


Abstract
The Crithidia fasciculata RNH1 gene encodes an RNase H, an enzyme that specifically degrades the RNA strand of RNA–DNA hybrids. The RNH1 gene is contained within an open reading frame (ORF) predicted to encode a protein of 53.7 kDa. Previous work has shown that RNH1 expresses two proteins: a 38 kDa protein and a 45 kDa protein which is enriched in kinetoplast extracts. Epitope tagging of the C-terminus of the RNH1 gene results in localization of the protein to both the kinetoplast and the nucleus. Translation of the ORF beginning at the second in-frame methionine codon predicts a protein of 38 kDa. Insertion of two tandem stop codons between the first ATG codon and the second in-frame ATG codon of the ORF results in expression of only the 38 kDa protein and the protein localizes specifically to the nucleus. Mutation of the second methionine codon to a valine codon prevents expression of the 38 kDa protein and results in exclusive production of the 45 kDa protein and localization of the protein only in the kinetoplast. These results suggest that the kinetoplast enzyme results from processing of the full-length 53.7 kDa protein. The nuclear enzyme appears to result from translation initiation at the second in-frame ATG codon. This is the first example in trypanosomatids of the production of nuclear and mitochondrial isoforms of a protein from a single gene and is the only eukaryotic gene in the RNase HI gene family shown to encode a mitochondrial RNase H.
INTRODUCTION
Crithidia fasciculata is a protozoan parasite which has a novel form of mitochondrial DNA (kinetoplast DNA or kDNA) composed of 5000 minicircles and approximately 25 maxicircles. Both the minicircles and the maxicircles are interlocked in a compact network structure at the base of the flagellum (1). Within the mitochondrial matrix the kDNA network is associated with histone-like proteins and is condensed into a disc structure ∼1 µm in diameter and 0.4 µm thick (2,3). When viewed by fluorescence microscopy of fixed cells on a microscope slide the disc is visualized from its edge (2). Each minicircle in the network is interlocked on average with three other minicircles (4). Minicircles are replicated free of the network via θ intermediates and are subsequently rejoined to the network by a mitochondrial DNA topoisomerase (5–7). Synthesis of minicircle light strands are RNA primed and continuous, whereas minicircle heavy strand synthesis is discontinuous and is also likely to involve RNA-primed Okazaki-like intermediates (8). Nuclear DNA replication intermediates have not been characterized in trypanosomatids but, as in prokaryotes and other eukaryotes, these must require RNA priming as well. RNA primers must be removed from both mitochondrial and nuclear DNA prior to cell division.
RNase H has been implicated in removing RNA primers laid down during DNA replication. Most organisms studied to date, including Crithidia fasciculata, have at least two such enzymes (9–11). RNase H enzymes have been shown to be involved in RNA primer removal in both eukaryotic and prokaryotic cells. RNase H was found to be required for RNA primer removal in DNA synthesis reconstitution experiments in mammalian systems (12–14) and an RNase H from Drosophila embryos was also shown to interact with the polymerase–primase and to remove primers synthesized and subsequently elongated by the polymerase–primase (15). In Escherichia coli, RNase HI facilitates removal of long RNA primers by DNA polymerase I but is not essential for primer removal (16). The potent 5′-exonuclease of DNA polymerase I appears to play the major role in RNA primer removal in E.coli (17). Plasmids bearing the unique chromosomal origin oriC require RNase HI for specific initiation of replication at oriC (18). RNase HI is suggested to possibly remove non-specific RNA primers elsewhere on the DNA. Another role for RNase HI in DNA replication is revealed in replication of the Col E1 plasmid, in which processing of an RNA transcript by RNase HI produces an RNA primer for initiation by DNA polymerase I (19).
In C.fasciculata an RNase H gene (RNH1) was cloned by complementation of a conditional lethal E.coli mutant (20). Analysis of the open reading frame (ORF) indicated that the RNH1 gene has the potential to express a 53.7 kDa protein. However, disruption of the RNH1 gene revealed that the gene encodes two protein products of 45 and 38 kDa (11). Further work demonstrated that the 45 kDa peptide is enriched in wild-type kinetoplasts (21), suggesting that these proteins represent sorting isozymes, enzymes encoded by a single gene but distributed to different subcellular compartments (22). Our current work shows that the 38 kDa isoform is the nuclear form of RNase H1 and the 45 kDa isoform is the kinetoplast form. Our results also address the mechanism by which the two isoforms are produced.
MATERIALS AND METHODS
Isolation of the 5′ flanking sequence of RNH1
A λ GEM11 genomic library (23) was screened with a labeled 792 bp NcoI–NotI fragment of the RNH1 coding sequence. Positive clones were picked and used for subcloning. An additional 400 bp of the 5′ flanking sequence of RNH1 was cycle sequenced using oligonucleotide E2 (5′-GTCTGTGAAATGCAGCACTC) and an Applied Biosystems automated sequencer at the UCLA DNA Sequencing Facility. The additional sequence has been added to GenBank (accession no. L18916).
Construction of epitope-tagged RNH1
A Bsp120I site was introduced close to the C-terminus of RNH1 by PCR mutagenesis using oligonucleotide A89 (5′-GCCGACGTGCTAGCCGTCGCTGGCGCGCGTATGCACGGGCCCAGTGAGTG) and oligonucleotide A94 (5′-CTACGGCGTTTCACTTCTGAGTTCGGCA) with template p6HIS-1 (20). Six copies of the hemagglutinin influenza tag were cloned into the Bsp120I site to create p6HIS1.2. A NcoI–DraI fragment of p6HIS1.2 was cloned into pX63HYG (24), which contains the hygromycin phosphotransferase gene flanked by the 5′- and 3′-untranslated regions (UTRs) of Leishmania major dihydrofolate reductase thymidylate synthase, to create pRNH1-HYG. An additional 364 bp of RNH1 5′ flanking sequence was cloned into pRNH1-HYG as a XhoI–NotI fragment to create pRNH1-HA.
To create plasmid pRNH1-Phleo the hygromycin phosphotransferase cassette was excised from pRNH1-HA by XhoI and BglII digestion and replaced with a XhoI–BamHI fragment from pXGPHLEO (25), which contains the phleomycin cassette flanked by the dihydrofolate reductase thymidylate synthase 5′- and 3′-UTRs.
Oligonucleotide mutagenesis
Two stop codons were created just upstream of the second methionine codon in RNH1 in plasmid pRNH1-Phleo by oligonucleotide mutagenesis using oligonucleotide F6 (5′-GCGAGCGGCGAAACGCGCAACTAGTAAGCACCGCCGCCCGCGTCG) and the Promega Gene Editor kit according to the manufacturer’s instructions, to create pRNH1-STOP. The second methionine codon in the RNH1 ORF in pRNH1-Phleo was mutated to a valine codon using the strategy outlined above and oligonucleotide F40 (5′-CCCGCGTCGCGGGTGAAGCCGTCG), creating plasmid pRNH1-GTG. A second vector containing a hygromycin selectable marker was created by cutting pRNH1-GTG with BglI and XhoI to remove the phleomycin cassette. A BglI–XhoI fragment containing the hygromycin cassette from pX63HYG was cloned into the vector to create pRNH1-GTGH.
C.fasciculata transformation
Wild-type C.fasciculata or Cf rnh1Δ cells (11), a cell line in which both alleles of the RNH1 gene have been disrupted, were transformed as described (26) except that cells carrying constructs containing the phleomycin cassette were selected on plates contained 80 µg/ml Zeocin (Invitrogen) and grown in liquid culture containing 40 µg/ml Zeocin.
Immunolocalization
Immunofluorescent localization of the HA epitope tag was done essentially as described (27). Mouse 12CA5 monoclonal antibody (BabCO) was used at a 1:500 dilution. FITC-conjugated goat anti-mouse antibody (Sigma) was used as secondary antibody at 1:50 dilution. Images were captured on Kodak Elite chrome ASA 400 film (Eastman Kodak, Rochester, NY) with a Nikon FX-35DX camera using a Nikon Optiphot microscope and a 100× oil immersion lens. Slides were subsequently scanned and images were processed using Adobe Photoshop.
RNase H activity gels
SDS–polyacrylamide activity gels containing [32P]rA:dT substrate were poured and run as described (21). Cell extracts were made with 4 × 107 cells resuspended in 96 µl of SDS–PAGE loading buffer and boiled for 5 min. Five microliters were loaded in each lane.
Mapping of RNH1 splice acceptor sites
cDNA was synthesized using poly(A)+ RNA, oligonucleotide B24 (5′-AGCGTCGAAGTCCTACTTGAGGTTC) and Superscript II Reverse Transcriptase (Gibco BRL). Following synthesis the cDNA was purified on a 4 ml S400 column (Pharmacia) equilibrated with 10 mM Tris–HCl, pH 8.3, and 1 mM EDTA. The cDNA was used in PCR reactions with a spliced leader oligonucleotide (5′-AACGCTATATAAGTATCAGTTTCTG) and oligonucleotide B11 (5′-TGTGCCGACGATGTTGATGGAATT). Forty cycles of PCR of 1 min at 95°C, 2 min at 55°C and 2 min at 72°C were performed with AmpliTaq (Perkin Elmer), followed by 7 min at 72°C. Specific fragments were isolated from a 1.8% agarose gel, cloned into pGEM 7 (Promega) and sequenced by dideoxynucleotide chain termination with modified T7 DNA polymerase (Sequenase) according to the manufacturer’s instructions (US Biochemical).
For primer extension analysis oligonucleotide B11 was hybridized with poly(A)+ RNA at 60 or 67°C. Reverse transcription was carried out at 45°C with M-MLV reverse transcriptase (Gibco BRL). Reactions were phenol/chloroform/isoamyl alcohol extracted, ethanol precipitated and analyzed on a 6% denaturing polyacrlyamide gel. Markers were prepared by 5′-32P-radiolabeling of a 1 kb ladder (Gibco BRL).
RESULTS
Localization of the RNase H1 proteins
In order to investigate the subcellular localization of the RNase H1 proteins we constructed a plasmid for episomal expression of epitope-tagged versions of the RNase H1 proteins. Six copies of a hemagglutinin epitope tag were inserted just upstream of the C.fasciculata RNH1 stop codon in a plasmid containing the hygromycin phosphotransferase gene as selectable marker in C.fasciculata. The construct pRNH1-HA (Fig. 1A) includes 364 bp of 5′ untranslated sequence upstream of the RNH1 coding region and contains the –235 splice acceptor site (Fig. 6). Wild-type C.fasciculata cells were transformed with pRNH1-HA and immunolocalization performed with an antibody to the hemagglutinin epitope tag. The protein localized specifically to both the nucleus and the kinetoplast (Fig. 2B). The cells were also stained with DAPI (4′,6-diamidino-2-phenylindole) to visualize the kinetoplast (brightly staining body) and the nucleus (diffusely staining body) (Fig. 2A). A similar immunolocalization was observed in the rnhΔ strain (Fig. 2D), in which both chromosomal alleles of RNH1 are disrupted (11).
Figure 1.

HA epitope-tagged RNH1 vectors. The vectors contain selectable markers flanked by the L.major 5′ and 3′ dihydrofolate reductase-thymidylate synthase gene flanking regions for expression of the drug resistance marker. Amino acids are represented by standard nomenclature; black circles indicate stop codons. Sequences are not to scale. (A) pRNH1-HA has a hygromycin phosphotransferase gene as selectable marker and wild-type C.fasciculata RNH1 with six copies of the influenza HA tag cloned just upstream of the stop codon. pRNH1-Phleo has a phleomycin selectable marker but is otherwise the same as pRNH1-HA. To create pRNH1-GTG the second methionine was mutated to a valine, but is otherwise the same as pRNH1-Phleo. (B) pRNH1-STOP is pRNH1-Phleo with two stop codons created by oligonucleotide mutagenesis indicated in the inset. The amino acids altered are underlined. In the wild-type protein the underlined amino acids are TSC.
Figure 6.
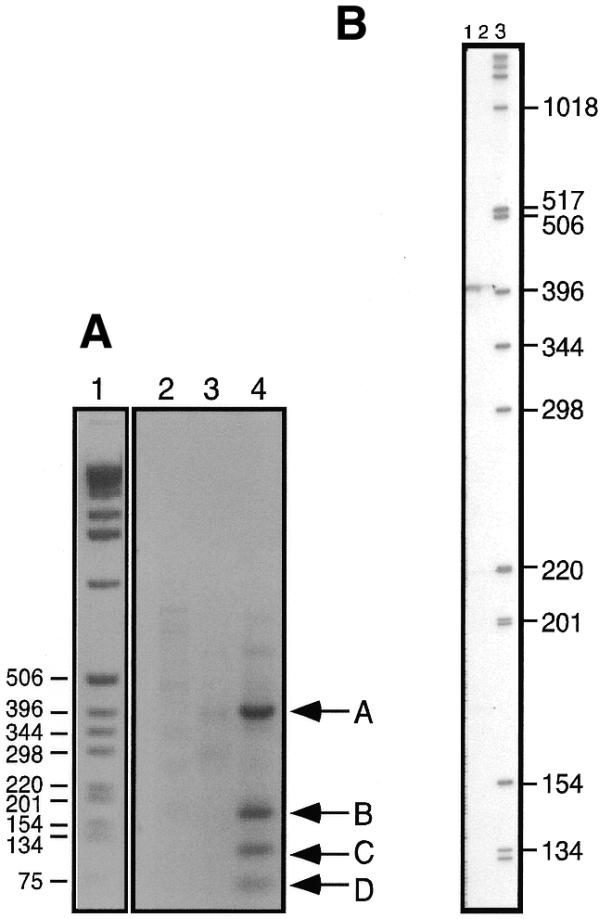
RT–PCR and primer extension analysis of the RNH1 5′ splice acceptor sites. (A) Agarose gel analysis of PCR products. The molecular weight markers (lane 1) are from the same gel as the reaction products but intervening lanes have been removed. Numbers indicate molecular masses (in bp). Lanes 2–4 contain DNA synthesized by RT–PCR using oligonucleotides B24 and poly(A)+ RNA for reverse transcription followed by PCR amplification with Taq polymerase using the spliced leader oligonucleotide (lane 2), B11 oligonucleotide, (lane 3) or oligonucleotides B11 and the spliced leader oligonucleotide (lane 4). Product A corresponds to the splice acceptor site at –235 in Figure 7 (the first nucleotide in the ORF is +1); B, sites –4 and +8; C, sites +58 and +65; D, site +109. (B) Denaturing polyacrylamide gel analysis of primer extension products. Lane 1, primer extension analysis using oligonucleotide B11; lane 2, 50% of the same extension products as in lane 1; lane 3, molecular weight markers. Numbers indicate marker sizes in nucleotides.
Figure 2.
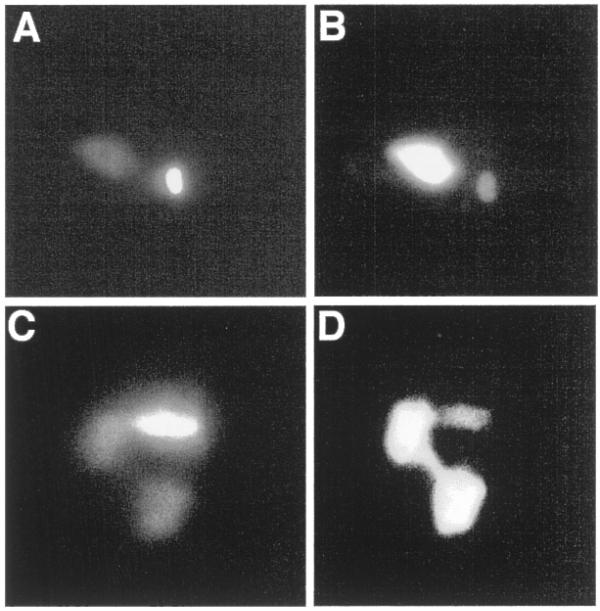
Epitope-tagged RNase H1 is present in both the nucleus and kinetoplast. (A) Wild-type C.fasciculata cells transformed with pRNH1-HA were stained with DAPI to visualize the nuclear and kinetoplast DNA. The faintly staining body is the nucleus; the brightly staining body is the kinetoplast. (B) Immunolocalization of RNH1-HA in the same cell as in (A) using a monoclonal antibody (12CA5) to the HA tag. The brightly fluorescing body is the nucleus and the faintly fluorescing body is the kinetoplast. (C) rnh1Δ strain cells transformed with pRNH1-Phleo were stained with DAPI. In this case the cell was in the process of dividing. The two faintly staining bodies are nuclei and the elongated brightly staining body is the kinetoplast. (D) Immunolocalization of RNH1-HA in the dividing cell shown in (C). Both nuclei and the kinetoplast show immunofluorescence.
Insertion of termination codons between the first and second ATG codons
One form of the RNase H1 protein is localized to the kinetoplast and data from RNase H activity gel experiments employing kinetoplast extracts indicate that the kinetoplast form of the protein is the 45 kDa species (21). Cell fractionation experiments also suggest that the 38 kDa species is the nuclear form of the enzyme (16). The smaller RNase H species has a molecular mass of 38 kDa, in agreement with that predicted from translation initiation at the second AUG codon. To investigate this possibility an RNH1 construct was made which contained a C-terminal HA tag and two tandem stop codons upstream of the second ATG codon in the ORF (Fig. 1B). rnh1Δ strain cells transformed with pRNH1-STOP were stained with DAPI and used for immunolocalization of the HA tag (Fig. 3A and B) The HA-tagged protein was observed only in the nucleus, consistent with production of the nuclear isoform by internal translation initiation. Elimination of targeting of the tagged enzyme to the kinetoplast in cells expressing pRNH1-STOP implies that the 45 kDa protein results from initiation at the first AUG codon. If both proteins were translated from the second AUG codon then the epitope-tagged protein expressed by pRNH1-STOP should have localized to both the nucleus and kinetoplast. However, if the 45 kDa protein is translated from the first AUG codon, only the smaller protein should be produced by the mutant construct and it would be predicted to localize to the nucleus, as observed. In order to reveal even a low level of localization to the kinetoplast, the exposure in Figure 3B relative to that in Figure 2B was adjusted to bring out the background staining of the entire cell. (Similar background staining is observed with untransformed wild-type cells.) Blocking of translation of the N-terminal sequence clearly prevents localization of the protein to the kinetoplast.
Figure 3.
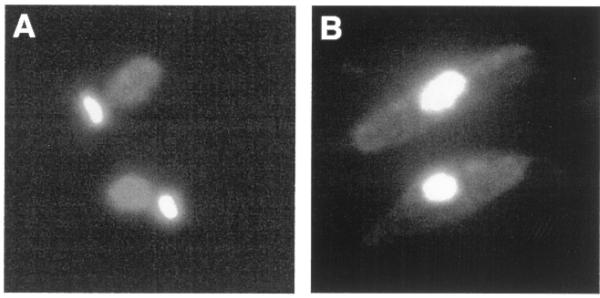
Translation of sequence between the first and second ATG codons in the RNH1 ORF is not necessary for production of the nuclear isoform of RNase H1. Immunolocalization of epitope-tagged RNase H1 in cells transformed with pRNH1-STOP. rnh1Δ cells transformed with pRNH1-STOP were observed by indirect fluorescence microscopy. (A) Cells stained with DAPI to visualize the kinetoplast and nuclear DNA. (B) Immunolocalization of epitope-tagged RNase H1 with monoclonal antibodies to the HA epitope tag.
Mutation of the second ATG codon
To address the possibility that the 45 kDa isoform of the RNase H1 protein might be present in both the kinetoplast and the nucleus, a second mutant construct was made in which the second methionine codon in the RNH1 ORF was mutated to a valine codon. The construct (pRNH1-GTG) was transformed into the rnh1Δ strain and the resulting cells were used for immunolocalization of the epitope tag. In cells stained with DAPI (Fig. 4A) and with antibody to the epitope tag (Fig. 4B) the epitope-tagged protein was only detected in the kinetoplast, demonstrating that the 45 kDa isoform is localized exclusively to the kinetoplast. As in Figure 3B, the long exposure required to visualize immunofluorescence from the kinetoplast enzyme in Figure 4B shows background staining of the whole cell. The significance of the slightly punctate staining elsewhere in the cell has not been determined. It should also be noted that fluorescence from DAPI-stained kinetoplasts as seen in Figures 2–4 is always much more intense than that from the nucleus, even though kDNA represents only ∼25% of total cell DNA. This intense DAPI fluorescence from the kinetoplast (Figs 2A, 3A and 4A) exaggerates the actual size of the kinetoplast. The thinner disc of immunofluorescence from the kinetoplast-localized enzyme (Fig. 4B) more nearly reflects the true size of the kinetoplast.
Figure 4.
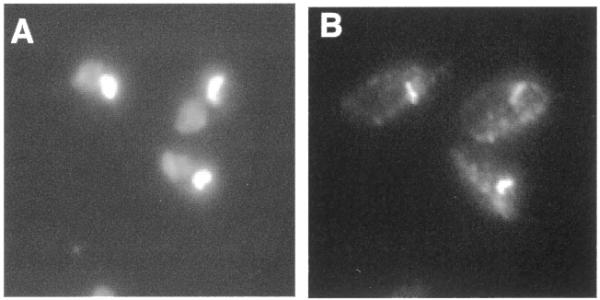
Mutation of the second ATG codon in the RNH1 ORF to a valine codon abolishes expression of the nuclear isoform of RNase H1. Immunolocalization of epitope-tagged RNase H1 in wild-type C.fasciculata cells transformed with pRNH1-GTGH. (A) DAPI staining to visualize the kinetoplast and nuclear DNA. (B) Immunolocalization of the HA-tagged RNase H1.
Activity gel analysis
To further investigate cells transformed with the pRNH1-STOP and pRNH1-GTG constructs, RNase H activity gel analysis was performed on whole cell extracts from wild-type cells, from rnh1Δ cells and from rnh1Δ cells carrying either pRNH1-Phleo, pRNH1-STOP or pRNH1-GTG (Fig. 5). The wild-type 45 and 38 kDa proteins encoded by the RNH1 gene can be seen in the wild-type extract (Fig. 5, lane 1). The RNH1 gene has been disrupted in the rnh1Δ strain and consequently no RNase H activity can be detected in whole cell extracts (Fig. 5, lane 2). The 32 kDa protein showing the RNase H activity of the DNA structure-specific endonuclease SSE1 is not visible in whole cell extracts unless a larger quantity of protein is electrophoresised on the gel (21). When whole cell extracts from the rnh1Δ strain containing pRNH1-Phleo were analyzed, two RNase H activities were observed (Fig. 5, lane 3). The increase in size of the two activities is due to the addition of six copies of the HA tag, which adds 7.7 kDa to each protein. When rnh1Δ cells expressing pRNH1-STOP were analyzed by activity gel (Fig. 5, lane 4) only one activity was detected, corresponding to the smaller protein (compare with Fig. 5, lane 3). Similarly, when rnh1Δ cells expressing pRNH1-GTG were analyzed (Fig. 5, lane 5) only one activity was visible, corresponding to the larger protein. These results, together with the immunolocalizations, indicate that the 38 kDa RNase H1 protein is the nuclear isoform and the 45 kDa RNase H1 protein is the kinetoplast isoform of RNase H1 protein.
Figure 5.

RNase H activity gel analysis of epitope-tagged RNH1 constructs. Lane 1, wild-type C.fasciculata whole cell extract. The wild-type 45 and 38 kDa activities are indicated by arrows. Lane 2, rnh1Δ whole cell extract. Lane 3, whole cell extract from the rnh1Δ strain transformed with pRNH1-Phleo. Lane 4, whole cell extract from the rnh1Δ strain transformed with pRNH1-STOP showing an activity migrating at ∼46 kDa. Lane 5, whole cell extract from the rnh1Δ strain transformed with pRNH1-GTG showing an activity migrating at ∼53 kDa. The RNase H activity of the SSE1 nuclease is not visible in this exposure.
Alternative splicing of the RNH1 transcripts
In an effort to understand how the RNH1 gene produces two proteins having different subcellular localizations, we analyzed the pattern of RNA splicing of the RNH1 transcript. Genes are transcribed in trypanosomes as polycistronic transcripts that are then processed by trans-splicing a spliced leader onto all mRNAs (28,29). We used a spliced leader oligonucleotide together with an oligonucleotide from the coding sequence to determine the 5′ splice acceptor sites of transcripts by reverse transcription–PCR (RT–PCR) (Fig. 6A). Four specific products, labeled A–D (Fig. 6A, lane 4), were produced by PCR with a spliced leader primer and primer B11 using template cDNA made with primer B24, shown in Figure 7. Each PCR product was cloned and sequenced to identify the location of each of the splice acceptor sites (Fig. 7). Two classes of transcripts are produced by trans-splicing. In one class splicing occurs upstream of the first methionine codon and in the second class trans-splicing occurs between the first and second methionine codons. The second methionine is marked with a circle. The splice acceptor site at –235 is the predominant product by primer extension (Fig. 6B, lane 1). No additional splice acceptor sites were found closer to the second methionine codon when oligonucleotide primers downstream of B11 and B24 were used for RT–PCR (data not shown). Although representing minor transcripts detected by PCR, the existence of splice sites downstream of the first ATG at least raises the possibility that such transcripts could also give rise to the 38 kDa nuclear enzyme. Further studies will be required to distinguish this possibility from that of internal translation initiation at the second AUG on the full-length mRNA. However, the relatively much higher level of the 38 kDa nuclear protein compared to that of the 45 kDa protein and the low level of these shorter transcripts, as indicated by their detection only by PCR and not by primer extension, strongly suggests that the 38 kDa protein results primarily from internal translation initiation on the full-length transcript. The possibility that these minor transcripts could be more abundant under some other physiological conditions and therefore contribute significantly to the production of the nuclear enzyme is an interesting posibility and cannot be excluded.
Figure 7.

Map of RNH1 splice acceptor sites. Partial sequence of the RNH1 gene is shown. The underlined regions indicate sequences complementary to oligonucleotides used for cDNA synthesis or for PCR with miniexon oligonucleotide. The arrows indicate the splice acceptor sites mapped by sequencing the PCR products in Figure 6. The circled methionine is the second methionine in the ORF. The sequence is numbered from the first nucleotide of the ORF.
DISCUSSION
We have shown here that the two proteins encoded by the RNH1 gene localize to different organelles. Mutational analysis demonstrated that the smaller 38 kDa protein is nuclear and is translated from the second AUG codon of the ORF. The 45 kDa kinetoplast-associated protein appears to be derived from the predicted 53.7 kDa precursor protein resulting from initiation at the first AUG codon in the RNH1 ORF. These results suggest that an approximately 80 amino acid cleavable presequence would need to be removed to generate the 45 kDa enzyme. While this is an extremely large presequence, mitochondrial import signals close to this size have been reported previously. In peas (Pisum sativum) the P subunit of glycine decarboxylase has an 85 amino acid presequence (30) and an 80 amino acid presequence is removed from yeast cytochrome b2 upon import into the mitochondrion (31). Of some 56 yeast precursor proteins with known mature N-termini, only three contained leader peptides longer than 50 amino acids (32). Interestingly, the only mitochondrial import signals recognized to date in C.fasciculata have been nine amino acid cleavable presequences seen on the H1 histone-like KAP proteins and the polymerase β protein (3,33,34). The nine N-terminal amino acids of the predicted 53.7 kDa protein share some features with these cleavable presequences. The sequence lacks acidic residues and contains hydrophobic residues and an Arg-Arg dipeptide. However, it lacks a leucine at the second residue, which is present in four of the five nine-amino acid cleavable presequences. Three other C.fasciculata mitochondria-localized proteins, structure-specific endonuclease 1, topoisomerase II and cytochrome c1, lack N-terminal cleavable presequences (23,27,35).
There are many examples in other eukaryotes of a single gene encoding proteins that are distributed to multiple subcellular compartments (22). Such proteins are termed sorting isozymes. Signals responsible for sorting isozyme distribution have been revealed by comparisons of eukaryotic sorting isozymes to their counterpart proteins in non-eukaryotic organisms, on the assumption that sequences important for catalytic function will be conserved. From phylogenetic comparisons and sequence alignments of five different families of proteins containing at least one confirmed sorting isozyme it was found that the eukaryotic proteins contained additional sequences not present in the eubacterial or archaeal counterparts. These additional sequences have been named ADEPTs (additional domains for eukaryotic protein targeting) and can be present on the N- or C-termini or located internally (22).
Mitochondrial targeting sequences have often been found at the N-terminus as a cleavable presequence. Such sequences are rich in basic residues and hydroxylated amino acids and are generally devoid of acidic amino acids (36). In some cases such sequences have been successfully fused to ‘passenger’ proteins, resulting in their targeting to the mitochondrial matrix (37). Some genes encoding isozymes have been found to target isozymes to as many as three intracellular compartments. In addition to having an N-terminal sequence necessary for mitochondrial targeting, the Saccharomyces cerevisiae MOD5 and CCA1 genes encoding tRNA processing enzymes express isozymes that target to the nucleus as a consequence of translation initiation at in-frame AUGs (38,39). In both cases the nuclear and mitochondrial forms are also found in the cytoplasm.
In the case of RNH1 the major RNH1 mRNA is transpliced upstream of the first AUG codon in the ORF and is predicted to produce a 53.7 kDa protein upon translation from the first AUG. The 45 kDa kinetoplast form of RNase H is proposed to result from processing of the 53.7 kDa protein in the course of import into the mitochondrion. Crithidia fasciculata RNase H and other eukaryotic RNases H have sequence and structural conservation with prokaryotic RNase HI (40–42). The RNase H domain is located in the C-terminal part of the protein in RNase H proteins from S.cerevisiae, Schizosaccharomyces pombe and a chicken cDNA encoding a protein that is likely to be a chicken homolog of RNase HI. These eukaryotic RNase H enzymes all have an N-terminal extension not present in the related prokaryotic enzymes (43) and which is characterized by the presence of a conserved sequence of approximately 40 residues. This sequence has been shown to confer binding to double-stranded RNA and is not required for RNase H activity. The C.fasciculata RNH1 ORF encodes an additional N-terminal extension of approximately 150 amino acids which we have shown here to be necessary for production of the 45 kDa mitochondrial isozyme. So far, the C.fasciculata RNH1 gene is the only known example of a eukaryotic gene in the RNase HI family that encodes a mitochondrial RNase H. An RNase H activity has been identified in mitochondrial lysates from beef heart and from Xenopus oocytes, but it is not known whether this enzyme represents a sorting isoform nor has the enzyme been immunolocalized to the mitochondrion (44). The only other member of the RNase HI gene family identified in a trypanosomatid is the TbbRNHI gene from Trypanosoma brucei (45,46). The TbbRNHI gene expresses a 33.1 kDa protein closely related to the C.fasciculata 38 kDa protein and is localized to the nucleus. The N-terminal domain of the Tbb RNase HI protein shows a high degree of sequence similarity to the N-terminal domain of the C.fasciculata 38 kDa protein and was shown to be essential for nuclear localization. So far, a mitochondrial isoform of the T.brucei protein has not been identified.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Stephen Beverly for the gift of pXGPHLEO and pX63HYG. This work was funded by NIH grants GM53254 and AI45536 to D.S.R. and by an Edwin W. Pauley Fellowship to M.L.E.
DDBJ/EMBL/GenBank accession no. L18916
References
- 1.Shapiro T.A. and Englund,P.T. (1995) The structure and replication of kinetoplast DNA. Annu. Rev. Microbiol., 49. 117–143. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson M., Torri,A.F., Ward,D.C. and Englund,P.T. (1992) In situ hybridization to the Crithidia fasciculata kinetoplast reveals two antipodal sites involved in kinetoplast DNA replication. Cell, 70, 621–629. [DOI] [PubMed] [Google Scholar]
- 3.Xu C.W., Hines,J.C., Engel,M.L., Russell,D.G. and Ray,D.S. (1996) Nucleus-encoded histone H1-like proteins are associated with kinetoplast DNA in the trypanosomatid Crithidia fasciculata. Mol. Cell. Biol., 16, 564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen J., Rauch,C.A., White,J.H., Englund,P.T. and Cozzarelli,N.R. (1995) The topology of the kinetoplast DNA network. Cell, 80, 61–69. [DOI] [PubMed] [Google Scholar]
- 5.Englund P.T. (1979) Free minicircles of kinetoplast DNA networks in Crithidia fasciculata. J. Biol. Chem., 254, 4895–4900. [PubMed] [Google Scholar]
- 6.Sheline C., Melendy,T. and Ray,D.S. (1989) Replication of DNA minicircles in kinetoplasts isolated from Crithidia fasciculata: structure of nascent mini circles. Mol. Cell. Biol., 9, 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitchin P.A., Klein,V.A. and Englund,P.T. (1985) Intermediates in the replication of kinetoplast DNA minicircles. J. Biol. Chem., 260, 3844–3851. [PubMed] [Google Scholar]
- 8.Birkenmeyer L., Sugisaki,H. and Ray,D.S. (1987) Structural characterization of site-specific discontinuities associated with replication origins of minicircle DNA from Crithidia fasciculata. J. Biol. Chem., 262, 2384–2392. [PubMed] [Google Scholar]
- 9.Vonwirth H., Köck,J. and Büsen,W. (1991) Class I and class II ribonuclease H activities in Crithidia fasciculata (Protozoa). Experientia, 47, 92–95. [DOI] [PubMed] [Google Scholar]
- 10.Crouch R.J. (1990) Ribonuclease H: from discovery to 3D structure. New Biol., 2, 771–777. [PubMed] [Google Scholar]
- 11.Ray D.S. and Hines,J.C. (1995) Disruption of the Crithidia fasciculata RNH1 gene results in the loss of two active forms of ribonuclease H. Nucleic Acids Res., 23, 2526–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goulian M., Richards,S.H., Heard,C.J. and Bigsby,B.M. (1990) Discontinuous DNA synthesis by purified mammalian proteins. J. Biol. Chem., 265, 18461–18471. [PubMed] [Google Scholar]
- 13.Huang L., Kim,Y., Turchi,J.J. and Bambara,R.A. (1994) Structure-specific cleavage of the RNA primer from Okazaki fragments by calf thymus RNase HI. J. Biol. Chem., 269, 25922–25927. [PubMed] [Google Scholar]
- 14.Ishimi Y., Claude,A., Bullock,P. and Hurwitz,J. (1988) Complete enzymatic synthesis of DNA containing the SV40 origin of replication. J. Biol. Chem., 263, 19723–19733. [PubMed] [Google Scholar]
- 15.DiFrancesco R.A. and Lehman,I.R. (1985) Interaction of ribonuclease H from Drosophila melanogaster embryos with DNA polymerase-primase. J. Biol. Chem., 260, 14764–14770. [PubMed] [Google Scholar]
- 16.Ogawa T. and Okazaki,T. (1984) Function of RNase H in DNA replication revealed by RNase H defective mutants of Escherichia coli. Mol. Gen. Genet., 193, 231–237. [DOI] [PubMed] [Google Scholar]
- 17.Kurosawa Y., Ogawa,T., Hirose,S., Okazaki,T. and Okazaki,R. (1975) Mechanism of DNA chain growth. XV. RNA-linked nascent DNA pieces in Escherichia coli strains assayed with spleen exonuclease. J. Mol. Biol., 96, 653–664. [DOI] [PubMed] [Google Scholar]
- 18.Kaguni J.M. and Kornberg,A. (1984) Replication initiated at the origin (oriC) of the E. coli chromosome reconstituted with purified enzymes. Cell, 38, 183–190. [DOI] [PubMed] [Google Scholar]
- 19.Dasgupta S., Masukata,H. and Tomizawa,J. (1987) Multiple mechanisms for initiation of ColE1 DNA replication: DNA synthesis in the presence and absence of ribonuclease H. Cell, 51, 1113–1122. [DOI] [PubMed] [Google Scholar]
- 20.Campbell A.G. and Ray,D.S. (1993) Functional complementation of an Escherichia coli ribonuclease H mutation by a cloned genomic fragment from the trypanosomatid Crithidia fasciculata. Proc. Natl Acad. Sci. USA, 90, 9350–9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engel M.L. and Ray,D.S. (1998) A structure-specific DNA endonuclease is enriched in kinetoplasts purified from Crithidia fasciculata. Nucleic Acids Res., 26, 4733–4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stanford D.R., Martin,N.C. and Hopper,A.K. (2000) ADEPTs: information necessary for subcellular distribution of eukaryotic sorting isozymes resides in domains missing from eubacterial and archaeal counterparts. Nucleic Acids Res., 28, 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pasion S.G., Hines,J.C., Aebersold,R. and Ray,D.S. (1992) Molecular cloning and expression of the gene encoding the kinetoplast-associated type II DNA topoisomerase of Crithidia fasciculata. Mol. Biochem. Parasitol., 50, 57–67. [DOI] [PubMed] [Google Scholar]
- 24.Cruz A., Coburn,C.M. and Beverley,S.M. (1991) Double targeted gene replacement for creating null mutants. Proc. Natl Acad. Sci. USA, 88, 7170–7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Freedman D.J. and Beverley,S.M. (1993) Two more independent selectable markers for stable transfection of Leishmania. Mol. Biochem. Parasitol., 62, 37–44. [DOI] [PubMed] [Google Scholar]
- 26.Pasion S.G., Brown,G.W., Brown,L.M. and Ray,D.S. (1994) Periodic expression of nuclear and mitochondrial DNA replication genes during the trypanosomatid cell cycle. J. Cell Sci., 107, 3515–3520. [DOI] [PubMed] [Google Scholar]
- 27.Engel M.L. and Ray,D.S. (1999) The kinetoplast structure-specific endonuclease I is related to the 5′ exo/endonuclease domain of bacterial DNA polymerase I and colocalizes with the kinetoplast topoisomerase II and DNA polymerase beta during replication. Proc. Natl Acad. Sci. USA, 96, 8455–8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson P.J., Kooter,J.M. and Borst,P. (1987) Inactivation of transcription by UV irradiation of T. brucei provides evidence for a multicistronic transcription unit including a VSG gene. Cell, 51, 273–281. [DOI] [PubMed] [Google Scholar]
- 29.Muhich M.L. and Boothroyd,J.C. (1988) Polycistronic transcripts in trypanosomes and their accumulation during heat shock: evidence for a precursor role in mRNA synthesis. Mol. Cell. Biol., 8, 3837–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turner S.R., Ireland,R. and Rawsthorne,S. (1992) Cloning and characterization of the P subunit of glycine decarboxylase from pea (Pisum sativum). J. Biol. Chem., 267, 5355–5360. [PubMed] [Google Scholar]
- 31.Guiard B. (1985) Structure, expression and regulation of a nuclear gene encoding a mitochondrial protein: the yeast L(+)-lactate cytochrome c oxidoreductase (cytochrome b2). EMBO J., 4, 3265–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Branda S.S. and Isaya,G. (1995) Prediction and identification of new natural substrates of the yeast mitochondrial intermediate peptidase. J. Biol. Chem., 270, 27366–27373. [DOI] [PubMed] [Google Scholar]
- 33.Hines J.C. and Ray,D.S. (1998) The Crithidia fasciculata KAP1 gene encodes a highly basic protein associated with kinetoplast DNA. Mol. Biochem. Parasitol., 94, 41–52. [DOI] [PubMed] [Google Scholar]
- 34.Torri A.F. and Englund,P.T. (1995) A DNA polymerase β in the mitochondrion of the trypanosomatid Crithidia fasciculata. J. Biol. Chem., 270, 1–3. [DOI] [PubMed] [Google Scholar]
- 35.Priest J.W., Wood,Z.A. and Hajduk,S.L. (1993) Cytochromes c1 of kinetoplastid protozoa lack mitochondrial targeting presequences. Biochim. Biophys. Acta, 1144, 229–231. [DOI] [PubMed] [Google Scholar]
- 36.Attardi G. and Schatz,G. (1988) Biogenesis of mitochondria. Annu. Rev. Cell. Biol., 4, 289–333. [DOI] [PubMed] [Google Scholar]
- 37.Van Steeg H., Oudshoorn,P., Van Hell,B., Polman,J.E. and Grivell,L.A. (1986) Targeting efficiency of a mitochondrial pre-sequence is dependent on the passenger protein. EMBO J., 5, 3643–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolfe C.L., Lou,Y.C., Hopper,A.K. and Martin,N.C. (1994) Interplay of heterogeneous transcriptional start sites and translational selection of AUGs dictate the production of mitochondrial and cytosolic/nuclear tRNA nucleotidyltransferase from the same gene in yeast. J. Biol. Chem., 269, 13361–13366. [PubMed] [Google Scholar]
- 39.Boguta M., Hunter,L.A., Shen,W.C., Gillman,E.C., Martin,N.C. and Hopper,A.K. (1994) Subcellular locations of MOD5 proteins: mapping of sequences sufficient for targeting to mitochondria and demonstration that mitochondrial and nuclear isoforms commingle in the cytosol. Mol. Cell. Biol., 14, 2298–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanaya S., Kohara,A., Miura,Y., Sekiguchi,A., Iwai,S., Inoue,H., Ohtsuka,E. and Ikehara,M. (1990) Identification of the amino acid residues involved in an active site of Escherichia coli ribonuclease H by site-directed mutagenesis. J. Biol. Chem., 265, 4615–4621. [PubMed] [Google Scholar]
- 41.Katayanagi K., Miyagawa,M., Matsushima,M., Ishikawa,M., Kanaya,S., Ikehara,M., Matsuzaki,T. and Morikawa,K. (1990) Three-dimensional structure of ribonuclease H from E. coli. Nature, 347, 306–309. [DOI] [PubMed] [Google Scholar]
- 42.Yang W., Hendrickson,W.A., Crouch,R.J. and Satow,Y. (1990) Structure of ribonuclease H phased at 2 Å resolution by MAD analysis of the selenomethionyl protein. Science, 249, 1398–1405. [DOI] [PubMed] [Google Scholar]
- 43.Cerritelli S.M., Fedoroff,O.Y., Reid,B.R. and Crouch,R.J. (1998) A common 40 amino acid motif in eukaryotic RNases H1 and caulimovirus ORF VI proteins binds to duplex RNAs. Nucleic Acids Res., 26, 1834–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pileur F., Toulme,J.J. and Cazenave,C. (2000) Eukaryotic ribonucleases HI and HII generate characteristic hydrolytic patterns on DNA-RNA hybrids: further evidence that mitochondrial RNase H is an RNase HII. Nucleic Acids Res., 28, 3674–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kobil J.H. and Campbell,A.G. (2000) Trypanosoma brucei RNase HI requires its divergent spacer subdomain for enzymatic function and its conserved RNA binding motif for nuclear localization. Mol. Biochem. Parasitol., 107, 135–142. [DOI] [PubMed] [Google Scholar]
- 46.Hesslein D.G. and Campbell,A.G. (1997) Molecular cloning and expression of a ribonuclease H from the kinetoplastid, Trypanosoma brucei. Mol. Biochem. Parasitol., 86, 121–126. [DOI] [PubMed] [Google Scholar]