP. We identified six cell lines (OVCA 429, OVCA 432, ES-2, OVCA-4, OVCA 10 and SKOV3) that demonstrated diminished ability to activate and cleave pro-caspase-9 in response to cytochrome c/dATP (Figure 2B). Consistent with the inability of the six cell lines to cleave pro-caspase-9 in response to cytochrome c/dATP, these cells also display diminished Apaf-1/caspase-9 binding (Figure 2A) and are resistant to cisplatin induced apoptosis (Figure 1C).
Caspase-9 function is normal in ovarian carcinoma cells
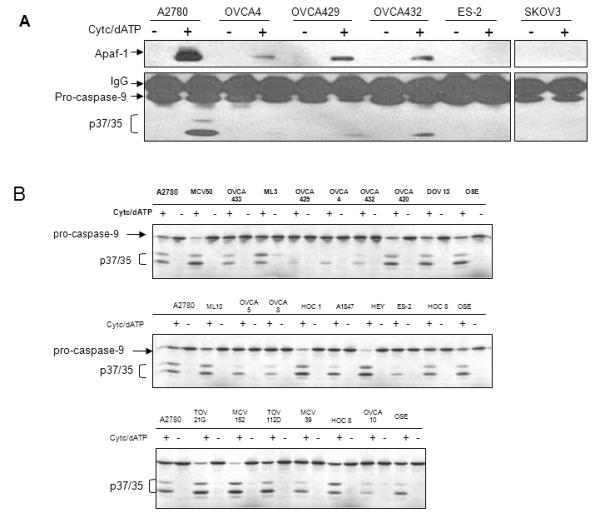
Reduced binding of Apaf-1 with caspase-9 in chemoresistant cells is not associated with reduced levels of Apaf-1 or caspase-9. This suggests that Apaf-1 or caspase-9 in these cells is dysfunctional, or that these cells contain inhibitors that suppress the interaction between Apaf-1 and caspase-9, and potentially the activation of caspase-9. To test caspase-9 function in chemoresistant ovarian cancer cells, we immunodepleted caspase-9 from the chemosensitive cell line A2780 (A2780-IPC9). In these assays, cleavage of [35S]-procaspase-3 was used as a marker of apoptosome activity. Pro-caspase-3 is a substrate of activated caspase-9 and its cleavage in response to cytochrome c and dATP is exclusively dependent on caspase-9. As expected, A2780 lysates immunodepleted of caspase-9, as well as chemoresistant OVCA 429, and SKOV3 lysates were unable to activate and cleave in vitro translated [35S]-pro-caspase-3 (Figure 3A, lanes 1-2, 5-6, and 11-12 respectively). We next determined that cytochrome c/dATP-dependent apoptosome activity could be completely restored in A2780-IPC9 lysates with the addition of lysates from A2780, OVCA 429, and SKOV3 (Figure 3A, lanes 7-8, 9-10 and 13-14 as compared to lanes 1-2). These results show that procaspase-9 is equivalent in chemosensitive (A2780) and chemoresistant (OVCA 429, and SKOV3) cells, and suggest that Apaf-1 function is altered in the chemoresistant cell lines.
Figure 3. Caspase-9 function is normal and Apaf-1 function is abnormal in chemoresistant cells.
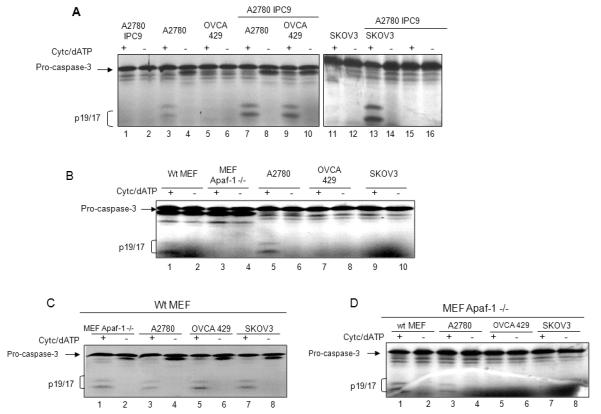
A. S100 cytosolic lysates from A2780 ovarian cancer cells immunodepleted of caspase-9 (A2780 IP C9), A2780, OVCA 429, and SKOV3 cells alone, or mixed in a 1:1 ratio as indicated. Caspase-9 function was assayed by incubating extracts with in vitro translated [35S] pro-caspase-3 in the presence or absence of cytochrome c/dATP. After 30 minutes of incubation, 35S-pro-caspase-3 and its cleavage products, p19/17, were separated by SDS-PAGE. Gels were dried and exposed for autoradiography. B. S100 cytosolic lysates from wt MEF, MEF Apaf-1 −/−, A2780, OVCA 429, or SKOV3 cells were prepared. Apoptosome function was assayed as indicated in (A). C. Cytosolic lysates from wt MEF were mixed in a 1: 1 ratio with MEF Apaf-1 −/−, A2780, OVCA 429, or SKOV3 cells, and apoptosome function was tested as indicated in (A). D. Cytosolic lysates from MEF Apaf-1 −/− were mixed in a 1: 1 ratio with wt MEF, A2780, OVCA 429, or SKOV3 cells, and apoptosome function was tested as indicated in (A).
Apaf-1 function is impaired in ovarian carcinoma
To test Apaf-1 function in the chemoresistant ovarian cell lines, we used cytosolic lysates made from mouse embryonic fibroblasts containing wild-type Apaf-1 (wt MEF), or that were Apaf-1 null (MEF Apaf-1 −/−). As expected, cytosolic extracts from wt MEF, and A2780 cells could be induced to cleave in vitro translated [35S] pro-caspase-3 by adding cytochrome c and dATP (Figure 3B, lanes 1-2 and 5-6), while extracts from MEF Apaf-1 −/−, OVCA 429, and SKOV3 were unable to activate and cleave in vitro translated [35S]-pro-caspase-3 (Figure 3B, lanes 3-4, 7-8, and 9-10 respectively). The addition of wt MEF cytosolic extracts to MEF Apaf-1 −/−, OVCA 429 and SKOV3 extracts completely restored apoptosome activity (Figure 3C, lanes 1-2, 5-6, and 7-8 respectively. Lysates from wt MEF or A2780 cells could restore apoptosome function in MEF Apaf-1 −/− cells (Figure 3D, lanes 1-2, 3-4). However, lysates from chemoresistant cells OVCA 429 and SKOV3 did not restore apoptosome function in MEF Apaf-1 −/− extracts (Figure 3D, lanes 5-6, and 7-8 respectively). These results show that Apaf-1 in OVCA 429 and SKOV3 extracts could not complement the specific deficiency in Apaf-1 in the MEF Apaf-1 −/− cells, indicating that the Apaf-1 in these cells is not functional. Although lower levels of Apaf-1 in SKOV3 cells can potentially explain the findings with these cells, normal levels of Apaf-1 in OVCA429 cells suggest that Apaf-1 activity is either inhibited due to post-translational modifications or by the presence of an inhibitor.
An alternative interpretation of the results shown in Figure 3 is that addition of wt MEF lysates to lysates prepared from chemoresistant cells (OVCA 429 and SKOV3) resulted in dilution of inhibitory factors, thus preventing activation of the apoptosome. To rule out the possibility of an inhibitor against Apaf-1 or Caspase-9 in the chemoresistant cells, we tested cytochrome c/dATP-induced apoptosome function in lystates after supplementing with purified, functional recombinant Apaf-1 (rApaf-1). Cytosolic lysates prepared from MEF Apaf-1 −/− cells required the addition of 3.5-7.5 nanomoles of recombinant Apaf-1 to reconstitute cytochrome c / dATP- triggered caspase-3 cleavage (Figure 4A). We found that adding this same amount of rApaf-1 restored apoptosome activation in lysates from SKOV3 (Figure 4B), OVCA4, OVCA429, OVCA432, and ES2 cells (Figure 4C), indicated by caspase-3 processing in response to cytochrome c and dATP. These results argue against the presence of a soluble inhibitor that interferes with caspase-9-Apaf-1 binding or apoptosome function in the chemoresistant cells. Thus, although a relative deficiency of Apaf-1 may be important for chemoresistance in certain ovarian cells (e.g. SKOV3), for other chemoresistant cells with normal Apaf-1 expression levels, our data are most consistent with Apaf-1 function being disrupted by post-translational protein modifications.
Figure 4. Exogenous Apaf-1 restores apoptosome function in ovarian cancer cells.
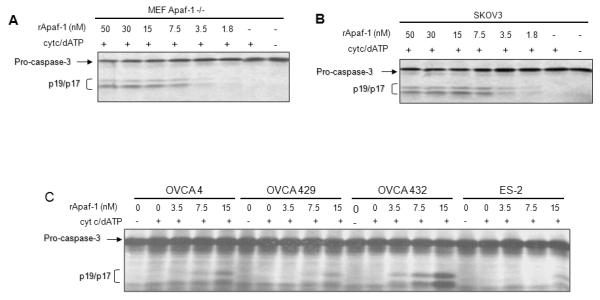
Cytosolic S100 lysates from MEF Apaf-1 −/− (A), SKOV3 (B), OVCA 4, OVCA 429, OVCA 432, and ES-2 cells (C) were incubated with various amounts of purified recombinant human Apaf-1 as indicated, and apoptosome function was tested by incubating with cytochrome c/dATP and in vitro translated [35S] pro-caspase-3. After 30 minutes of incubation, [35S] pro-caspase-3 and its cleavage products, p19/17, were separated by SDS-PAGE. Gels were dried and exposed for autoradiography.
HDACi modulates Apaf-1 expression
Apaf-1 expression has recently been shown to be induced by the histone deacetylase inhibitor (HDACi ) suberoylanilide hydroxamic acid (SAHA) in CEM cells, a human lymphoid cell line (9). Other investigators found that SAHA or TSA treatment did not result in increased Apaf-1 levels in melanoma cells, suggesting that HDACi effects on apoptotic factors may be cell type specific (10). We first evaluated apoptotic response to the HDACi TSA in several representative ovarian carcinoma cell lines. As shown in Figure 5A, TSA induced apoptosis in 60-75% of cells after 48 hours of incubation in both the chemosensitive (A2780) and chemoresistant (SKOV3 and OVCA 4) cells, as compared to untransformed ovarian surface epithelial cells (OSE) which remained viable.
Figure 5. TSA induces apoptosome activation in ovarian carcinoma cells.
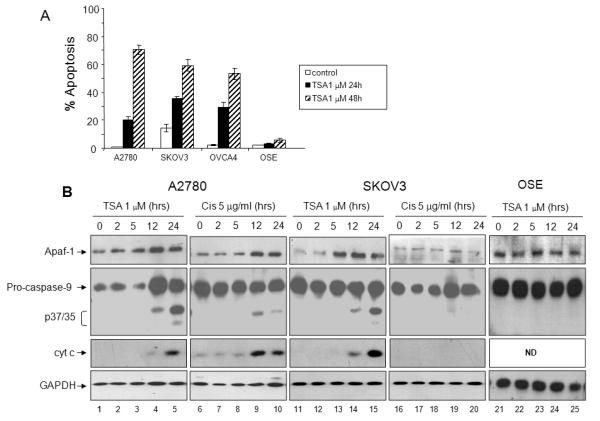
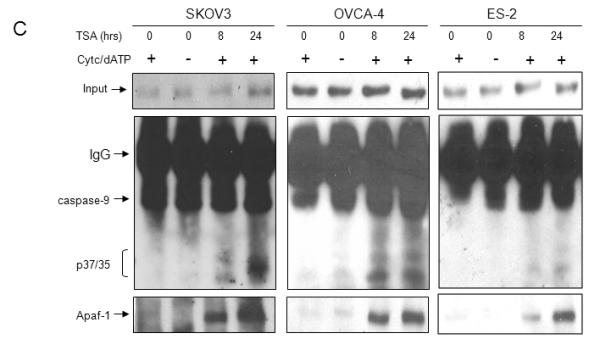
A. Ovarian cancer cells (A2780, OVCA4, SKOV3) and OSE cells were treated with TSA (1 μM) for 24 or 48 hours as indicated. Percentage of apoptotic cells after treatment was determined by propidium iodide staining and subGo analysis. Values are expressed as mean ± SD (N=3). B. Equal amounts of cytosolic extracts from A2780, SKOV3, and OSE cells were incubated with TSA (1 μM) or cisplatin (5 μg/ml) (for A2780 and SKOV3 cells) for 0-24 hours as indicated. Expression of cytoplasmic Apaf-1, caspase-9, and cytochrome c was determined by immunoblotting. C. Representative ovarian cancer cell lines with dysfunctional Apaf-1 activity (SKOV3, OVCA4, ES-2) were treated with vehicle control (DMSO) or TSA. Cytosolic S100 lysates were prepared, and normalized for Apaf-1 expression levels (40 μg protein for DMSO treated cells and 20 μg protein for TSA treated cells). Lysates were immunoprecipitated using anti-caspase-9 antibody, followed by immunoblotting with anti-Apaf-1 and anti-caspase-9.
Because the low levels of Apaf-1 expression in SKOV3 cells are potentially the cause of chemoresistance in this cell line, we tested whether TSA increases Apaf-1 expression and restores apoptosome function in SKOV3 cells. We also examined the response of the chemosensitive cell line A2780 to treatment with TSA. TSA (1 μM) treatment resulted in increased Apaf-1 expression levels in both A2780 and SKOV3 cells within 5 hours of exposure (Figure 5B, lanes 1-5 and 11-15, top panel, respectively), whereas Apaf-1 expression following TSA treatment remained unchanged in non-transformed ovarian surface epithelial cells (Figure 5B, lanes 21-25, top panel). Concurrent with the increase in Apaf-1 expression, pro-caspase-9 expression increased in the A2780 cell line, proteolytic fragments of processed caspase-9 (p37/35) appeared, and cytochrome c was released into the cytoplasm in both the A2780 and SKOV3 cell lines, but not in normal ovarian epithelial cells (Figure 5B, lanes 1-5, 11-15, and 21-25 respectively). Thus, TSA not only increased Apaf-1 expression but also triggered functional activation of caspase-9 in ovarian cancer cells. Surprisingly, in the chemosensitive A2780 cell line, Apaf-1 expression also increased following treatment with cisplatin after 12 hours of treatment, as compared to the chemoresistant SKOV3 cell line, where Apaf-1 expression remained unchanged following cisplatin treatment (Figure 5A, lanes 6-10 and 16-20 respectively). As expected, cisplatin treatment also resulted in activation of pro-caspase-9, and release of cytochrome c into the cytoplasm in A2780 cells, and not in the chemoresistant SKOV3 cells (Figure 5B, lanes 6-10 and 16-20 respectively).
TSA enhances Apaf-1 activity
We next evaluated the effect of TSA treatment on Apaf-1/caspase-9 binding. Representative ovarian cancer cell lines with dysfunctional apoptosomes (SKOV3, OVCA4, ES-2) were treated with vehicle control (DMSO) or TSA. Cytosolic lysates were prepared, and input was normalized for Apaf-1 expression (Figure 5C, top panel). Cytosolic expression of Apaf-1 was equivalent in 40 μg of DMSO treated cell lysates as compared to 20 μg of TSA treated cell lysates. Lysates were imunoprecipitated with anti-caspase-9 antibody, followed by immunoblotting for Apaf-1 (lower panel) and caspase-9 (middle panel). As shown in Figure 5C, the ability of Apaf-1 to bind caspase-9 was markedly enhanced in TSA treated cells as compared to DMSO treated cells. Likewise, after controlling for Apaf-1 expression levels, TSA treated cells exhibited activation and cleavage of pro-caspase-9 as compared to DMSO treated cells, which could not activate/cleave pro-caspase-9, even in the presence of exogenous cytochrome c/dATP.
We next sought to confirm the effect of TSA on Apaf-1 activity independent of its effect on Apaf-1 or caspase-9 expression. First, cells with similar endogenous expression levels of Apaf-1 but divergent Apaf-1 function were treated with TSA (1μM) for 0, 12 and 24 hours. As expected, in the chemosensitive A2780 cell line, Apaf-1 expression increased, and pro-caspase-9 was activated/cleaved (Figure 6A, lanes 1-3). In the chemoresistant ES-2 cell line, Apaf-1 expression increased slightly, and pro-caspase-9 was also activated/cleaved (Figure 6B, lanes 4-6). Both cell lines were immunodepleted of caspase-9. As expected, A2780-IP-C9 and ES-2-IPC9 cells were devoid of caspase-9 and remained viable following TSA treatment (A2780-IPC9, Figure 6B, lanes 7-9 and ES-2-IPC9 lanes 10-12 respectively). Next, lysates from TSA treated A2780-IP-C9 and ES-2-IP-C9 cells were normalized for Apaf-1 expression and were incubated with in vitro translated caspase-9. Apaf-1 function was determined by adding cytochrome c/dATP and assessing caspase-9 activation/cleavage. As shown in Figure 6B, TSA enhanced Apaf-1 activity independent of increasing Apaf-1/caspase-9 expression levels, manifested in increased cleavage of in vitro translated pro-caspase-9 cleavage in the presence of cytochrome c/dATP.
Figure 6. TSA treatment increases Apaf-1 activity.
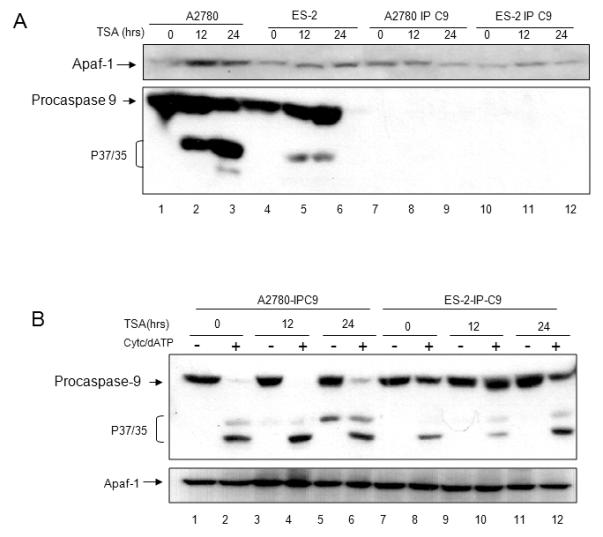
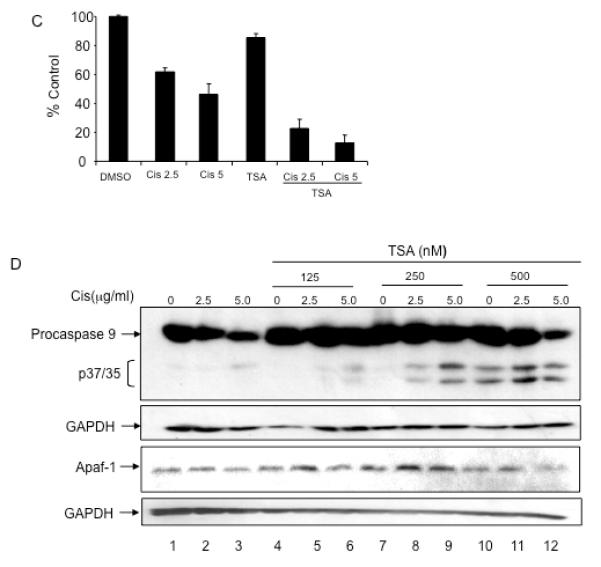
A. A2780 and ES-2 cells were treated with TSA (0.5 μM) for 0, 12, and 24 hours. Cellular lysates were immunodepleted of caspase-9 (A2780-IP-C9, ES-2-IP-C9). Apaf-1 and caspase-9 expression levels were determined by immunoblotting. B. A2780-IP-C9 and ES-2-IP-C9 lysates were normalized for Apaf-1 expression (bottom panel). Lysates were incubated with in vitro translated pro-caspase-9 in the presence or absence of cytochrome c/dATP. Apaf-1 activity was determined by assessing caspase-9 cleavage by immunoblotting. C. ES-2 cells were pre-incubated with TSA at indicated concentrations for 12 hours, followed by a 24 hour incubation with cisplatin. Viability was assessed using Sulforhodamine B assay. All samples were prepared in triplicate, and data are presented as means +/− S.D. Representative data of three independent experiments are shown. D. Caspase-9 activation was determined by immunoblotting. Apaf-1 expression levels remained stable in these conditions.
Because TSA treatment increased Apaf-1 activity in apoptosome deficient cells, independent of its effect on Apaf-1 and/or caspase-9 expression, we reasoned that pre-treating chemoresistant cells with TSA may sensitize cells to cisplatin induced apoptosis. Representative ES-2 cells were pre-treated with a sublethal dose of TSA (250 nM) for 12 hours, followed by treatment with cisplatin. Pre-treatment with TSA enhanced cisplatin induced cell death (Figure 6C). Cisplatin treatment alone and sublethal doses of TSA (125 and 250 nM) alone did not induce activation/cleavage of pro-caspase-9 (Figure 6D, lanes 1-3, 4, and 7). Pre-incubating cells with TSA for 12 hours followed by cisplatin for 24 hours resulted in increased activation and cleavage of pro-caspase-9 (Figure 6D, lanes 5-6, 8-9, and 11-12) as compared to cisplatin treatment alone (Figure 6D, lanes 2-3). Pre-incubating cells with sublethal doses of TSA did not affect Apaf-1 expression (Figure 6D, bottom panel). These data suggest that TSA enhances tumor cell sensitivity to cisplatin induced apoptosis in chemoresistant ovarian cancer cells.
Discussion
The development of chemoresistance is the major factor limiting long term treatment success for patients with epithelial ovarian cancer. However, the molecular mechanisms underlying chemoresistance are poorly understood. Recent data suggest that dysregulation of apoptosis is a key contributor to chemoresistance. We previously showed that the majority of primary ovarian tumors and chemoresistant ovarian cell lines are deficient in Apaf-1 activity, suggesting that Apaf-1 dysfunction may contribute toward the aggressive, chemoresistant phenotype associated with this disease (3).
Inactivation of Apaf-1 and subsequently chemoresistance has been attributed to several factors in other tumor types. Apaf-1 gene methylation and loss of expression in melanoma and acute myeloblastic leukemia have been associated with resistance to chemotherapy induced apoptosis (11, 12). Low expression levels of Apaf-1 have also been described in Burkitt lymphomas (BL), where the bulk of Apaf-1 was found to associate with discrete domains in the plasma membrane. Disruption of lipid rafts sensitized BL cells to apoptosis induced by chemotherapy, suggesting that ectopic (noncytosolic) localization of Apaf-1 may play a role in chemoresistance in B lymphoma (13). Overexpression of Apaf-1 in melanoma (11), leukemia (13), and cervical (14) cancer cells resulted in activation of caspase-9 indicating that apoptosome function was restored. Furthermore, expression of Apaf-1 was recently identified as a predictive marker of response to preoperative radiation therapy in patients with rectal carcinoma (6).
We have shown here that Apaf-1 expression does not correlate with apoptosome activity or response to cisplatin based chemotherapy in ovarian carcinoma. Despite relatively high expression levels of Apaf-1, several chemoresistant ovarian carcinoma cell lines fail to assemble functional apoptosomes. We have previously demonstrated the lack of Apaf-1 mutations or expression of non-functional isoforms of Apaf-1 in ovarian carcinoma (3). Other possibilities that may contribute towards Apaf-1 inactivation include the presence of an Apaf-1 inhibitor, or the lack of an Apaf-1 enhancer in ovarian carcinoma cells. However, because we could restore apoptosome function in ovarian carcinoma extracts by mixing them with extracts prepared from cells with wt Apaf-1, our data suggest the absence of an Apaf-1 inhibitor. The fact that apoptosome function was restored following addition of purified recombinant human Apaf-1 to ovarian carcinoma extracts also suggests that an Apaf-1 enhancer is not required to stimulate apoptosome function in ovarian carcinoma cells.
Because recent data suggest that several HDACi in the hydroxamic acid class can regulate expression of apoptotic genes (9, 15), we next investigated the possibility that TSA could alter Apaf-1 expression and function. Incubation of ovarian carcinoma cells with TSA resulted in an increase in Apaf-1 expression along with activation and cleavage of pro-caspase-9. Unexpectedly, incubation of the chemosensitive cell line A2780 with cisplatin also resulted in increased Apaf-1 expression, whereas Apaf-1 expression in SKOV3 cells remained unchanged. Cell lysates used for these experiments were prepared from cytosolic extracts to avoid contamination with endogenous cytochrome c. Therefore, it is possible that the increase in Apaf-1 expression seen following cisplatin and TSA treatment in A2780 cells is due to relocalization of Apaf-1 to the cytoplasmic compartment. However, the fact that cytoplasmic expression of Apaf-1 is not predictive of apoptosome function in the chemoresistant cell lines argues against the theory that ectopic (noncytosolic) localization of Apaf-1 may play a role in chemoresistance in ovarian cancer. Although treatment with TSA resulted in increased Apaf-1 expression in ovarian carcinoma cell lines, this finding does not preclude the possibility that TSA treatment also alters the function of Apaf-1. TSA induced growth inhibition and cytotoxicity may be attributed to acetylation of both histone and non-histone proteins. Thus, it is possible that TSA treatment results in post-translational modifications of apoptosome factors such as Apaf-1. Our data suggests that TSA treatment enhances the ability of Apaf-1 to bind pro-casepase-9, independent of increasing Apaf-1 expression levels. Furthermore, TSA treatment results in increased caspase-9 activation independent of its effects on Apaf-1 and/or caspase-9 expression. Preliminary experiments indicate that recombinant human Apaf-1 is acetylated in an in vitro acetylation assay using purified histone acetyltransferases, CBP and p300 (data not shown). Experiments are currently underway to determine the functional significance of Apaf-1 acetylation in vivo. Other groups have also shown that normal, untransformed cells have a strikingly reduced sensitivity to HDACi induced cell death as compared to tumor cells (16). Differences in the acetylome in normal versus tumor cells may account for differential response to HDACi, and Apaf-1 may play an important role in modulating response to treatment.
HDACi from multiple classes have clinical activity in several tumor types (4). HDACi can increase the expression and/or activity of proteins that directly transmit an apoptotic signal through death receptor pathways such as death receptors, death receptor ligands, and downstream caspases (caspase-8 and -3), and can downregulate proteins that negatively regulate death-receptor signaling (i.e., c-FLIP, XIAP, survivin, c-IAP1/2)(17). The mechanisms through which HDACi activates the intrinsic apoptotic pathway is not entirely clear. There is some evidence that HDACi treatment results in alterations in gene expression of pro-apoptotic (BH3-only genes) and anti-apoptotic (pro-survival Bcl-2 family genes), favoring an overall apoptotic response(18). There is also some evidence that HDACi can mediate post-translational modification of pro-apoptotic proteins such as the cleavage and activation of Bid, resulting in activation of the intrinsic pathway(19). Our work represents the first report of HDACi activation of the intrinsic pathway by enhancing Apaf-1 function.
Alterations in acetylation status can affect protein-protein interactions among non-histone substrates. Although HDACi likely have pleiotropic effects on non-histone substrates, it is possible that HDACi induced apoptosis may be associated with modulation of acetylation status of apoptosome factors. Modulation of Apaf-1 function using HDACi may be an effective means of overcoming chemoresistance in ovarian cancer.
Condensed abstract: Resistance of ovarian cancer cells to the pro-apoptotic effects of chemotherapy is due in part to deficient Apaf-1 activity. TSA restores Apaf-1 function independent of alterations in Apaf-1 expression.
Acknowledgments
Supported by the Ovarian Cancer Research Foundation and CA106868 (JRL).
Abbreviations
- (Apaf-1)
Apoptotic protease activating factor 1
- (HDACi)
histone deactylase inhibitor
- (MEF)
mouse embryonic fibroblasts
- (TMA)
tissue microarray
- (TSA)
trichostatin A
Footnotes
There are no financial disclosures from any of the authors.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Hajra KM, Liu JR. Apoptosome dysfunction in human cancer. Apoptosis. 2004;9:691–704. doi: 10.1023/B:APPT.0000045786.98031.1d. [DOI] [PubMed] [Google Scholar]
- 3.Liu JR, Opipari AW, Tan L, et al. Dysfunctional apoptosome activation in ovarian cancer: implications for chemoresistance. Cancer Res. 2002;62:924–31. [PubMed] [Google Scholar]
- 4.Garcia-Manero G, Issa JP. Histone deacetylase inhibitors: a review of their clinical status as antineoplastic agents. Cancer Invest. 2005;23:635–42. doi: 10.1080/07357900500283119. [DOI] [PubMed] [Google Scholar]
- 5.Ashburner BP, Westerheide SD, Baldwin AS., Jr. The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Molecular & Cellular Biology. 2001;21:7065–77. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zlobec I, Vuong T, Compton CC. The predictive value of apoptosis protease-activating factor 1 in rectal tumors treated with preoperative, high-dose-rate brachytherapy. Cancer. 2006;106:284–6. doi: 10.1002/cncr.21600. [DOI] [PubMed] [Google Scholar]
- 7.Zlobec I, Minoo P, Baker K, et al. Loss of APAF-1 expression is associated with tumour progression and adverse prognosis in colorectal cancer. Eur J Cancer. 2007;43:1101–7. doi: 10.1016/j.ejca.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 8.Merino R, Grillot DA, Simonian PL, et al. Modulation of anti-IgM-induced B cell apoptosis by Bcl-xL and CD40 in WEHI-231 cells. Dissociation from cell cycle arrest and dependence on the avidity of the antibody-IgM receptor interaction. J Immunol. 1995;155:3830–8. [PubMed] [Google Scholar]
- 9.Peart MJ, Smyth GK, van Laar RK, et al. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:3697–702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Facchetti F, Previdi S, Ballarini M, Minucci S, Perego P, La Porta CA. Modulation of pro- and anti-apoptotic factors in human melanoma cells exposed to histone deacetylase inhibitors. Apoptosis. 2004;9:573–82. doi: 10.1023/B:APPT.0000038036.31271.50. [DOI] [PubMed] [Google Scholar]
- 11.Soengas MS, Capodieci P, Polsky D, et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature. 2001;409:207–11. doi: 10.1038/35051606. [DOI] [PubMed] [Google Scholar]
- 12.Furukawa Y, Sutheesophon K, Wada T, Nishimura M, Saito Y, Ishii H. Methylation silencing of the Apaf-1 gene in acute leukemia. Mol Cancer Res. 2005;3:325–34. doi: 10.1158/1541-7786.MCR-04-0105. [DOI] [PubMed] [Google Scholar]
- 13.Sun Y, Orrenius S, Pervaiz S, Fadeel B. Plasma membrane sequestration of apoptotic protease-activating factor-1 in human B-lymphoma cells: a novel mechanism of chemoresistance. Blood. 2005;105:4070–7. doi: 10.1182/blood-2004-10-4075. [DOI] [PubMed] [Google Scholar]
- 14.Kamarajan P, Sun NK, Sun CL, Chao CC. Apaf-1 overexpression partially overcomes apoptotic resistance in a cisplatin-selected HeLa cell line. FEBS Lett. 2001;505:206–12. doi: 10.1016/s0014-5793(01)02817-4. [DOI] [PubMed] [Google Scholar]
- 15.Maiso P, Carvajal-Vergara X, Ocio EM, et al. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 2006;66:5781–9. doi: 10.1158/0008-5472.CAN-05-4186. [DOI] [PubMed] [Google Scholar]
- 16.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 17.Bots M, Johnstone RW. Rational combinations using HDAC inhibitors. Clin Cancer Res. 2009;15:3970–7. doi: 10.1158/1078-0432.CCR-08-2786. [DOI] [PubMed] [Google Scholar]
- 18.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 19.Frew AJ, Johnstone RW, Bolden JE. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett. 2009;280:125–33. doi: 10.1016/j.canlet.2009.02.042. [DOI] [PubMed] [Google Scholar]