

Abstract
We examined how crowding of the surfaces of lipid vesicles with either grafted polyethyleneglycol (PEG) chains or bilayer-anchored protein molecules affects the binding of soluble proteins to the vesicle surface. Escherichia coli dihydrofolate reductase (DHFR, 18 kDa) or a larger fusion protein, NusA-DHFR (72 kDa), binds reversibly but with high affinity to a methotrexate-modified lipid (MTX-PE) incorporated into large unilamellar vesicles. Incorporation of phosphatidylethanolamine-PEG5000 into the vesicles strongly decreases the affinity of binding of both proteins, to a degree that varies roughly exponentially with the lateral density of the PEG chains. Covalently coupling maltose-binding protein (MBP) to the vesicle surfaces also strongly decreases the affinity of binding of NusDHFR or DHFR, to a degree that likewise varies roughly exponentially with the surface density of anchored MBP. Surface-coupled MBP strongly decreases the rate of binding of NusDHFR to MTX-PE-incorporating vesicles but does not affect the rate of NusDHFR dissociation. The large magnitudes of these effects (easily exceeding an order of magnitude for moderate degrees of surface crowding) support previous theoretical analyses and suggest that surface-crowding effects can markedly influence a variety of important aspects of protein behavior in membranes.
Introduction
The surfaces of biological membranes are crowded structures in which a large portion of the total surface area is occupied by protein molecules and the free area fraction is correspondingly limited (1–6). This feature of membrane structure may affect diverse aspects of the behavior of both permanently membrane-bound and reversibly membrane-associating proteins, and theoretical treatments have suggested that the magnitude of these effects may be large for surfaces incorporating proteins at the lateral densities found in biological membranes (7–9). However, only a few studies to date have examined experimentally the thermodynamics of crowding effects in model or biological membrane systems. Polyethyleneglycol (PEG) chains anchored to bilayer-intercalated lipid molecules have been shown to antagonize the adsorption of macromolecules to lipid surfaces, but only a few studies have examined this effect in a quantitative manner (10–12). Still fewer experimental data on the thermodynamics of steric interactions between proteins at membrane surfaces have been reported (2,13,14).
We here describe an experimental system to assess quantitatively how surface crowding modulates protein binding to a membrane surface. Escherichia coli dihydrofolate reductase (DHFR) binds with high affinity, but reversibly, to lipid vesicles incorporating MTX-PE, a lipid derivative of the DHFR inhibitor methotrexate (MTX). Using centrifugation- and fluorescence-based assays, we measured the affinity and kinetics of binding of DHFR and a larger DHFR-containing fusion protein, NusDHFR, to MTX-PE incorporated into vesicles whose surfaces were crowded with varying lateral densities of either flexible polymer (PEG) chains or an anchored globular protein. In agreement with previous theoretical analyses, our results demonstrate that crowding effects alter both the affinity and the kinetics of binding of proteins to the bilayer surface, by factors of an order of magnitude or larger, when the density of macromolecules present at the surface approaches that observed in biological membranes. As we discuss further below, the steric effects quantified here are relevant for understanding how membrane surface crowding affects not only binding of soluble proteins to specific membrane lipids, as examined in our measurements, but also a variety of other important aspects of the behavior of both reversibly and permanently membrane-associated proteins.
Materials and Methods
Materials
1-Palmitoyl-2-oleoylphosphatidylcholine (POPC),-phosphatidylethanolamine (POPE), and -phosphatidylglycerol (POPG) were obtained from Avanti Polar Lipids (Alabaster, AL). N-(lissamine rhodaminesulfonyl)-POPE (RhoPE) was synthesized from dioleoyl POPE according to the method of Struck et al. (15). All common chemicals and synthetic reagents were of reagent grade or better.
POPE-PEG3-NHBoc was prepared by activating 25 μmol 8-(N-t-Boc-amino-3,6-dioxaoct-1-yl)-succinamic acid (16) overnight at 20°C with 1 equivalent dicyclohexylcarbodiimide and 1.1 equivalent N-hydroxysuccinimide in 0.2 mL 1:1 dry CH2Cl2/dimethylformamide, then adding the soluble fraction to 20 μmol POPE in 350 μL dry CH2Cl2 containing 100 μmol diisopropylethylamine and 20 μmol trifluoroacetic acid. After stirring for 4 h at 37°C, the reaction products were partitioned between CH2Cl2 and 1:1 methanol/0.5 M Mes, pH 6.0. The CH2Cl2 phase was concentrated in vacuo and the products were purified by preparative TLC in 50:20:7.5:7.5:4 CH2Cl2/acetone/methanol/CH3COOH/water. N-deprotection with HCl in dry CH2Cl2 (17) gave POPE-PEG3-NH2 in an overall yield of 72%. N-(4-maleimidobutyroyl)-POPE was prepared by reacting 20 μmol of well-dried POPE with 30 μmol 4-maleimidobutyric acid succinimidyl ester (Toronto Research Chemicals, Toronto, Ontario, Canada) in 1 mL anhydrous CH2Cl2 containing 100 μmol diisopropylethylamine and 20 μmol trifluoroacetic acid (4 h, 22°C), purified by preparative thin-layer chromatography as described above and coupled to γ-cysteamidomethotrexate as described previously (18) to yield MTX-PE. N-(4-maleimidobutyroyl)-PEG3-POPE was prepared from POPE-PEG3-NH2 as described above for N-(4-maleimidobutyroyl)-POPE. POPE-PEG5000 was synthesized from POPE and the succinic half-ester of monomethoxy-PEG5000 (19) using the procedure described above for synthesis of POPE-PEG3-NHBoc.
Plasmid constructs
A polymerase chain reaction (PCR) product encoding the mature form of E. coli maltose-binding protein (MBP) with a C-terminal–SerCys extension was prepared using pMAL-c4x (New England Biolabs, Pickering, Ontario, Canada) as the template and the 5′- and 3′-primers CATATGAAAATCGAAGAAGGTAAACTGGTAATC and TCTAGATTAACAACTGAATTCTGAAATCCTTCCCTCG. The product was subcloned between the NdeI and XbaI sites of pMAL-c4x to give the plasmid pMAL(MBPCys). A PCR fragment incorporating the complete coding sequence of E. coli NusA (replacing the C-terminal stop codon with an -ITSLYK-encoding linker sequence) and flanked by NdeI and KpnI sites was prepared using E. coli JM109 chromosomal DNA as the template, and ligated between the NdeI and KpnI sites of pET-42a-c(+) (Invitrogen Canada, Burlington, Ontario, Canada). Between the KpnI and BamHI sites of the resulting plasmid, a PCR fragment was ligated that comprised the complete coding sequence of E. coli DHFR flanked by the 5′-sequence -GGTACCGAAACCTGTACTTCCAGGGA- and a 3′-XbaI restriction sequence, to give the plasmid pET(NusDHFR). A PCR fragment encoding the complete coding sequence of E. coli DHFR and flanked by a 5′-NdeI site and a 3′-BamHI site was prepared using E. coli chromosomal DNA as the template, and ligated between the NdeI and BamHI sites of pMAL-c4x to produce the plasmid pMAL(DHFR).
Protein preparation and labeling
All protein purification steps were carried out at 0–4°C. NusDHFR was expressed in E. coli strain BL21 transformed with pET(NusDHFR) using the autoinduction method of Studier (20). Twenty-two hours after the culture (500 mL) was shifted to 20°C, the cells were harvested and disrupted via a French press in 50 mL of 137 mM NaCl, 2.7 mM KCl, 4.3 mM NaH2PO4, 1.47 mM K2HPO4, pH 7.4, containing 5 mM DTT, 1 mM phenylmethanesulfonyl fluoride (PMSF), and 2.5 μg/mL each of pepstatin, leupeptin, and aprotinin. After centrifugation (13,000 × g, 20 min) NusDHFR was purified from the supernatant by chromatography on MTX-agarose (21), yielding 60–80 mg of purified protein per liter of culture. DHFR was prepared in a similar manner from E. coli BL21 transformed with pMAL(DHFR). MBPCys was expressed in pMAL(MBPCys)-transformed E. coli by incubating cultures first at 37°C in LB medium containing 0.2% glucose and 100 μg/mL ampicillin to OD660 nm = 0.6, then adding isopropylthiogalactoside to 0.3 mM and culturing for 3 h more at 30°C. The cell pellet harvested from 1 L of culture was resuspended in 60 mL of 200 mM NaCl, 20 mM NaH2PO4, 1 mM EDTA, pH 7.4, containing 5 mM DTT, 1 mM PMSF, and 2.5 μg/mL each of pepstatin, leupeptin, and pepstatin, then disrupted with a French press. After centrifugation (13,000 × g, 20 min), the supernatant was applied to a 10-mL column of amylose resin (New England Biolabs) that was washed with 60 mL of 200 mM NaCl, 20 mM NaH2PO4, 1 mM each of EDTA and DTT, pH 7.4. MBPCys was then eluted with 60 mL of 10 mM maltose in the same buffer. The protein was further purified on a 25-mL column of DEAE-Sepharose Fast Flow (Sigma-Aldrich Canada, Oakville, Ontario, Canada), eluting with a gradient of 20–400 mM NaCl in 10 mM MOPS, 1 mM DTT, pH 7.0, to yield 50–60 mg of protein per liter of culture.
For fluorescence labeling of NusDHFR and DHFR, the pentafluorophenyl ester of 5(6)-carboxyfluorescein was first prepared by incubating 1.0 mg carboxyfluorescein, 0.64 mg pentafluorophenol, and 0.55 mg dicyclocarbodiimide overnight in the dark in 60 μL anhydrous dimethylformamide. A 7.8-μL aliquot of the supernatant from this reaction was added to 4 mg of DHFR or NusDHFR dissolved in 400 μL 150 mM KCl, 10 mM KH2PO4, 0.1 mM EDTA, 3 mM folate, pH 7.0, plus 50 μL of 1 M NaHCO3. The mixture was stirred for 1 h at 22°C in the dark, then chromatographed on Sephadex G-50 in 150 mM KCl, 10 mM KH2PO4, 1 mM DTT, 0.1 mM EDTA to remove uncoupled dye.
Liposome and proteoliposome preparations
Lipid mixtures were dispensed from stocks in CH2Cl2 or CH2Cl2/methanol 9:1, dried under nitrogen, and further dried under high vacuum for at least 2 h. The lipids were then vortexed (at 10–20 mM lipid) in sucrose/KCl buffer (176 mM sucrose, 50 mM KCl, 5 mM KH2PO4, 1 mM EDTA, pH 7.0) and extruded through 0.1-μm pore size polycarbonate filters using a hand-held extrusion device (Avestin, Ottawa, Canada) to generate large unilamellar vesicles. To conjugate MBPCys to vesicles, aliquots of protein (0.2–6 mg), freshly incubated for 1 h in sucrose/KCl buffer containing 1 mM DTT, were passed through 1-mL BioGel P-6 spin columns (22) to remove the DTT and then immediately mixed with 4 μmol of lipid vesicles. The protein/vesicle mixtures were gently agitated for 1–16 h under argon and with exclusion of light, then further incubated with cysteine (5 mM, 30 min) and finally passed through a 10-mL column of Sepharose 6B-CL in sucrose/KCl buffer to remove uncoupled protein. Protein was assayed by means of the Lowry protein assay (BioRad Laboratories, Mississauga, Ontario, Canada) using bovine plasma γ-globulin as a standard. The concentrations of MBP determined by this method agreed closely with those calculated from protein absorbance based on the reported value of A280 nm = 1.46 for a 1 mg mL−1 solution of the protein (23). Phospholipid was assayed according to the method of Lowry and Tinsley (24) for lipid stock solutions and for solvent extracts of vesicle lipids (the latter prepared by the method of Bligh and Dyer (25)).
Equilibrium protein-vesicle binding measurements
For equilibrium protein-vesicle binding measurements, 1.5-μg or 5-μg portions of fluorescein-labeled DHFR or NusDHFR, respectively, were incubated for 1–2 h at 22°C in the dark with lipid or lipid/MBPCys vesicles (0–200 μM lipid), incorporating the indicated molar percentage of MTX-PE, in 4 mL of 150 mM KCl, 5 mM KH2PO4, 1 mM EDTA, 1 mM DTT, pH 7.0, then centrifuged for 1 h in polycarbonate tubes (Beckmann 75Ti rotor, 120,000 × g, 4°C). The bulk of the supernatant (upper fraction, volume = Vupper) was recovered, as was the pellet together with a small, accurately measured volume of residual supernatant (lower fraction, volume = Vlower). The fluorescence in both fractions was determined at λex/λem = 495/517 nm and 525/596 nm (excitation/emission slits 5/5 nm) to measure fluorescein-labeled DHFR/NusDHFR and RhoPE (the latter as a lipid marker to quantify the completeness of vesicle sedimentation), respectively. After appropriate blank and spectral-spillover corrections, these readings (designated below as FLc(protein)upper, etc.) were used to calculate the percentage of labeled protein bound to the vesicles using the following equations:
| (1a) |
| (1b) |
| (2) |
The percentage of vesicle-bound DHFR/NusDHFR thereby determined for each sample was plotted versus the concentration of protein-accessible (surface-exposed) MTX-PE calculated from the total phospholipid concentration in the sample, the molar fraction of MTX-PE in the phospholipid mixture, and the fraction of vesicle lipid exposed at the outer surface (determined by the method of Nordlund et al. (26) to be 0.48 ± .03 for equivalent vesicles incorporating 5 mol % POPE). The value of Kd for binding of DHFR/NusDHFR to vesicle-incorporated MTX-PE was then determined from the resulting hyperbolic curves as illustrated in Fig. 1.
Figure 1.
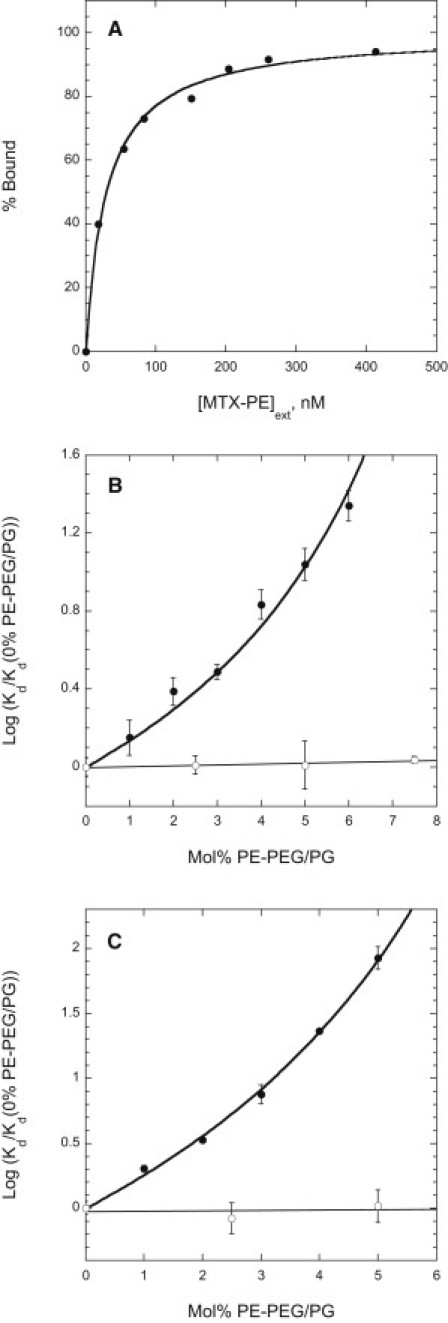
(A) Representative binding curve for association of fluorescein-labeled NusDHFR with POPC/MTX-PE/RhoPE (99.5:0.5:0.02 molar proportions) vesicles, determined using the ultracentrifugation-based assay described in Materials and Methods. Data are fit to an equation of the form (% Bound) = A+B∗[MTX-PE]ext/([MTX-PE]ext + Kd), where [MTX-PE]ext is the concentration of MTX-PE exposed at the vesicles' outer surface, and A, B and Kd are adjustable parameters. (B and C) Dissociation constants determined for binding of DHFR (B) or NusDHFR (C) to vesicles prepared from POPC, 0.5 mol % MTX-PE, 0.02 mol % RhoPE, and the indicated molar percentages of POPE-PEG5000 (solid circles) or POPG (open circles). The Kd values shown (normalized to the value of Kd determined in the same experiment for vesicles lacking POPE-PEG5000/POPG) represent the mean (± SE) of values determined in three independent experiments. Data are fitted to theoretical curves as described in the text.
Kinetic measurements
For kinetic measurements, 3 μg of fluorescein-labeled NusDHFR were dispensed into a stirred fluorimeter cuvette containing 3 mL of 150 mM KCl, 5 mM KH2PO4, 1 mM EDTA, 1 mM DTT, pH 7.0, at 22°C. POPC/MTX-PE/RhoPE vesicles (95:4:1 molar proportions, prepared as described above) were added, and the fluorescence was monitored continuously as the fluorescent-labeled protein gradually bound to the quencher-containing vesicles. Free MTX or a 20-fold excess of sonicated POPC/MTX-PE (96:4 mol/mol) vesicles was then added, and the resulting rise in fluorescence was recorded to monitor the kinetics of dissociation of the protein from the vesicles.
Results
Association of DHFR and NusDHFR with lipid vesicles incorporating MTX-PE
Fluorescein-labeled E. coli trimethoprim-sensitive DHFR (M = 18.0 kDa) or a DHFR-NusA fusion protein (NusDHFR, M = 73.8 kDa) binds reversibly (as shown below) but with high affinity to large unilamellar vesicles incorporating a MTX-conjugated derivative of POPE (MTX-PE; structure shown in Fig. S1 of the Supporting Material). We used a centrifugation-based method as described in Materials and Methods to measure the binding of tracer amounts of these proteins to varying concentrations of vesicles incorporating MTX-PE, and from these data we determined the dissociation constants Kd for interaction of the proteins with the vesicle-associated MTX-PE. Binding curves were determined and Kd-values are expressed with reference to the concentration of MTX-PE exposed at the outer surfaces of the lipid vesicles (and hence accessible to added DHFR/NusDHFR), calculated as described in Materials and Methods. As illustrated in Fig. 1 A, in this manner we determined a dissociation constant of 31 ± 5 nM for binding of NusDHFR to MTX-PE incorporated into large unilamellar POPC vesicles. As expected, the measured Kd-value was independent of the molar percentage of MTX-PE incorporated in the vesicles. A very similar Kd-value (36 ± 3 nM) was measured for binding of DHFR to vesicle-incorporated MTX-PE. Neither protein bound detectably to vesicles lacking MTX-PE.
Effects of PE-PEG5000 on binding of DHFR and NusDHFR to MTX-PE-incorporating vesicles
PE-PEGs have been shown to reduce the binding of proteins to lipid surfaces by creating unfavorable steric interactions between surface-associated protein molecules and the surface-tethered PEG chains (10,11). However, the effect of PE-PEG on the affinity of binding of proteins to lipid bilayers has not previously been examined quantitatively. We therefore determined the affinities of DHFR and NusDHFR for vesicles containing a fixed molar proportion of MTX-PE (0.5 mol %) and variable levels of PE-PEG5000. As shown in Fig. 1, B and C, the affinity of either protein for the vesicles decreases strongly (i.e., the dissociation constant Kd increases) as the level of PE-PEG5000 in the bilayer is increased. The magnitude of the effect is larger for NusDHFR than for the smaller DHFR; incorporation of 5 mol % PE-PEG5000, for example, reduces the affinity of binding by 85-fold for NusDHFR, compared with 11-fold for DHFR.
To test whether the above results were affected by electrostatic effects due to the charge of the incorporated PE-PEG5000 (net charge −1), we also examined the binding of DHFR and NusDHFR to vesicles containing MTX-PE and increasing amounts of POPG (also a monoanionic lipid) in place of PE-PEG5000. As shown in Fig. 1, B and C, incorporation of POPG had no significant effect on the affinity of binding of either protein over the range of mol fractions of anionic lipid examined.
The relationships between the dissociation constant Kd for DHFR or NusDHFR and the mol % PE-PEG5000 incorporated into the MTX-PE-containing vesicles are roughly exponential (Fig. 1, B and C), but they can be fit equally well or better to an equation derived by treating the bound PEG5000 chains as a van der Waals gas, which we previously found to describe the behavior of the flexible grafted PEG chains better than a hard-disc model (12):
| (3) |
where R is the ratio of the average surface area occupied by one molecule of bound DHFR or NusDHFR to that occupied by one tethered PEG5000 chain, Kdo is the value of Kd measured for equivalent vesicles lacking PE-PEG5000, and B is the ratio of the average surface area occupied by one PEG5000 chain to the area of one host phospholipid molecule. Fitting the data shown for NusDHFR in Fig. 1 C to the above equation yields estimates of B = 8.7 ± 0.7 and R = 5.0 ± 0.8. The estimated value of B, the average number of phospholipid molecules covered by one PEG5000 chain, agrees well with previous estimates based on both simulations and experimental data (7.7–8.6 (12)). The estimated surface area occupied by one NusDHFR molecule, B × R = 44 ± 8 lipid molecules or 30 ± 6 nm2 for a POPC molecular area of 0.67 nm2 (27), agrees well with the value estimated (∼30 nm2) for a roughly spherical protein of this molecular mass with a partial specific volume (0.73 cm3 g−1) and degree of hydration (0.30 g H2O/g protein) typical for a soluble protein. Fitting the relationship between the dissociation constant Kd and the molar fraction of PE-PEG5000 for binding of DHFR to vesicles containing MTX-PE and PE-PEG (Fig. 1 C) did not permit independent estimation of the parameters B and R in Eq. 3 with acceptable accuracy. However, fitting this equation to the DHFR-binding data with the value of B set to 8.7, as determined from the NusDHFR-binding data, gave a value of R = 2.3 ± 0.2 for DHFR, indicating that one vesicle-bound DHFR molecule occupies on average B × R = 19 ± 5 lipid molecules, or 13 ± 3 nm2. This estimate is consistent with that expected (11.4 nm2) for a roughly spherical protein with the molecular mass of DHFR (18 kDa).
Effects of surface-coupled protein on binding of DHFR and NusDHFR to MTX-PE-incorporating vesicles
To assess how protein molecules present on a lipid surface at varying lateral densities affect the association of other proteins with the surface, we examined the binding of DHFR and NusDHFR to MTX-PE-incorporating vesicles on the surfaces of which we coupled varying amounts of MBPCys (molecular mass = 43.2 kDa), a modified form of the normally cysteine-free E. coli MBP bearing a C-terminal –SerCys extension. MBP was chosen for this purpose because it is monomeric, highly soluble, and resistant to aggregation, but also carries only a modest net negative charge at neutral pH. Preparations of vesicles bearing MBPCys bound to their surface at varying lateral densities were obtained by incubating vesicles incorporating 0.5 mol % MTX-PE and 2.5 mol % N-(4-maleimidobutyroyl)-PEG3-PE with MBPCys for varying times and at varying input protein/lipid ratios, after which the vesicles were treated with excess cysteine to quench residual maleimido groups and separated from uncoupled protein by gel filtration. We then examined the affinity of binding of fluorescein-labeled NusDHFR or DHFR to the resulting MBP-coupled, MTX-PE-incorporating vesicles (and to a parallel sample of mock-treated, protein-free vesicles in each experiment) using the centrifugation-based assay described above.
In Fig. 2 A the value of the dissociation constant Kd measured for binding of NusDHFR and DHFR to MBPCys-coupled vesicles is shown as a function of the surface density of bound MBPCys. For both DHFR and NusDHFR, the value of Kd rises roughly exponentially with the density of surface-coupled MBPCys, showing a steeper variation for the larger NusDHFR. For vesicles bearing 0.35 mg MBPCys/μmol lipid coupled to their surfaces (corresponding to roughly one coupled protein molecule per 60 lipid molecules exposed at the vesicle outer surface), the values of Kd for DHFR and NusDHFR binding are roughly 9- and 50-fold higher, respectively, than those measured for binding of these proteins to MTX-PE incorporated in bare control vesicles.
Figure 2.
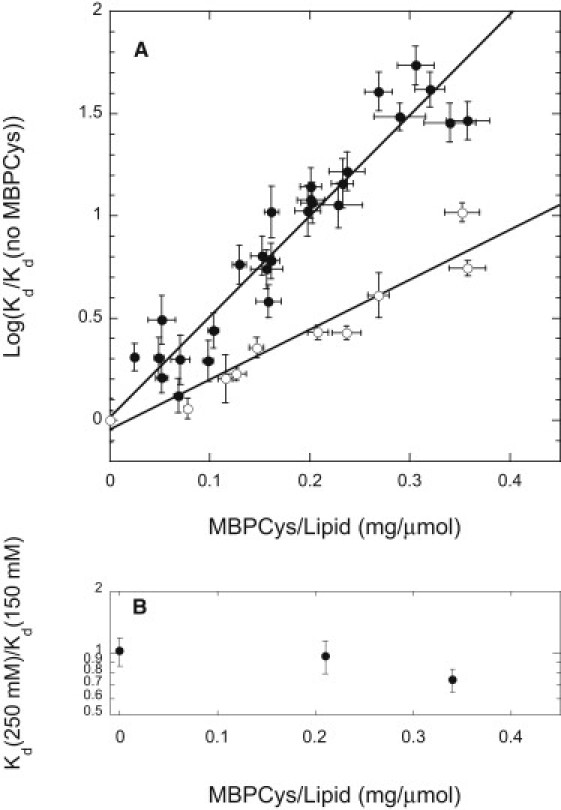
(A) Kd values (normalized to the Kd values measured using protein-free vesicles in the same experiment) for binding of DHFR (open circles) and NusDHFR (solid circles) to POPC/MTX-PE/RhoPE/N-(4-maleimidobutyroyl)-PEG3-POPE (97/0.5/0.02/2.5 molar proportions) vesicles coupled to MBPCys at the indicated ratios of bound protein to lipid. Data are plotted and fitted as log values to ensure appropriate weighting, as the calculated errors of estimation of Kd increased in direct proportion to the value of Kd. (B) Effect of increased ionic strength (250 mM KCl in place of the normal 150 mM KCl) on binding of NusDHFR to vesicles bearing coupled MBPCys at varying surface densities. Data are presented as the ratio of Kd-values measured at 250 mM vs. 150 mM KCl and are scaled to the same axis dimensions as for panel A.
The results shown in Fig. 2 A were obtained in a buffer of physiological ionic strength (150 mM KCl, 5 mM phosphate, 1 mM EDTA and DTT, pH 7.0). As shown in Fig. 2 B, increasing the concentration of KCl to 250 mM had no significant effect on NusDHFR binding to MTX-PE on protein-free vesicles or vesicles bearing intermediate levels of MBCys, and only slightly decreased the value of Kd for binding of NusDHFR to MTX-PE on vesicles bearing MBP at the highest surface density examined. This result, together with our observation above that the presence of a negative surface charge (due to incorporated POPG) has no effect on the binding affinity of either DHFR or NusDHFR for MTX-PE-incorporating vesicles, implies that steric rather than electrostatic interactions contribute most importantly to the weakened binding of these proteins to vesicles bearing surface-anchored MBPCys.
Using a hard-disk model and scaled-particle theory, Chatelier and Minton (7) predicted how the value of Kd for binding of low levels of a tracer protein A (here, DHFR or NusDHFR) to a surface should be affected by steric repulsions due to the presence at the surface of a second, more abundant protein B (here, MBPCys). The general equation derived by these authors contains several particle shape-dependent parameters that cannot be estimated independently and with reasonable accuracy by fitting data like those presented above. However, these authors also found that unless species A and B are highly asymmetric particles, the effects of species B on the affinity of binding of the tracer protein A under the conditions just outlined depend much more strongly on the area fraction occupied by B than on the detailed shapes of the two proteins. We can therefore use the following simpler equation (rigorously applicable for spherical particles (7)) to extract from the data in Fig. 2 A an approximate estimate of the surface area occupied per molecule of surface-coupled MBPCys:
| (4) |
where xB is the molar ratio of species B (MBPCys) to lipid at the vesicle surface, Kd(A) is the value of the dissociation constant measured for binding of A (DHFR or NusDHFR) to vesicles with a given ratio of MBPCys to lipid at the surface, Kdo(A) is the value of Kd for binding of species A to equivalent bare lipid vesicles, B, is the mean surface area occupied by one molecule of B (expressed as a multiple of the area per lipid molecule), and fR = (MA/MB)1/3, where M is the molecular mass, for particles of similar shape. Fitting the data shown for DHFR in Fig. 2 A to this equation, using a value of fR = 0.75 for DHFR/MBPCys, yields a value of B = 23.8 ± 0.7 for MBPCys, or 15.9 nm2 assuming a mean area of 0.67 nm2 per lipid molecule (27). Fitting the data for NusDHFR to the same equation using a value of fR = 1.20 for NusDHFR/MBPCys yields a value of ΦB = 28.8 ± 0.7 or 19.3 nm2 for MBPCys. These values are consistent with the dimensions of MBP (∼7.0 × 4.5 × 3.5 nm; calculated average projected area (28) = ∼19 nm2)) determined from published crystal structures (29,30). These estimates of the average surface area occupied per molecule of MBPCys may be distorted to a small degree by the approximations discussed above, but they demonstrate that the observed experimental data are consistent with theoretical predictions of the magnitudes of steric interactions between surface-associated protein molecules like those examined here.
Kinetics of interaction of NusDHFR with protein-free and protein-coupled vesicles
The kinetics of binding of fluorescein-labeled NusDHFR to vesicles containing MTX-PE and the energy-transfer quencher RhoPE was examined by monitoring the time-dependent change of protein fluorescence after vesicle addition. Addition of vesicles to the labeled protein produces an instantaneous fluorescence decrease due to inner-filter effects (measured in parallel runs using control vesicles containing RhoPE but not MTX-PE), followed by a slower decline of fluorescence due to protein binding to the vesicles (Fig. 3 A). As illustrated in Fig. 3 B, the time course of the latter fluorescence change is well described by an exponential expression of the form F(t) = Fmin + (Fo − Fmin) × exp(−kQt). Subsequent addition of excess free MTX leads to recovery of the fluorescence, reflecting the kinetics of dissociation of the protein from the quencher-containing vesicles (Fig. 3 A). As illustrated in the inset to Fig. 3 B, the time course of this fluorescence change is also well described by an exponential expression, of the form F(t) = Fmax - (Fmax - Fo) × exp(- krect). Essentially identical values for the rate constant krec were determined for dequenching induced by addition of either 15 or 60 μM free MTX, or a 20-fold excess of quencher-free vesicles incorporating MTX-PE (not shown), indicating that these agents do not participate directly in the dissociation process, but rather trap NusDHFR after it has dissociated from the quencher-containing vesicles.
Figure 3.
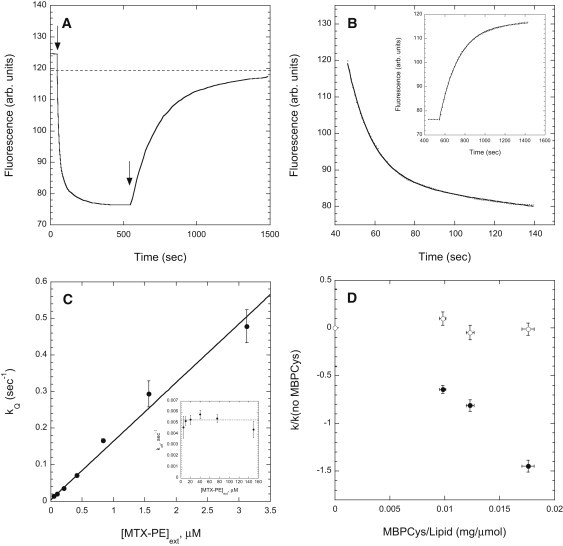
(A) Fluorescence measurement of the time course of association of NusDHFR with, and subsequent dissociation of the protein from, lipid vesicles containing MTX-PE. To fluorescein-NusDHFR (1 μg/mL) in a stirred cuvette, POPC/MTX-PE/RhoPE vesicles (95:4:1 molar proportions, 21 μM for the sample trace shown) were added at the time indicated by the first arrow. At the time indicated by the second arrow, 15 μM free MTX was added. The dashed line indicates the level to which the protein fluorescence falls due to inner-filter effects immediately upon vesicle addition. (B) Fit of the decrease in fluorescence following vesicle addition in panel A (omitting the instantaneous decrease due to inner-filter effects) to an exponentially falling function with rate constant kQ. (Inset) Fit of the increase in fluorescence after addition of MTX in panel A to an exponentially rising function with rate constant krec. (C) Variation of the measured rate constant kQ with the concentration of added vesicles (indicated as the concentration of externally exposed MTX-PE). (Inset) Rate constant krec measured at varying concentrations of added vesicles. (D) Variation of ka (closed circles) and kd (open circles) with the density of MBPCys bound to POPC/MTX-PE/RhoPE vesicles. Values of ka and kd were determined as illustrated in panel C, and in each case are scaled to the values of these parameters determined for protein-free vesicles in the same experiment. Other experimental details were as described in the text.
Solution of the appropriate kinetic equation for the first phase of the above experiment predicts that the effective rate constant kQ for the exponential fall in protein fluorescence upon addition of vesicles is equal to (ka[MTX-PE]ext + kd), where ka is the second-order rate constant for binding of NusDHFR to vesicle-incorporated MTX-PE, [MTX-PE]ext is the concentration of MTX-PE exposed at the external surface of the vesicles (and hence accessible to NusDHFR), and kd is the first-order rate constant for dissociation of the protein from the vesicles. In agreement with this prediction, the rate constant kQ measured for the quenching phase increases linearly with the concentration of added vesicles (Fig. 3 C). The slope and y-intercept of this relationship yield estimates of ka = 1.56 ± 0.08 × 10−1 s−1 (μM MTX-PE)-1 and kd = 4.1 ± 1.9 × 10−3 s−1, respectively. The rate constant krec measured for the fluorescence recovery observed upon addition of free MTX (which is predicted to equal kd, the rate constant for dissociation of NusDHFR from the vesicles) does not depend on the vesicle concentration and gives an average estimated value of kd = 5.3 ± 0.5 × 10−3 s−1 (Fig. 3 B, inset). Using these values of ka and kd we can calculate a value for the dissociation constant Kd = kd/ka = 34 ± 4 nM for NusDHFR binding to vesicle-incorporated MTX-PE, in excellent agreement with the value of Kd estimated using the centrifugation-based assay described above (31 ± 5 nM).
To assess the effects of surface-bound MBPCys on the kinetics of NusDHFR interaction with vesicles containing MTX-PE, we repeated the above experiments using vesicles to which MPBCys was coupled at different protein/lipid ratios. As shown in Fig. 3 D, increasing the density of surface-coupled protein strongly reduces the association rate constant ka without significantly altering the dissociation rate constant kd. The presence of MBPCys on the vesicle surface thus retards the binding of NusDHFR to vesicles incorporating MTX-PE but does not influence its rate of dissociation from the vesicles. This behavior is consistent with that predicted (9) if we assume that the transition-state complex through which NusDHFR binds to MTX-PE at the vesicle surface occupies an amount of surface area equal to that occupied by the final, stable form of the complex.
Discussion
The findings presented here confirm predictions from previous theoretical studies, and extend the limited experimental data available, which suggest that steric interactions between macromolecules at the surfaces of model and biological membranes contribute substantially to the overall free energies of membrane-associated proteins (2,7–14). Our results demonstrate that for a given protein, the magnitude of such effects depends strongly on both the overall lateral density of macromolecules at the membrane surface and the size of the protein species of interest. For the experimental system utilized here, we quantitated these effects through the increase in Kd for binding of soluble proteins (DHFR or NusDHFR) to a specific lipid (MTX-PE) incorporated in the vesicle bilayer. However, our findings are equally relevant (quantitatively as well as qualitatively) to proteins that bind reversibly by other means to the lipid bilayer surface—for example, via generalized electrostatic interactions with anionic lipids or by intercalation of a lipidic tail or hydrophobic amino acid side chains into the bilayer (in which case the binding may be more accurately characterized as a partitioning equilibrium). The origin of the steric effects measured here (the fractional reduction in the area on a surface that is accessible for occupation by a protein X due to the simultaneous presence of other macromolecules at the surface) is in fact quite general and is independent of the specific nature of the interaction between X and the bilayer interface. As discussed further below, because of the generality of such steric phenomena, our results also have potential implications for the behavior of proteins that are permanently membrane-associated.
The lateral densities of surface-associated protein examined here (and hence the magnitudes of the steric effects that we observe) for the MBPCys/POPC vesicle system span a range that extends up to values approaching, but not exceeding, those predicted for biological membranes. Dupuy and Engelman (5), for example, determined that the ratio of extramembrane protein mass to lipid mass in human erythrocyte membranes is 1.0 mg protein/mg lipid. Combining this value with the estimate by these authors that roughly 23% and 77%, respectively, of the intramembrane area in the erythrocyte membrane is occupied by protein and lipid, and assuming a 48:52 molar ratio of cholesterol to polar lipids in the erythrocyte membrane (31,32), a mean molecular mass of 775 for the membrane polar lipids and the partial molecular areas estimated by Pandit et al. (27) for cholesterol and POPC (as a representative membrane polar lipid) in POPC/cholesterol bilayers, we calculate that for the erythrocyte membrane the average of the mass densities of extramembrane protein at either face is ∼200 ng/cm2 of surface. A comparable mass density of MBPCys on POPC vesicles equates to a protein/lipid molar ratio of 0.0185 at the vesicles' outer surface, a value that slightly exceeds the highest protein/lipid ratios examined in our study. Compositional and biophysical analyses of rat brain synaptic vesicles (4) suggest that the mass density of protein at the outer surface of these membranes is still higher. Of course, these comparisons do not take into account the fact that the proteins associated with biological membranes constitute a heterogeneous population whose extramembranous portions vary in shape and molecular mass, whereas in our experimental system a single protein species is coupled to the vesicle surfaces. Nonetheless, our results strongly suggest that surface crowding effects at the surfaces of biological membranes are large and need to be considered when assessing diverse aspects of the behavior of membranes and membrane proteins. To cite an obvious example, they suggest that measurements of the rates and affinities of reversible binding of proteins to lipids on bare lipid vesicles may substantially overestimate the magnitudes of these parameters for binding to biological membranes. Steric crowding at membrane surfaces may also influence numerous other important aspects of membrane and membrane protein behavior, including lateral diffusion of membrane proteins (33–35), membrane deformation (36), and the thermodynamic potential for membrane-associated proteins to form oligomeric complexes (1).
Our results suggest a further prediction that is relevant to both integral and peripheral membrane proteins, namely that within membranes the lateral distributions of different types or classes of proteins may exhibit substantial coupling arising from steric effects alone. In accord with previous theoretical predictions (7) we find that the free energy of a given membrane-associated protein molecule varies strongly, in a linear or supralinear manner, with the total lateral density of macromolecules in the protein's local environment. It can readily be shown that the magnitude of this effect is the same for a protein that associates reversibly with membranes, as in the examples studied here, and for a permanently anchored (in the simplest case, monotopic) membrane protein with an extramembrane domain of the same size. We can thus predict that even a modest increase or decrease in the overall surface density of proteins in a given region of a membrane, driven, for example, by constraints imposed by the cytoskeleton or other juxtamembrane structures, may produce a reciprocal variation in the local concentrations of (mobile) membrane proteins that are not subject to such constraints. Glycosylphosphatidylinositol (GPI-) anchored proteins, for example, are typically found to be excluded from clathrin-coated pits (37). We recently reported experimental evidence (18) to support the proposal of Pearse and Bretscher (38) that this phenomenon is a direct result of steric factors. The results observed here suggest that coated pits could efficiently exclude GPI proteins (and other membrane proteins lacking appropriate targeting signals) via steric effects even if the lateral density of membrane proteins in coated pits only moderately exceeds that found elsewhere in the membrane.
A subtler example of the potential for steric coupling between the lateral distributions of different membrane proteins may arise in cases where one class of abundant membrane proteins is depleted in particular regions of a membrane. In such cases, it is predicted that other (mobile) membrane proteins will become enriched in these regions in response to the locally decreased steric pressure. Since the magnitude of steric repulsions between membrane proteins varies strongly with the overall local lateral density of proteins, such effects may be substantial. Consistent with this prediction, Discher et al. (13) found that within projections of the erythrocyte plasma membrane formed by aspiration into micropipettes, the lateral densities of endogenous GPI-anchored and artificial lipid-anchored proteins were markedly increased, in a manner that correlated strongly but inversely with the degree of local depletion of cytoskeletally anchored membrane proteins. Under physiological conditions, regions of a membrane in which one class of abundant proteins (e.g, cytoskeletally associated proteins or proteins excluded from ordered-lipid microdomains) is depleted may likewise, via steric effects, exhibit significant local enrichment of proteins that are not subject to the same constraint. Such possibilities illustrate the variety of means by which steric effects may influence the behavior of membrane-associated proteins, and the need to assess such effects and their potential consequences for membrane protein behavior in a broad range of contexts.
Acknowledgments
This work was supported by an operating grant (MOP-7776) from the Canadian Institutes of Health Research to J.R.S.
Supporting Material
References
- 1.Grasberger B., Minton A.P., Metzger H. Interaction between proteins localized in membranes. Proc. Natl. Acad. Sci. USA. 1986;83:6258–6262. doi: 10.1073/pnas.83.17.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryan T.A., Myers J., Webb W.W. Molecular crowding on the cell surface. Science. 1988;239:61–64. doi: 10.1126/science.2962287. [DOI] [PubMed] [Google Scholar]
- 3.Engelman D.M. Membranes are more mosaic than fluid. Nature. 2005;438:578–580. doi: 10.1038/nature04394. [DOI] [PubMed] [Google Scholar]
- 4.Takamori S., Holt M., Jahn R. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 5.Dupuy A.D., Engelman D.M. Protein area occupancy at the center of the red blood cell membrane. Proc. Natl. Acad. Sci. USA. 2008;105:2848–2852. doi: 10.1073/pnas.0712379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou H.-X., Rivas G., Minton A.P. Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu. Rev. Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chatelier R.C., Minton A.P. Adsorption of globular proteins on locally planar surfaces: models for the effect of excluded surface area and aggregation of adsorbed protein on adsorption equilibria. Biophys. J. 1996;71:2367–2374. doi: 10.1016/S0006-3495(96)79430-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minton A.P. Effects of excluded surface area and adsorbate clustering on surface adsorption of proteins. I. Equilibrium models. Biophys. Chem. 2000;86:239–247. doi: 10.1016/s0301-4622(00)00151-4. [DOI] [PubMed] [Google Scholar]
- 9.Minton A.P. Effects of excluded surface area and adsorbate clustering on surface adsorption of proteins. II. Kinetic models. Biophys. J. 2001;80:1641–1648. doi: 10.1016/S0006-3495(01)76136-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noppl-Simson D.A., Needham D. Avidin-biotin interactions at vesicle surfaces: adsorption and binding, cross-bridge formation, and lateral interactions. Biophys. J. 1996;70:1391–1401. doi: 10.1016/S0006-3495(96)79697-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du H., Chandaroy P., Hui S.W. Grafted poly-(ethylene glycol) on lipid surfaces inhibits protein adsorption and cell adhesion. Biochim. Biophys. Acta. 1997;1326:236–248. doi: 10.1016/s0005-2736(97)00027-8. [DOI] [PubMed] [Google Scholar]
- 12.Rex S., Zuckermann M.J., Silvius J.R. Experimental and Monte Carlo simulation studies of the thermodynamics of polyethyleneglycol chains grafted to lipid bilayers. Biophys. J. 1998;75:2900–2914. doi: 10.1016/S0006-3495(98)77732-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Discher D.E., Mohandas N., Evans E.A. Molecular maps of red cell deformation: hidden elasticity and in situ connectivity. Science. 1994;266:1032–1035. doi: 10.1126/science.7973655. [DOI] [PubMed] [Google Scholar]
- 14.Heimburg T., Marsh D. Protein surface-distribution and protein-protein interactions in the binding of peripheral proteins to charged lipid membranes. Biophys. J. 1995;68:536–546. doi: 10.1016/S0006-3495(95)80215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Struck D.K., Hoekstra D., Pagano R.E. Use of resonance energy transfer to monitor membrane fusion. Biochemistry. 1981;20:4093–4099. doi: 10.1021/bi00517a023. [DOI] [PubMed] [Google Scholar]
- 16.Collet M., Lenger J., Sewald N. Molecular tools for metalloprotease sub-proteome generation. J. Biotechnol. 2007;129:316–328. doi: 10.1016/j.jbiotec.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Silvius J.R., Gagné J. Lipid phase behavior and calcium-induced fusion of phosphatidylethanolamine-phosphatidylserine vesicles. Calorimetric and fusion studies. Biochemistry. 1984;23:3232–3240. [Google Scholar]
- 18.Bhagatji P., Leventis R., Silvius J.R. Steric and not structure-specific factors dictate the endocytic mechanism of glycosylphosphatidylinositol-anchored proteins. J. Cell Biol. 2009;186:615–628. doi: 10.1083/jcb.200903102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferruti P., Tanzi M.C., Cecchi R. Succinic half-esters of poly(ethyleneglycol)s and their benzotriazole and imidazole derivatives as oligomeric drugbinding matrices. Makromol. Chem. 1981;182:2183–2192. [Google Scholar]
- 20.Studier F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh P., Cheng J., Wagner C.R. Expression, purification and characterization of recombinant mouse translation initiation factor eIF4E as a dihydrofolate reductase fusion protein. Protein Expr. Purif. 2008;60:132–139. doi: 10.1016/j.pep.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nath S., Brahma A., Bhattacharyya D. Extended application of gel-permeation chromatography by spin column. Anal. Biochem. 2003;320:199–206. doi: 10.1016/s0003-2697(03)00302-6. [DOI] [PubMed] [Google Scholar]
- 23.Ganesh C., Shah A.N., Varadarajan R. Thermodynamic characterization of the reversible, two-state unfolding of maltose binding protein, a large two-domain protein. Biochemistry. 1997;36:5020–5028. doi: 10.1021/bi961967b. [DOI] [PubMed] [Google Scholar]
- 24.Lowry R.R., Tinsley I.J. A simple, sensitive method for lipid phosphorus. Lipids. 1974;9:491–492. doi: 10.1007/BF02534277. [DOI] [PubMed] [Google Scholar]
- 25.Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 26.Nordlund J.R., Schmidt C.F., Thompson T.E. Transbilayer distribution of phosphatidylethanolamine in large and small unilamellar vesicles. Biochemistry. 1981;20:3237–3241. doi: 10.1021/bi00514a039. [DOI] [PubMed] [Google Scholar]
- 27.Pandit S.A., Chiu S.-W., Scott H.L. Cholesterol packing around lipids with saturated and unsaturated chains: a simulation study. Langmuir. 2008;24:6858–6865. doi: 10.1021/la8004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vickers G.T., Brown D.J. The distribution of projected area and perimeter of convex, solid particles. Proc. R. Soc. Lond. A. 2001;457:283–306. [Google Scholar]
- 29.Spurlino J.C., Lu G.-Y., Quiocho F.A. The 2.3-Å resolution structure of the maltose- or maltodextrin-binding protein, a primary receptor of bacterial active transport and chemotaxis. J. Biol. Chem. 1991;66:5202–5219. doi: 10.2210/pdb1mbp/pdb. [DOI] [PubMed] [Google Scholar]
- 30.Telmer P.G., Shilton B.H. Insights into the conformational equilibria of maltose-binding protein by analysis of high affinity mutants. J. Biol. Chem. 2003;278:34555–34567. doi: 10.1074/jbc.M301004200. [DOI] [PubMed] [Google Scholar]
- 31.Hui S.W., Stewart C., Stewart T.P. Effects of cholesterol on lipid organization in human erythrocyte membrane. J. Cell Biol. 1980;85:283–291. doi: 10.1083/jcb.85.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aloia R.C., Jensen F.C., Gordon L.M. Lipid composition and fluidity of the human immunodeficiency virus. Proc. Natl. Acad. Sci. USA. 1988;85:900–904. doi: 10.1073/pnas.85.3.900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peters R., Cherry R.J. Lateral and rotational diffusion of bacteriorhodopsin in lipid bilayers: experimental test of the Saffman-Delbruick equations. Proc. Natl. Acad. Sci. USA. 1982;79:4317–4321. doi: 10.1073/pnas.79.14.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saxton M.J. Lateral diffusion in an archipelago. The effect of mobile obstacles. Biophys. J. 1987;52:989–997. doi: 10.1016/S0006-3495(87)83291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frick M., Schmidt K., Nichols B.J. Modulation of lateral diffusion in the plasma membrane by protein density. Curr. Biol. 2007;17:462–467. doi: 10.1016/j.cub.2007.01.069. [DOI] [PubMed] [Google Scholar]
- 36.Stachowiak J.C., Hayden C.C., Sasaki D.Y. Steric confinement of proteins on lipid membranes can drive curvature and tubulation. Proc. Natl. Acad. Sci. USA. 2010;107:7781–7786. doi: 10.1073/pnas.0913306107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bretscher M.S., Thomson J.N., Pearse B.M.F. Coated pits act as molecular filters. Proc. Natl. Acad. Sci. USA. 1980;77:4156–4159. doi: 10.1073/pnas.77.7.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pearse B.M.F., Bretscher M.F. Membrane recycling by coated vesicles. Annu. Rev. Biochem. 1981;50:85–101. doi: 10.1146/annurev.bi.50.070181.000505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.