

Abstract
We present a method, fastIBD, for finding tracts of identity by descent (IBD) between pairs of individuals. FastIBD can be applied to thousands of samples across genome-wide SNP data and is significantly more powerful for finding short tracts of IBD than existing methods for finding IBD tracts in such data. We show that fastIBD can detect facets of population structure that are not revealed by other methods. In the Wellcome Trust Case Control Consortium bipolar disorder case-control data, we find a genome-wide excess of IBD in case-case pairs of individuals compared to control-control pairs. We show that this excess can be explained by the geographical clustering of cases. We also show that it is possible to use fastIBD to generate highly accurate estimates of genome-wide IBD sharing between pairs of distant relatives. This is useful for estimation of relationship and for adjusting for relatedness in association studies. FastIBD is incorporated in the freely available Beagle software package.
Introduction
Haplotypes are identical by descent if they are identical and inherited from a common ancestor. Tracts of identity by descent (IBD) are broken up by recombination during meiosis, so expected length of IBD depends on the number of generations since the common ancestor at the locus. If the common ancestor lived a great many generations ago (ancient IBD), the individuals share very short tracts of genetic material. In the sense of ancient IBD, identical single nucleotide polymorphism (SNP) alleles are often assumed to be identical by descent; it is assumed that there is no recurrent mutation. At the other extreme, in families, individuals who have IBD typically share very long tracts (>10 cM), and IBD is only defined with respect to documented common ancestry. Familial IBD can be detected with linkage programs.1,2 Recent IBD3 is IBD between individuals of possibly undocumented relationship, and it results from common ancestry within approximately the past 30 generations. Using high-density SNP genotype data, one can detect the majority of recent IBD tracts with lengths greater than 2 cM in data from north-western Europeans.3
IBD is fundamental to genetic mapping. Association mapping methods rely on linkage disequilibrium (LD), which is due to ancient IBD between unrelated individuals. Pedigree-based linkage methods use familial IBD. Recent IBD can be used for population-based linkage analysis in founder populations.4–6 Detection of close relationships by means of familial and recent IBD (Witherspoon et al., abstract 367, ASHG annual meeting, November 5, 2010 and Han and Abney, abstract 1105, ASHG annual meeting, November 4, 2010) is useful for correcting the variance of association statistics.7–11 Ancient IBD is also useful in detecting and measuring population structure.4,12,13
There are several existing methods for detecting IBD. Some methods are based on detecting long segments of identity by state (IBS) (Nelson, S., et al. abstract 1530, ASHG annual meeting, October 11, 2006. and references5,14–16). Other methods calculate probabilities of IBD.3,4,6 Some probability-based methods require that SNPs be in linkage equilibrium,4 which requires prior thinning of SNPs and thus reduces power. Beagle allows SNPs to be in LD by modeling haplotype frequencies. We previously developed an IBD detection method (Beagle IBD) and showed that it had higher power than several other methods when we controlled for the false-positive rate.3 However, Beagle IBD is computationally intensive and cannot be applied to all pairs of individuals genome-wide in large-scale genome-wide association studies.
We present an alternative IBD detection method, fastIBD, which accounts for haplotype frequencies and uncertain haplotype phase while enabling fast computation on genome-wide SNP data. The fastIBD method is more than 1000 times faster than the existing Beagle IBD method, and this increased speed permits it to be applied to genome-wide data on thousands of samples. One can use the fastIBD method to find IBD directly. Alternatively, one can combine the fastIBD method with the probabilistic Beagle IBD method by using fastIBD as a filter to find pairs of individuals who are likely to have IBD in a genomic region and then applying the full IBD probability calculation on those pairs.
The fastIBD method is based on estimating frequencies of shared haplotypes. Haplotype frequency is critical because a shared common haplotype is unlikely to reflect recent IBD, whereas a shared haplotype that is very rare is likely to be identical by descent. The fastIBD method allows for uncertain haplotype phase by sampling multiple realizations of haplotype phase given the data, then allowing for some switching between alternative phasings; there is, however, a switch penalty to prevent excessive switching. The extent of haplotype sharing is measured by a score that is the frequency of the shared haplotype modified by the penalties assessed at each switch between alternate phasings. Thus, a small score (close to zero) for a pair indicates that the two individuals share a low-frequency haplotype and are thus likely to be identical by descent. We use sampled haplotypes with a sliding marker window, as is done in GERMLINE,5 which permits rapid computation. A critical difference between our method and GERMLINE is that our method is based on shared haplotype frequency rather than shared haplotype length.
Material and Methods
The fastIBD algorithm starts by sampling a fixed number of haplotype pairs (four pairs by default) for each individual from the posterior haplotype distribution. Each sampled haplotype corresponds to a sequence of hidden Markov model (HMM) states. The fastIBD algorithm searches for pairs of sampled haplotypes sharing the same sequence of HMM states for a set of consecutive markers. If the pair of sampled haplotypes belongs to two distinct individuals, the shared haplotype tract is recorded. For each pair of individuals, overlapping shared haplotype tracts are merged, and the merged shared haplotype tract is a mosaic of pairs of sampled haplotypes (see Figure 1). A fastIBD score is calculated for each merged tract, and if the score is below a user-specified threshold, the tract is printed to an output file. We now describe in detail the algorithm for finding shared haplotype tracts, the calculation of fastIBD scores for those tracts, and the algorithmic details that allow for efficient computation. Pseudocode is available as supplemental data.
Figure 1.

Merging of Shared Haplotype Tracts
Four pairs of haplotypes have been sampled from individuals 1 and 2. Two shared haplotype tracts have been found (denoted by patterned regions). The two tracts are merged into a single shared haplotype tract.
Shared Haplotype Tracts
A shared haplotype tract T consists of a pair of sampled haplotypes (T.H1 and T.H2), a starting marker index (T.start), an ending marker index (T.end), and a fastIBD score (T.score). We use the convention that the starting marker index is inclusive and the ending marker index is exclusive. When shared haplotype tracts are first discovered, the fastIBD score is equal to the pairwise haplotype score defined below for the two haplotypes in the marker interval. However, after shared haplotype tracts are found, overlapping shared haplotype tracts are merged, and the merging algorithm defines a new fastIBD score for the merged tract. In general, the fastIBD score roughly approximates the frequency of the shared haplotype.
Pairwise Haplotype Scores
For any pair of haplotypes H1 and H2 and any interval of markers m1 < m2, we define a pairwise haplotype score S(H1, H2, m1, m2). The Beagle model defines a unique sequence of HMM states for each haplotype. If both haplotypes have the same sequence of HMM states in the marker interval, the pairwise haplotype score is the haplotype frequency or, more precisely, the frequency of the shared sequence of HMM states. As a consequence of the LD model's being a HMM, the frequency of a sequence of HMM states sm, sm+1, …, sm+k can be expressed as a product of state and transition probabilities:
In the preceding equation, there is a term corresponding to each marker: P(sm) for marker m and P(sm+j | sm+j−1) for marker m + j (j > 0). If the two haplotypes do not have the same HMM state at one or more markers in the marker interval, one obtains the pairwise haplotype score by replacing the corresponding state P(sm) or transition probability P(sm+j | sm+j−1) with 100 at each marker for which the two haplotypes have different HMM states. This penalizes the pairwise haplotype score by inflating the estimated shared haplotype frequency.
Merging Shared Haplotype Tracts
Two shared haplotype tracts T and U can be merged to create a merged shared haplotype tract M if the pair of sampled haplotypes in each tract corresponds to a single pair of individuals and if either the marker intervals for the two shared haplotype tracts overlap or the starting marker for one tract is the ending marker for the other tract. When merging overlapping shared haplotype tracts for a pair of individuals, we merge tracts with the smallest starting marker indices first.
The marker interval for the merged tract is the union of the two marker intervals. The fastIBD score of the merged tract is defined to be less than or equal to the two component fastIBD scores. For the purposes of further computation, we only need to keep track of the haplotypes at the right end of the merged tract, so the two notated haplotypes of the merged tract are the haplotypes from the tract with the largest ending index. For example, if the marker interval in shared haplotype tract T is a subset of the marker interval in shared haplotype tract U, we say tract U covers tract T, and we define the merged tract as M.H1 = U.H1, M.H2 = U.H2, M.start = U.start, M.end = U.end, and M.score = min{T.score, U.score}.
If shared haplotype tracts T and U can be merged, and if one tract does not cover the other tract, then either T.start ≤ U.start and T.end ≤ U.end or U.start ≤ T.start and U.end ≤ T.end. If we assume the former configuration, the merged tract haplotypes are M.H1 = U.H1 and M.H2 = U.H2, the merged tract marker interval is M.start = T.start, M.end = U.end, and the merged-tract fastIBD score M.score is the minimum of a left score and a right score.
The left score is defined as
The right score is defined as
The left score is the left-tract fastIBD score multiplied by the minimum of 1 and (100 × the fastIBD score from the extra markers contributed by the right tract). The right score is the right-tract fastIBD score multiplied by the minimum of 1 and (100 × the fastIBD score from the extra markers contributed by the left tract). The minimum function in the definition of left and right scores ensures the merged tract score is equal to or smaller than the fastIBD scores for the left and right shared haplotype tracts. The penalty term is needed to prevent the discovered IBD tracts from switching arbitrarily often between an individual's haplotypes. Such switching would allow the algorithm to find excessive numbers of false-positive IBD tracts. On the other hand, it is important to allow the possibility of some switching between haplotypes in a long IBD tract because phased haplotypes do contain switch errors.17 We tried other penalty values (25 and 400) and found very little difference in performance in terms of power and false-positive rates (results not shown).
Marker Windows
The algorithm reduces computer memory requirements by storing only a window of marker data in memory at any time. The window is a set of consecutive markers, and the window is moved down the chromosome during the analysis. The window contains two smaller windows of markers, which are adjacent. We refer to the two smaller windows as the leading window and the trailing window. The number of markers in each window is chosen on the basis of properties of the Beagle HMM in the region (as described below). This enables the number of markers in each window to adapt to the local haplotypic structure as the windows advance along the chromosome.
The fastIBD algorithm moves this pair of smaller windows along the chromosome. At the first step, only the leading window is defined. At the next step, the old leading window becomes the new trailing window, and the new leading window starts adjacent to the new trailing window. This process is repeated along the chromosome.
Suppose that the trailing window is from markers m0 (inclusive) to m1 (exclusive) and that the leading window is from markers m1 (inclusive) to m2 (exclusive). We use the leading window to find shared haplotype tracts from pairs of sampled haplotypes that share the same sequence of HMM states in the window. For each distinct sequence of HMM states in the leading window, all pairs of sampled haplotypes that have the sequence and that correspond to distinct individuals are identified. Although the number of pairs of samples grows quadratically with sample size, one can efficiently identify the pairs of sampled haplotypes with identical HMM states in the leading window by using a dictionary data structure, as noted by Gusev et al.5 For each identified pair of haplotypes, H1 and H2, that share the same sequence of HMM states in the leading window, the beginning and ending markers of the shared haplotype are determined as follows: First, we search backward from the start of the leading window and find the minimal marker m∗ such that the two sampled haplotypes have the same sequence of HMM states for the markers from m∗ (inclusive) to m2 (exclusive). Second, we search forward from the end of the leading window, m2, and find the marker m∗∗, which minimizes S(H1, H2, m∗, m∗∗) and satisfies S(H1, H2, m∗, m) ≤ 1 for all m∗ < m ≤ m∗∗. Note that we permit the shared haplotype tract to contain markers beyond the leading window for which the candidate pair of haplotypes have different HMM states if including these markers will minimize the total pairwise haplotype score. Once the starting and ending markers m∗ and m∗∗ are discovered, we record a shared haploytpe tract for the pair of sampled haplotypes. This shared haplotype tract starts with marker m∗, ends with marker m∗∗, and has a fastIBD score of S(H1, H2, m∗, m∗∗).
After finding the shared haplotype tracts from the leading window, we identify all pairs of individuals with shared haplotype tracts, and for each pair of individuals, we merge all covered shared haplotype tracts with their covering tract.
Extending Shared Haplotype Tracts
After identifying and recording shared haplotypes tracts by using the leading window, we next attempt to extend all previously recorded shared haplotype tracts that end within the trailing window. If a shared haplotype tract detected in the leading window overlaps with the tract that ends in the trailing window, we extend the tract that ends in the trailing window by merging it with the tract detected in the leading window. Otherwise, if a shared haplotype tract for a pair of haplotypes H1 and H2 ends at marker m in the trailing window, we identify the pair of individuals corresponding to the sampled haplotypes H1 and H2. For this pair, we look among the sampled haplotypes for haplotypes that have the same HMM state at marker m. If there is no pair of sampled haplotypes with the same HMM state at marker m, then the tract cannot be extended. For each pair of haplotypes (if any) with the same HMM state at marker m, we calculate the pairwise haplotype score from marker m to the last marker for which the two sampled haplotypes share the same HMM state for all markers in the interval. We select the extending haplotype pair with the minimal pairwise haplotype score and create a shared haplotype tract that is merged with the original haplotype pair ending at marker m. We repeat this process as long as there is a shared haplotype tract that ends in the trailing window and can be extended. If a shared haplotype tract terminates in the trailing window and cannot be extended, we remove the shared haplotype tract, after first printing it to an output file if its fastIBD score is less than the user-specified threshold.
Beagle Distance and Window Size
We use a model-based measure of distance along a chromosome that we call Beagle distance. This distance is defined in terms of the state and transition probabilities of the Beagle HMM model. We use Beagle distance to determine the size of each marker window (described below). Define tm as
where Sm is the set of HMM states at marker m, P(s) is the probability of being in state s at marker m, and P(s | ·) is the unique, non-zero probability of transitioning to state s conditional on being in a state at marker s − 1 that permits a transition to s. In the Beagle HMM, transition probabilities for transitions into a state (“edge” in the terminology of Browning and Browning17) are unique because all transitions to the state go through an intermediate node from which the transition probabilities are defined. This property does not apply to HMMs in general, but it is a convenient attribute of the Beagle HMM. The value tm is the average transition probability into states at marker m.
We define the Beagle distance d(m − 1, m) between marker m − 1 and marker m to be −ln tm and the distance d(m1, m2) between markers m1 and m2 (m1 < m2) to be the sum
The negative logarithm transformation converts products of probabilities to sums of non-negative distances. This definition of Beagle distance assigns 0 distance between markers that are completely correlated. If markers m − 1 and m are completely correlated, the HMM state at marker m − 1 completely determines the HMM state at marker m, and all non-zero transition probabilities for transitions into states at marker m are equal to 1. Consequently, if markers m − 1 and m are completely correlated, the distance between the markers is d(m − 1, m) = −ln 1 = 0. It can be shown that the smallest possible value of t for diallelic SNPs is 0.5, corresponding to a maximum distance of 0.69 between adjacent SNPs.
Figure 2 shows the relationship between position defined by Beagle distance and position defined by the usual cM genetic distance (taken from HapMap18). The correlation between these two measures is extremely high. Because the maximum Beagle distance is constrained, large gaps in the markers (such as a 1.1 cM gap at 28 cM in region 1A) do not correspond to large jumps in the Beagle distance. This is not important for the purposes of defining window size, for which we use Beagle distance. We use Beagle distance rather than cM distance because the Beagle distances are generated automatically rather than requiring input by the user. Also, Beagle distance can adapt to different relationships between LD and cM distance—for example, African data will tend to have lower LD than European data, and this will be represented in the Beagle distances.
Figure 2.
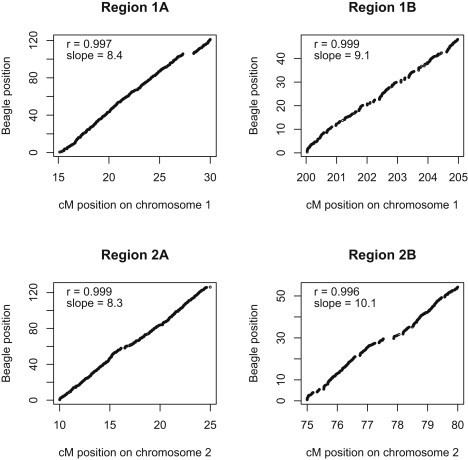
Beagle Distance versus cM Distance Calculated on the Four Genomic Regions Used in the Power Study
Data are from the UK 1958 birth cohort and HapMap CEU on Illumina 550K SNPs. Beagle positions are the Beagle distance from the start of the region. CentiMorgan (cM) positions are estimates from HapMap. For each genomic region, the correlation between the two measures (r) and the slope of the least-squares regression line fit to the data (slope) are given.
As a default criterion for window size, we use a distance of 1.6 (approximately 0.2 cM in European data; see Figure 2). We chose this window size empirically to achieve a reasonable balance between computational efficiency and sensitivity. Phasing errors increase with window length, so the use of larger window sizes runs the risk of missing detection of pairs of individuals who share haplotypes IBD. On the other hand, smaller windows are computationally inefficient because too many shared haplotypes are identified in the leading window.
FastIBD Computation
The process underlying a single fastIBD computational run involves phasing the data (using the usual ten iterations of the phasing algorithm), sampling four pairs of haplotypes per individual, creating a Beagle model from the sampled haplotypes to determine the underlying HMM states (used in detecting IBD), and finding the IBD tracts. When building the Beagle model from the sampled haplotypes, we use a parsimonious version of the Beagle model with a scale factor of 2.0 (as compared with the default scale of 1.0 used in phasing or, at the other extreme, the default scale of 4.0 used in haplotypic testing with Beagle; see Browning and Browning for description of scale factors19), which improves the power to detect IBD. In the results shown, we repeat the whole process ten times (except where otherwise noted), with different seeds for the random number generator, and we take the minimum score from the ten runs at each position where IBD is found.
Results
Power and False Discovery Rates
To investigate power, we used artificial IBD data described previously.3 In brief, we took phased HapMap Phase II CEU (Utah residents with ancestry from northern and western Europe) data18 and copied a segment of one individual's haplotype onto another individual to create artificial IBD in 30 pairs of individuals. In order to have a large sample size for building the haplotype frequency model, we added approximately 1500 individuals from the UK 1958 birth cohort20 genotyped on the Illumina 550K platform, restricting attention to SNPs genotyped in both data sets. We used the fastIBD method to find IBD between the HapMap pairs in whom artificial IBD had been created. We repeated the data creation and analysis for IBD tract sizes of 1, 2, 3, 4, and 5 cM. To estimate power, we average the proportion of artificial IBD that is detected by the method over pairs of individuals and SNP markers. In other words, power is the average proportion of an IBD tract that is detected as identical by descent. We applied the fastIBD method with various thresholds on the score. We also applied three other methods: shared segment detection from PLINK v1.07,4 GERMLINE v1.4,5 and Beagle IBD v3.3. We used default settings for these programs as much as possible. For PLINK, we first thinned the markers according to suggestions in the PLINK documentation. We ran PLINK both with default settings, which require IBD segments to be at least 1 megabase and 100 SNPs long, and with relaxed settings, which require IBD segments to be at least 200 kb and 20 SNPs. For GERMLINE, we set the minimum tract length to 0.5 cM, whereas the default is 5 cM. This allows GERMLINE a chance to find some of the small segments in the power comparison, but it would not be a recommendable setting in general because it would result in a high false-discovery rate. For Beagle IBD, we used the new default setting of ibdscale = 2.0, introduced in version 3.3, which increases power over the previous default setting of ibdscale = 1.0 used in results reported previously.3
In order to investigate false-positive rates, we used data constructed from the 1958 birth cohort chromosome 1 Illumina 550K data in such a way as to destroy any IBD tracts of length 0.2 cM or greater. We accomplished this by creating composite individuals, as described previously.3 The purpose of destroying IBD tracts is to avoid including true-positive results in the false-positive rates. (In contrast, in order to keep the power analysis as realistic as possible, we did not attempt to destroy existing IBD tracts in the power analysis.) We tested for IBD in these data. For a given size of tract, we used all detected tracts within 10% of this size (for example, tracts of detected size 0.9–1.1 for 1 cM and size 1.8–2.2 for 2 cM) and recorded the mean proportion of SNPs per pair of individuals at which IBD is detected in tracts within this size range. This is the false-positive rate for a given tract length.
Whereas the false-positive rate only measures the ability of the method to control type I error (false detection of IBD tracts), the false-discovery rate is a function of false-positive rate, power, and the rate of true IBD tracts in the data. Specifically, the false discovery rate is the proportion of SNPs that are reported to be identical by descent but that are not identical by descent within reported tracts of a given length. In order to estimate this quantity, one needs to have an estimate of the true rate, T, of IBD of estimated length L. We estimated the value T by using the rate of IBD detection by Beagle IBD in four regions in the 1958 birth cohort Illumina 550K data and the corresponding false-positive and power rates from our previous analyses.3 We also need F, the per-SNP, per-pair false-positive estimate described above, and P, the per-SNP, per-pair power estimate described above. The formula for estimating the false-discovery rate is (1 − T)F/[(1 − T)F + TP]. To derive this formula, note that (1 − T) is the proportion of SNPs that are not in an IBD tract of length L, F is the rate of estimating such SNPs to be IBD, so that (1 − T)F is the rate at which, per pair of individuals, SNPs that are not identical by descent are incorrectly estimated to be identical by descent (in a tract of estimated length L). Similarly, T is the rate of SNPs that are in an IBD tract of length L, and P is the rate at which such SNPs are estimated to be identical by descent, so that TP is the rate at which, per pair of individuals, SNPs that are identical by descent in a tract of length L are correctly estimated to be identical by descent. The denominator then is the rate, per pair of individuals, at which SNPs are estimated to be identical by descent (in a tract of length L).
Results are shown in Figure 3. We see that fastIBD and Beagle IBD are very effective at finding IBD tracts of size 2 or 3 cM with both high power and low false-discovery rate, whereas PLINK and GERMLINE have low power to detect tracts of this size or a high false-discovery rate. No method has very high power for finding tracts of size 1 cM, but fastIBD and Beagle IBD at least have a low false-discovery rate for this size tract. For large tracts (4 or 5 cM), all methods do well. Overall, a fastIBD-score threshold of 10−10 gives high power while keeping the false-discovery rate close to zero, so we use this threshold in all further analyses unless otherwise noted.
Figure 3.
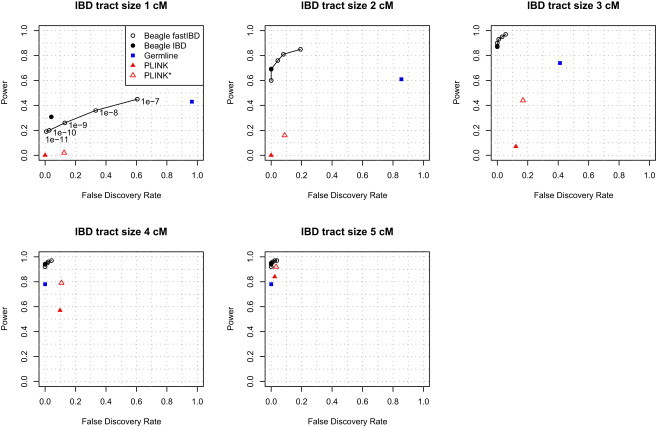
False Discovery Rate versus Power
Results for the fastIBD method at five values of score threshold and for Beagle IBD, germline, and PLINK with default and relaxed settings for five sizes of IBD tract. Results for PLINK with relaxed settings are denoted by PLINK∗.
The fastIBD results described above, and those used in the analyses below, are based on combined results of ten independent runs of the method (the minimum score over the ten runs at each position was used). The reason for combining results from multiple runs is that single runs can miss tracts of IBD as a result of stochastic variation in the estimated haplotypes. Figure 4 shows results from combining one, three, or ten run(s). Using ten runs gives significantly better results than using one run and somewhat better results than using three runs. Using five runs gives results that fall approximately midway between those for three and ten runs (data not shown). Thus, combining results from three or five runs would be reasonable if computing resources are limited.
Figure 4.
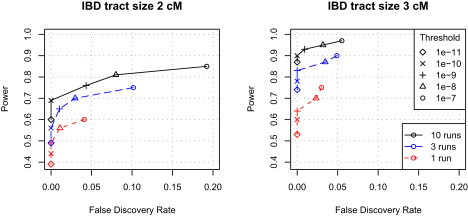
False-Discovery Rate versus Power for the FastIBD Method: Combined Results from Different Numbers of Runs
The results from ten runs are the same as those in Figure 2. “Threshold” is the score threshold applied to the fastIBD scores.
Bipolar Analysis
We applied the fastIBD method to the Wellcome Trust Case Control Consortium (WTCCC) bipolar disorder data20 genotyped on the Affymetrix 500K platform. High genotype accuracy is critically important when detecting IBD, so we re-called the SNP genotypes with BEAGLECALL,21 which incorporates LD to improve genotype call accuracy. After quality control filtering, there were 1868 cases and 2938 controls comprising individuals from the UK 1958 birth cohort (58C) and from the UK Blood Service (UKBS), and 459,983 autosomal SNPs. We found an excess of IBD in case-case pairs compared to control-control pairs in these data. Figure 5 shows that the excess IBD is spread across the genome. IBD proportions tend to drop at the ends of chromosomes because of the reduced information there. Table 1 shows case and control IBD proportions as well as average kinship coefficients estimated with PLINK (--genome option). Overall, there is approximately 10% excess IBD in cases relative to controls.
Figure 5.
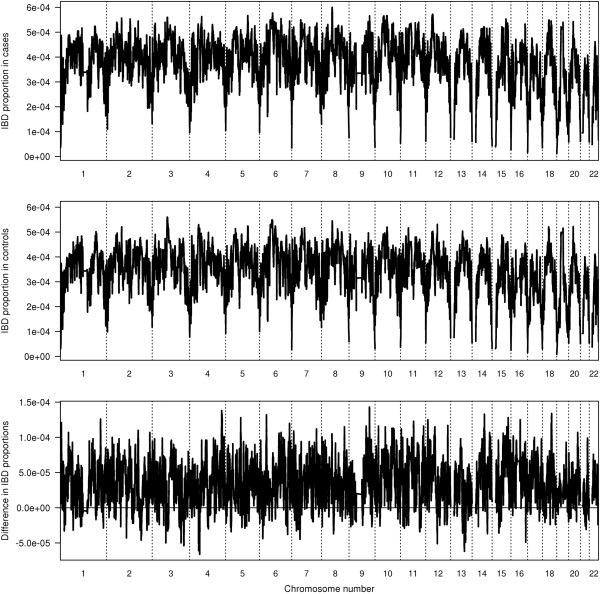
FastIBD Average Proportions of Case-Case IBD Sharing and Control-Control IBD Sharing along the Genome for WTCCC Bipolar Case-Control Data
The IBD proportion in cases (y axis on top panel) is the fraction of pairs, computed from all case-case pairs, that are estimated to be identical by descent at a given location in the genome. Similarly, the IBD proportion in controls (y axis in middle panel) is computed from all control-control pairs. The difference in IBD proportions (y axis in lower panel) is the IBD proportion in cases minus IBD proportion in controls. Dashed vertical lines mark chromosome boundaries (chromosomes 1–22). The horizontal line in the lowest panel is the difference = 0 line (i.e., IBD proportion in cases = IBD proportion in controls), for comparison.
Table 1.
Average IBD Sharing within Cohorts in the WTCCC Bipolar Data
|
FastIBD |
PLINK Kinship |
|||||
|---|---|---|---|---|---|---|
| Bipolar | 58C | UKBS | Bipolar | 58C | UKBS | |
| Overall | 0.00038 | 0.00035 | 0.00034 | 0.0045 | 0.0040 | 0.0045 |
| Within England | 0.00036 | 0.00035 | 0.00034 | 0.0046 | 0.0041 | 0.0044 |
| Within Wales | 0.00083 | 0.00093 | 0.00111 | 0.0044 | 0.0049 | 0.0043 |
| Within Scotland | 0.00075 | 0.00068 | 0.00070 | 0.0053 | 0.0040 | 0.0051 |
The WTCCC data include the geographical origin of each sample. Each individual is identified as originating from one of 12 regions of the UK. We stratified our analysis of IBD proportions by geographic region, and results are shown in Table 1. The levels of average IBD sharing within Wales and within Scotland are much higher than for other regions. The differences between cohorts within Wales could be due to population structure within Wales and unequal sampling of the different subregions of Wales in the three cohorts. In contrast, the other ten geographical (English) regions have lower average IBD proportions for each of the cohorts. The bipolar disorder recruitment effort included a large component from Cardiff, so that 21% of the bipolar samples are from Wales, whereas only 5% are from Wales for the 1958 birth cohort and UK Blood Service cohorts (see Table 2). This bias seems to be driving the overall difference in IBD proportions between cases and controls in the WTCCC study, although it is possible that polygenic disease-susceptibility factors22 also play a role in this difference.
Table 2.
Percentage of Sample from Each Geographic Region for WTCCC Bipolar Disorder and Control Cohorts
| England | Wales | Scotland | |
|---|---|---|---|
| Bipolar disorder | 69.7 | 20.6 | 9.7 |
| UK 1958 birth cohort | 85.0 | 5.1 | 9.9 |
| UK Blood Service | 86.5 | 4.9 | 8.6 |
We were interested in whether these geographic effects on IBD proportions would be seen with other approaches to population structure. We first looked at kinship coefficients estimated with PLINK's --genome option after removing all SNPs with minor-allele frequency of <5%.4 This approach does not utilize tracts of IBD but averages allelic sharing genome-wide. For direct comparison with fastIBD proportions, PLINK's kinship coefficients should be multiplied by two, but because PLINK's kinship coefficients are already significantly higher than the fastIBD proportions in these data, we did not make this adjustment; it is the relative differences between cohorts and regions that are of interest here. PLINK's average estimated kinship coefficients were 0.0040–0.0053 for any set of individuals whom we chose to compare, whether we created subsets by cohort or by geographic region (see Table 1). Thus, the maximum difference seen between regions or between cohorts is 30%, compared to 300% for the fastIBD proportions. The pattern in the PLINK kinship results is also less consistent; there is excess sharing in Wales for the 58C cohort only and excess sharing in Scotland for the other two cohorts only. Thus, although estimated kinship coefficients hint at the geographic differences that we found by IBD tract detection, they do not present as strong or clear a view of these differences. We also compared our approach to a principal-components analysis of these data (Figure S8 of the WTCCC's published analysis20). In the principal-components analysis, Scotland was significantly more of an outlier than was Wales; London was also an outlier. This is in contrast to our results, where Wales had slightly higher levels of IBD sharing than Scotland and London had similar levels of sharing to the other English regions (results not shown), but significantly less than that of Wales and Scotland. Thus, our approach based on IBD tracts reveals different aspects of population structure than do usual genomic estimates of IBD sharing, such as PLINK's kinship coefficients or principal-components analysis. Our approach would be expected to measure recent population dynamics rather than long-term allelic drift. Regional differences in levels of recent relationship could reflect population sizes and the extent of population movement (immigration from other regions) over the past 5–30 generations.
Detection of Relationships
We created artificial cousin data to investigate the utility of the fastIBD method for estimating overall genomic IBD sharing between pairs of individuals. Accurate estimation of genomic IBD sharing depends not only on detection of IBD tracts but also on accurate estimation of the ends of those tracts. To create the data, we simulated the IBD process for cousins of given degree by simulating the underlying inheritance vectors, which form a Markov process along the chromosome with distance measured in cM, if we assume no crossover interference.23 We then superimposed this process onto CEU haplotypes from the HapMap II data, as in the construction of artificial IBD for the power study above. As for the power study, we included 1958 birth cohort Illumina 550K genotypes when building the Beagle LD model. For each pair of individuals, we recorded the amount of actual IBD and the amount of estimated IBD. Out of thirty pairs of CEU individuals considered, two pairs were discarded from the results because they showed a relatively high degree of relatedness prior to the addition of artificial IBD (over 20 cM of detected IBD tracts). Thus, each data set included 28 pairs of cousins of given degree. We considered first to fifth cousins. First cousins are the children of aunts and uncles, second cousins are the children of first cousins, and so on. We also analyzed the same pairs of individuals without any added IBD (these pairs are “unrelated”). Figure 6 shows the relationship between actual and estimated IBD. Actual IBD is the amount of constructed IBD divided by the length of the genome. For distant cousins, some pairs might have no actual IBD. Estimates of the IBD proportion were obtained from fastIBD (the total length of detected IBD tracts was divided by the length of the genome) and from PLINK (twice the kinship coefficient obtained from PLINK's --genome option, after SNPs with a minor-allele frequency of <5% were removed). Estimates based on IBD tracts detected with fastIBD are much more accurate than kinship estimates, particularly for the more distant relationships. For first cousins, IBD proportion is slightly underestimated, which can be remedied by the use of a less stringent score threshold (e.g., 10−7; results not shown). For best results over a range of relationships, one could first apply the 10−10 threshold, and if the estimated IBD proportion is greater than 10%, one could reanalyze with the less stringent threshold. Estimation of genomic IBD sharing with fastIBD should in principal be possible for more distant relatives because there is good power to detect IBD segments of size 2 cM (corresponding to the expected length of IBD tracts in 24th cousins, when such tracts exist). However, distant cousins usually have no IBD tracts. Also, the background level of relatedness in populations will subsume specific distant relatedness. For example, we found IBD at a rate of 3.5 × 10−4 in the 1958 birth cohort data in analysis of the WTCCC bipolar study, whereas sixth cousins are expected to share 2 × 10−4 of their genome IBD.
Figure 6.
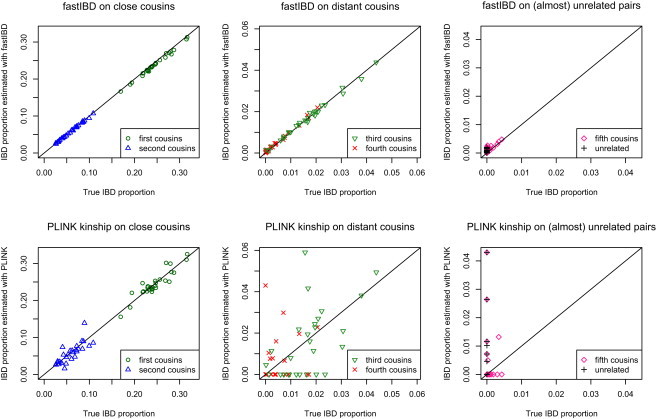
IBD Proportions Estimated against Constructed “true” IBD Proportions for Five Degrees of Cousins and Unrelated Pairs of Individuals via FastIBD and PLINK Kinship Coefficients
IBD proportion is the proportion of the genome containing an IBD tract, or twice the kinship coefficient.
Computation Times
Computation times for a single run of the fastIBD algorithm include the time to phase the data, plus an additional 10%–20% to detect the IBD tracts. In the results presented here, we combined results from ten runs of the fastIBD analysis. Computation is easily parallelized by run and by chromosome. As an example of computing time, a single run (one of the ten runs) on chromosome 1 of the WTCCC bipolar analysis, with 4806 individuals and 37,645 SNPs and for which IBD was estimated for all possible pairs of individuals across the chromosome, took approximately 17 hr on a single core of an Intel Xeon E5620 Quad-Core compute node running at 2.40GHz. Computing time for 10 runs of fastIBD is similar to the computing time for PLINK shared-segment detection for the same number of markers; however, thinning the marker set before running PLINK reduces the computing time to a corresponding extent. If the input data are phased, GERMLINE is between 2 and 3 orders of magnitude faster than ten runs of fastIBD. Phasing data with BEAGLE takes time that is similar to one run of fastIBD, so in practice, the computation time for ten runs of fastIBD is approximately one order of magnitude larger than the computation time for GERMLINE when the phasing step is included. However, the greatly improved accuracy of fastIBD compensates for the increased computing time.
Discussion
The fastIBD detection method is highly accurate for detecting IBD and yet is sufficiently fast to perform genome-wide analysis in large samples. The choice of threshold for the fastIBD score is a compromise between power and false-discovery rate and can be varied depending on the context. For the applications we considered, we wanted to have an approximately zero false-discovery rate, so we used a threshold of 10−10. When probabilities of IBD are required, the fastIBD method can be used as a filter to reduce the amount of computation needed by the original Beagle IBD method.
The fastIBD method reveals aspects of recent shared ancestry and population structure that have implications for statistical analysis. We found that bipolar disorder cases have more IBD than controls in the WTCCC data. The difference can be explained by uneven sampling of cases and controls from different regions in the UK. Imbalances in rates of IBD tracts have particular relevance for multilocus analyses that utilize information from genomic segments larger than the range of LD. For example, population-based linkage analysis,4 which looks for an excess of IBD tract sharing in cases compared to controls, will be severely affected by multiple false-positive results across the genome, unless adjustment for average rates of IBD tract sharing is made. Gene-wise analysis of rare variants will also be affected. Individuals who are identical by descent across a gene will share the same set of rare variants, and this will inflate the variance of tests for case-control differences. Variance correction utilizing detected IBD tracts should be possible using modified versions of existing methods for correcting the variance of single-marker association test statistics in the presence of relatedness in case-control studies.9–11
The fastIBD method allows for improved estimation of relationship. Accurate estimates of relationship will be useful for proper adjustment of association tests in case-control studies,8 for analysis of quantitative traits,24 for conservation studies, and for studies of population dynamics.25 The fastIBD method will also be useful in other applications, such as population-based linkage in founder populations26 and improving haplotype phase inference and imputation.14
Acknowledgments
This study makes use of data generated by the Wellcome Trust Case Control Consortium and the Wellcome Trust Sanger Institute. The Illumina 550K genotype data for individuals in the 1958 British birth cohort were generated by the Wellcome Trust Sanger Institute. A full list of the investigators who contributed to the generation of the Wellcome Trust Case Control Consortium data is available from www.wtccc.org.uk. Funding for the Wellcome Trust Case Control Consortium project was provided by the Wellcome Trust under award 076113. This work was supported by National Institutes of Health awards R01GM075091, R01HG005701, and R01004960. The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Wellcome Trust.
Contributor Information
Brian L. Browning, Email: browning@uw.edu.
Sharon R. Browning, Email: sguy@uw.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Beagle, http://faculty.washington.edu/browning/beagle/beagle.html
European Genotype Archive (repository of WTCCC genotype data), http://www.ebi.ac.uk/ega/
Welcome Trust Case Control Consortium, http://www.wtccc.org.uk
HapMap, http://www.hapmap.org
References
- 1.Kruglyak L., Daly M.J., Reeve-Daly M.P., Lander E.S. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am. J. Hum. Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 2.Abecasis G.R., Cherney S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nature Genetics. 2001;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 3.Browning S.R., Browning B.L. High-resolution detection of identity by descent in unrelated individuals. Am. J. Hum. Genet. 2010;86:526–539. doi: 10.1016/j.ajhg.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gusev A., Lowe J.K., Stoffel M., Daly M.J., Altshuler D., Breslow J.L., Friedman J.M., Pe'er I. Whole population, genome-wide mapping of hidden relatedness. Genome Res. 2009;19:318–326. doi: 10.1101/gr.081398.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Albrechtsen A., Sand Korneliussen T., Moltke I., van Overseem Hansen T., Nielsen F.C., Nielsen R. Relatedness mapping and tracts of relatedness for genome-wide data in the presence of linkage disequilibrium. Genet. Epidemiol. 2009;33:266–274. doi: 10.1002/gepi.20378. [DOI] [PubMed] [Google Scholar]
- 7.Choi Y., Wijsman E.M., Weir B.S. Case-control association testing in the presence of unknown relationships. Genet. Epidemiol. 2009;33:668–678. doi: 10.1002/gepi.20418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slager S.L., Schaid D.J. Evaluation of candidate genes in case-control studies: A statistical method to account for related subjects. Am. J. Hum. Genet. 2001;68:1457–1462. doi: 10.1086/320608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bourgain C., Hoffjan S., Nicolae R., Newman D., Steiner L., Walker K., Reynolds R., Ober C., McPeek M.S. Novel case-control test in a founder population identifies P-selectin as an atopy-susceptibility locus. Am. J. Hum. Genet. 2003;73:612–626. doi: 10.1086/378208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thornton T., McPeek M.S. ROADTRIPS: case-control association testing with partially or completely unknown population and pedigree structure. Am. J. Hum. Genet. 2010;86:172–184. doi: 10.1016/j.ajhg.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Browning S.R., Briley J.D., Briley L.P., Chandra G., Charnecki J.H., Ehm M.G., Johansson K.A., Jones B.J., Karter A.J., Yarnall D.P., Wagner M.J. Case-control single-marker and haplotypic association analysis of pedigree data. Genet. Epidemiol. 2005;28:110–122. doi: 10.1002/gepi.20051. [DOI] [PubMed] [Google Scholar]
- 12.Weir B.S., Cockerham C.C. Estimating F-statistics for the analysis of population-structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 13.Wright S. The genetical structure of populations. Ann. Eugen. 1951;15:323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 14.Kong A., Masson G., Frigge M.L., Gylfason A., Zusmanovich P., Thorleifsson G., Olason P.I., Ingason A., Steinberg S., Rafnar T. Detection of sharing by descent, long-range phasing and haplotype imputation. Nat. Genet. 2008;40:1068–1075. doi: 10.1038/ng.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leibon G., Rockmore D.N., Pollak M.R. A SNP streak model for the identification of genetic regions identical-by-descent. Stat. Appl. Genet. Mol. Biol. 2008;7:e16. doi: 10.2202/1544-6115.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas A., Camp N.J., Farnham J.M., Allen-Brady K., Cannon-Albright L.A. Shared genomic segment analysis. Mapping disease predisposition genes in extended pedigrees using SNP genotype assays. Ann. Hum. Genet. 2008;72:279–287. doi: 10.1111/j.1469-1809.2007.00406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Browning S.R., Browning B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Browning B.L., Browning S.R. Efficient multilocus association testing for whole genome association studies using localized haplotype clustering. Genet. Epidemiol. 2007;31:365–375. doi: 10.1002/gepi.20216. [DOI] [PubMed] [Google Scholar]
- 20.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Browning B.L., Yu Z. Simultaneous genotype calling and haplotype phasing improves genotype accuracy and reduces false-positive associations for genome-wide association studies. Am. J. Hum. Genet. 2009;85:847–861. doi: 10.1016/j.ajhg.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purcell S.M., Wray N.R., Stone J.L., Visscher P.M., O'Donovan M.C., Sullivan P.F., Sklar P., Ruderfer D.M., McQuillin A., Morris D.W., International Schizophrenia Consortium Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donnelly K.P. The probability that related individuals share some section of genome identical by descent. Theor. Popul. Biol. 1983;23:34–63. doi: 10.1016/0040-5809(83)90004-7. [DOI] [PubMed] [Google Scholar]
- 24.Yu J., Pressoir G., Briggs W.H., Vroh Bi I., Yamasaki M., Doebley J.F., McMullen M.D., Gaut B.S., Nielsen D.M., Holland J.B. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006;38:203–208. doi: 10.1038/ng1702. [DOI] [PubMed] [Google Scholar]
- 25.Lynch M., Ritland K. Estimation of pairwise relatedness with molecular markers. Genetics. 1999;152:1753–1766. doi: 10.1093/genetics/152.4.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kenny E.E., Gusev A., Riegel K., Lütjohann D., Lowe J.K., Salit J., Maller J.B., Stoffel M., Daly M.J., Altshuler D.M. Systematic haplotype analysis resolves a complex plasma plant sterol locus on the Micronesian Island of Kosrae. Proc. Natl. Acad. Sci. USA. 2009;106:13886–13891. doi: 10.1073/pnas.0907336106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.