

Abstract
Deregulation of the expression of p53R2, a p53-inducible homologue of the R2 subunit of ribonucleotide reductase, has been found in various human cancer tissues; however, the roles p53R2 plays in cancer progression and malignancy remain controversial. In present study, we examined changes in gene expression profiles associated with p53R2 in cancer cells using the analysis of cDNA microarray. Gene set enrichment analysis (GSEA) identified the gene set regulating cell cycle progression was significantly enriched in p53R2-silencing human oropharyngeal carcinoma KB cells. Attenuation of p53R2 expression significantly reduced p21 expression and moderately increased cyclin D1 expression in both wild-type p53 cancer cells: KB, MCF-7, and mutant p53 cancer cells: PC3 and MDA-MB-231. Conversely, overexpression of p53R2-GFP resulted in an increase in the expression of p21 and decrease in the expression of cyclin D1, which correlated with reduced cell population in S-phase in vitro and suppressed growth in vivo. Furthermore, the MEK inhibitor PD98059 partially abolished modulation of p21 and cyclin D1 expression by p53R2. Moreover, under the conditions of non-stress and adriamycin-induced genotoxic stress, attenuation of p53R2 in KB cells significantly increased phosphorylated H2AX, which indicates attenuation of p53R2 may enhance DNA damage induced by adriamycin. Overall, our study demonstrates that p53R2 may suppress cancer cells proliferation partially by up-regulation of p21 and down-regulation of cyclin D1; p53R2 plays critical roles not only in DNA damage repair but also in proliferation of cancer cells.
Keywords: p53R2, cDNA microarray, Gene set enrichments analysis (GSEA), ERK1/2, Cell proliferation
Introduction
Ribonucleotide reductase (RNR) plays an essential role in catalyzing conversion of ribonucleoside diphosphates to the corresponding 2′-deoxyribonucleoside diphosphates, a rate-limiting step in the production of 2′-deoxyribonucleoside 5′-triphosphates (dNTP) required for DNA synthesis and repair (1, 2). Human ribonucleotide reductase consists of two subunits: RRM1 and RRM2, both of which are required for enzymatic activity (3). The p53-inducible p53R2 encodes a peptide that showed striking similarity to RRM2 (4). Expression of the p53R2 can be induced by signals activating p53, such as DNA-damaging agents and ionizing radiation, UV-irradiation in a wild-type p53-dependent manner to synthesize the dNTP for DNA repair after DNA damage (5).
Elevated RNR activity and overexpression of RRM2 have been found to significantly increase the drug-resistant properties, and the angiogenic and invasive potential of human cancer cells (3, 6-8). Therefore, RRM2 is well accepted as an important therapeutic target for DNA replication-dependent diseases, such as cancer. However, the roles p53R2 plays in carcinogenesis and malignancy of human cancer remain largely controversial. Some studies support the idea that p53R2 is also a potential target for cancer gene therapy like RRM2. The idea is mainly based on the crucial role p53R2 plays in dNDP synthesis and DNA damage repair. Inhibition of p53R2 enhances 5-fluorouracil sensitivity of cancer cells in vitro (9). Several studies have also observed that elevated p53R2 expression was positively correlated with anticancer agent resistance of human malignancies, including oral cavity (10) and esophageal cancers (11). Recently, a study by Devlin et al showed that p53R2 was overexpressed in prostate tumor cell lines, while silencing p53R2 enhances the apoptotic effects of ionizing radiation and doxorubicin (12). On the other hand, another study identified that disruption of the p53R2 mediated DNA repair in ulcerative colitis initiated carcinogenesis of colon (13). Consistently, an early study also showed that p53R2−/− embryonic fibroblasts (MEFs) became immortal much earlier and were more susceptible to apoptosis induced by oxidative stress compared to p53R2+/+ MEFs. They also showed an increased rate of proliferation after seven passages (14). Some studies reported that positive p53R2 expression was significantly correlated with depth of invasion, lymph node metastasis, stage and poor prognosis in patients with esophageal squamous cell carcinoma and lung cancer (15, 16). However, another study showed that the expression of p53R2 did not correlated with stage; grade and histological types of gastric tumors (17). Controversially, Yoshida et al addressed that p53R2 expression significantly decreased with progression from ulcerative colitis-associated carcinogenesis dysphasia to carcinoma indicating an inverse relationship between p53R2 and cancer development (13). Our previous studies identified that p53R2 was negatively correlated with the metastasis of colon adenocarcinoma samples (18, 19).
In the present study, we reported the changes in gene expression profile associated with knockdown of p53R2 in human KB cancer cells and the impact of p53R2 on cancer cells proliferation. We identified the gene set regulating cell cycle progression was significantly enriched in p53R2-attenuated KB cells using gene set enrichment analysis (GSEA) with cDNA microarray (20). Overexpression of p53R2 significantly suppressed cancer cells proliferation regardless of p53 status of cells. Our results indicated that overexpression of p53R2 may suppress cancer cells proliferation through alterations in the expression of cell-cycle-regulating genes.
Materials and Methods
Cell culture and plasmid transfection
Human oropharyngeal carcinoma KB (wild-type p53) and breast adenocarcinoma MCF-7 cells (wild-type p53) were cultured in DMEM with 10% FBS. Human breast carcinoma MDA-231 (mutated p53) and prostate adenocarcinoma cells PC3 (truncated p53) were cultured in RPMI with same supplements. Cell lines were purchased from ATCC previously; they were tested and authenticated for genotypes by DNA fingerprinting (Figure S1). Adriamycin and PD98059 were purchased from Sigma-Aldrich (St. Louis, MO). The human p53R2 gene specific siRNA (sc-36338) and scramble siRNA were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Addgene plasmid 16451: p21-Luc promoter driven luciferase inserted with a WAF1 promoter region including a p53-binding element 2.4 kb upstream of WAF1, and Addgene Plasmid 16442: PG13-luc, a p53 response reporter plasmid containing 13 tandem repeats of the p53/TAp73 consensus DNA binding sites used to measure the transcriptional activity of p53/TAp73, were generous gifts from professor Vogelstein (21). The coding region of p53R2 cDNA was cloned into the unique EcoR I and BamH I of pEGFP-N1 (Clontech) to express p53R2 fused to the N-terminus of EGFP, named as p53R2-GFP.
Microarray analysis of p53R2-associated genes
siRNA transfection and total RNA from p53R2-silencing KB and control cells were performed as reported previously (8). Duplicated RNA samples were subjected to analysis of cDNA microarray. The Affymetrix GeneChip Human Gene 1.0-ST array (Affymetrix, Santa Clara, CA) was used to define gene expression profiles from the samples. Hybridization, data generation and analysis were done at Microarray Core of City of Hope according to the user manual of the kits. Genes that were differentially expressed between p53R2-attenuated KB cells and the control KB cells were selected with a cut-off of adjusted P < 0.05 and log2 ratio of 1.5. Gene set enrichment analysis (GSEA, v2.0) (20) was used to determine whether an a priori defined set of genes in C2 of GSEA/MSigDB shows statistically significant, concordant differences between control siRNA and p53R2 siRNA transferred Kb cells.
Quantitative reverse transcriptional PCR and Western blot analysis
The primers used in the study are listed in Table S1. Quantitative real time PCR (q-RT-PCR) was carried out in the ABI Prism 7900 HT Sequence Detection System (Applied Biosystems, Foster City, CA). The reaction mixture of 20 μl consisted of 1 × ABI SYBR Green PCR Master Mix, 0.25 μl cDNA and 0.2 μmol/L of each primer. Relative gene-expression quantification method as reported previously was used to calculate the fold change of mRNA expression according to the comparative Ct method using β-actin as an endogenous control (8). Data was represented as ratio or folds change to control sample. The antibodies against GAPDH, p53R2, p53, cyclin D1, phosphorylated-ATM, ERK and phosphorylated ERK1/2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA); Anti-phospho-gamma-Histone H2AX (Ser139, clone JBW301) was purchased from Millipore (Millipore, Billerica, MA). Signals were densitometrically assessed and normalized to the signals of GAPDH.
Dual Luciferase reporter assay
KB cells were seeded at a concentration of 5.0 × 104 cells per well of 24-well plate. Cells were transfected with 125 ng of p21-Luc or PG13 plus 10 ng renilla luciferase reporters with 250ng of GFP or p53R2-GFP plasmid/well by Lipofeactamin™-2000. Forty-eight hours after reporter plasmid transfection, cells were washed once with phosphate-buffered saline and lysed with reporter lysis buffer from Promega (Madison, WI). Firefly and renilla luciferase activities were determined according to the manufacturer’s instruction of dual luciferase assay (Promega, Madison, WI). Relative luciferase activity of each reporter was normalized to the value of renillla luminescence. The experiment was repeated three times. All experiments were done in triplicates. Data were reported as average ± SD.
Cell Cycle Analysis
One million cells were washed in cold PBS, fixed in 70% ethanol in PBS for at least 1 hour on ice, washed, and resuspended in PBS containing 25 μg/mL propidium iodide (PI) and 100 μg/mL ribonuclease A and incubated for 30 min at 37°C. Fluorescence was measured on a Becton Dickinson FACSCalibur flow cytometer (excitation 488 nm, measurement 564–607 nm) within 1 hour. Data were analyzed using the MODFIT 2.0 program (Verity Software). The mean and standard errors for the percentage of cells in each phase of the cell cycle were derived from at least three independent experiments, each in duplicate.
In vitro proliferation and in vivo tumor growth
2.5 × 103 KB/PC3-GFP and KB/PC3-p53R2-GFP cells were seeded into wells of 16-well devices compatible with a W200 real-time cell electronic sensing (RT-CES) analyzer and 16 × station (Acea Biosciences, San Diego, CA). Cell growth was monitored periodically (typically, every 0.5 or 1 h) for indicated durations via calculation of a “cell index” (normalized impedance) for each well. Unless otherwise indicated, cells from each well of the original six-well plates were reseeded into four replicate wells for cell index measurement (8). Six to eight-week old NOD/SCID/IL2Rgamma null mice (City of Hope) were subcutaneously inoculated in the right flank with either 5 × 106 KB/PC3-GFP or KB/PC3-p53R2-GFP cells. Tumor xenograft diameters were measured with digital calipers twice a week, and the tumor volume in mm3 was calculated by the formula: Volume = (width) 2 × length/2. Results were presented as mean tumor volume ± SD of two independent experiments (8). The animal experiments were performed by Animal Tumor Model Core Facility in City of Hope Beckman Research Institute.
Statistics
Data were collected using an MS-Excel spreadsheet. Data were analyzed using the JMP Statistical Discovery Software version 6.0 (SAS Institute, Cary, NC, Group comparisons for continuous data were done with student’s t-test for independent means or two-way ANOVA. Statistical significance was set at P < 0.05.
Results
GSEA with cDNA microarray data revealed that gene set regulating cell cycle progression was enriched in p53R2-attenuated KB cells
Gene set enrichment analysis (GSEA) with cDNA microarray identified that 17 out of 429 gene sets from the C2 inventory created by Molecular Signatures Database (MSigDB) (20) were significantly enriched and 24 were significantly reversely enriched in p53R2-attenuated KB cells compared with control cells (Nom P < 0.05). These top enriched gene sets (Nom P < 0.05) included the gene sets of cell cycle, ABC transporter-general and inhibition of matrix metalloproteinase etc, which are shown in Table S2.
As shown by the enrichment plot in Figure 1A, the gene set regulating cell cycle progression was significantly enriched in p53R2 attenuated KB cells. The heat map (Figure 1B) displayed names, positions and expression levels of the enriched genes in the set. Quantitative reverse transcriptional PCR (q-RT-PCR) analysis was performed to validate the differential expression of the 10 selected genes in the set (Figure 1C). Q-RT-PCR analysis validated that attenuation of p53R2 expression significantly up-regulated the mRNA expression of several cell cycle regulatory proteins such as cyclin A2, cyclin D1 and CDK1 etc, while simultaneously decreasing mRNA expression of several cell cycle inhibitors such as CDKN1A (p21WAF1/CIP1, p21), CDKN1C (KIP2) (Figure 1C). Additionally, we used an alternative method of ingenuity pathways analysis (IPA) to validate the GSEA pathway analysis result that cell cycle pathway was enriched by p53R2 knock-down. We selected the genes that are differentially expressed between p53R2 and control with P < 0.01 (equivalent to FDR < 0.1), which resulted in 1,997 probe sets. These genes were analyzed using IPA, and the significant functional categories (P < 0.01) were identified and shown in Figure S2. The original microarray data were submitted to GEO of NCBI, it will be obtained from website GEO of NCBI with accessing number of GSE25238.
Figure 1.

GSEA identified that the gene set regulating cell cycle progression was enriched in p53R2 attenuated KB cells. A, enrichment plot showed the gene set of cell cycle was significantly enriched in p53R2 attenuated KB. B, the heat map displayed the list of enriched genes, location and expression level of each individual gene. p53R2 for p53R2 siRNA transfected KB, and control for control siRNA transfected KB. C, cDNA microarray data was validated by q-RT-PCR analysis that attenuation of p53R2 up-regulated the mRNA expression of several cell cycle regulatory proteins such as cyclin D1 and CDK1 etc, simultaneously decreased the mRNA expression of several cell cycle inhibitors such as CDKN1B (p21WAF1/CIP1), CDKN1C (KIP2). The data for q-RT-PCR was the average of three independent experiments.
Attenuation of p53R2 decreased p21 expression and increased cyclin D1 in cancer cells
At gene level, cDNA microarray analysis identified that attenuation of p53R2 caused about 100 genes differentially expressed in KB cells (P < 0.05). These genes including p21 and cyclin D1 are listed in Table S3 of supplementary data. P21 and cyclin D1 are essential cell cycle regulators; we further examined the regulation of p53R2 on these two genes in various cancer cell lines. Additionally, since p21 is a well-known target of p53, we also questioned whether the regulation of p21 by p53R2 was wild-type p53-dependent or not. Attenuation of p53R2 expression by siRNA (Figure 2A) significantly decreased p21 and increased cyclin D1 expression in both p53 wild-type (KB and MCF-7) and p53 mutated (PC3, MDA-MB-231) cancer cells (Figure 2B and 2C), which indicates the regulation was wild-type p53 independent. Quantitative analysis of the triplicate data showed that the differential expression of RNA and protein of p21 and cyclin D1 was statistically significant (Figure 2B and 2D).
Figure 2.
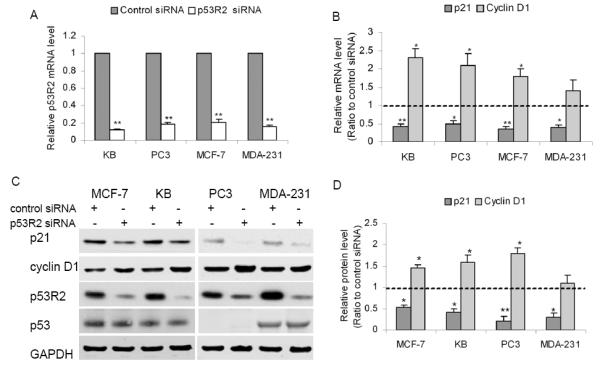
Attenuation of p53R2 decreased p21 expression and increased cyclin D1 in cancer cells. A, q-RT-PCR analysis validated that p53R2 mRNA expression was significantly decreased by at least ~92% in KB (p53 wild-type), PC3 (p53-trucated), MCF-7 (p53 wild-type) and MDA-MB-231 (p53-mutated) cells at 48 hour post-transfection with p53R2 siRNA. B, q-RT-PCR and C, Western blot analysis showed that p21 expression was significantly decreased by at least ~50% and cyclin D1 expression was moderately increased in the cancer cells after attenuation of p53R2 (Control: scramble siRNA transfected, p53R2: p53R2 siRNA transfected KB). D, quantitative levels of p21 protein and cyclin D1 in cancer cells transfected with p53R2 siRNA were determined by measuring the density of the band of p21/cyclin D1 and normalized to that of GAPDH. Data were presented as the ratio to control and were the mean ± SD of three experiments, *P < 0.05, **P < 0.01, compared with control cells.
Overexpression of p53R2 increased p21 and decreased cyclin D1 and phosphorylation of ERK1/2 in both KB and PC3 cells
To further elucidate the regulation of p53R2 on expression of cell-cycle regulatory genes, we constructed two cancer cell lines stably overexpressing p53R2-GFP. FACS technique was applied to enrich cancer cells highly expressing GFP/p53R2-GFP. As shown in Figure 3A, KB-p53R2-GFP and PC3-p53R2-GFP cells highly expressed p53R2-GFP, which was clearly observed by the florescent microscope (Figure 3A). Western blot assay further showed that the p53R2-GFP fused protein (around 76-KDa) was highly expressed in both KB-p53R2-GFP and PC3-p53R2-GFP cells (Figure 3B). Compared with KB-GFP and PC3-GFP, the p21 protein expression was significantly increased and cyclin D1 expression was moderately decreased in KB-p53R2-GFP and PC3-p53R2-GFP as shown in Figure 3C (P < 0.05). A most recent study showed that p53R2 negatively regulates MEK-ERK signal pathway through direct interaction with MEK2 (22). Using Western blot analysis, we also found that overexpression of p53R2-GFP significantly decreased phosphorylated ERK1/2 in KB and PC3 cancer cells (Figure 3B). Knockdown of p53R2 moderately increased the level of phosphorylated ERK1/2 in these cancer cells (Figure 3D). Furthermore, the MEK inhibitor PD98059 partially abolished the impact of p53R2 on expression of cell-cycle regulatory genes: p21 and cyclin D1 in KB cells (Figure 3D). The above observation indicates that p53R2 may regulate expression of cell-cycle regulatory genes partially through inhibition of ERK1/2 phosphorylation.
Figure 3.

Overexpression of p53R2 increased p21, decreased cyclin D1 and phosphorylated ERK1/2 in both KB and PC3 cells. A, the photos of fluorescence microscopy (×100) showed a high expression of p53R2-GFP fused protein in GFP and p53R2-GFP cancer cells. B, Western blot analysis showed that p21 protein was significantly upregulated; cyclin D1 and phosphorylated ERK1/2 were moderately downregulated in KB-p53R2-GFP/PC3-p53R2-GFP cells compared with KB-GFP/PC3-GFP. C, relative quantitative level of indicated proteins in cells was determined by measuring the density of the band of target protein and normalized to that of GAPDH. Data were presented as the ratio to control and were the mean ± SD of three experiments, *P <0.05, **P < 0.01, compared with control cells. D, knockdown of p53R2 moderately upregulated serum-induced phosphorylated ERK1/2. The MEK inhibitor PD98059 partially abolished the impact of p53R2 on expression of p21 and Cyclin D1 in KB cells. KB cells were transferred with p53R2 siRNA, serum-starved overnight after 24 hours, and followed by stimulation with 20% serum in the absence or presence of MEK inhibitor PD98059 (25 μmol/L).
p53R2-GFP reduced the percentage of cells in S-phase of both KB and PC3 cells
To test the impact of p53R2 on cell cycle progression, we first measured the impact of attenuation of p53R2 on cell cycle progression of KB and PC3 cancer cells. We found that transient attenuation of p53R2 by siRNA moderately increased the percentage of cells in S phase in both wild type KB (about ~6%) and PC3 cells (about ~4%). We further compared cell cycle profiles of KB-p53R2-GFP and PC3-p53R2-GFP with their controls. The representative cell cycle profiles of KB-GFP/p53R2-GFP and PC3-GFP/p53R2-GFP cells were shown in Figure 4A and 4B respectively. In comparison with KB-GFP cells (28.36 ± 3.12%), the percentage of KB-p53R2-GFP cells in S-phase (14.67 ± 4.49%) was significantly decreased (P < 0.01). Simultaneously, compared with KB-GFP (58.72 ± 5.43%), the percentage of KB-p53R2-GFP cells in G1-phase (74.52 ± 4.22%) was markedly increased (P < 0.01) (Figure 4C). Compared with PC3-GFP (S-phase: 23.43 ± 5.66%, G1-phase: 53.52 ± 3.28 %), the percentage of PC3-p53R2-GFP cells in S-phase (13.57 ± 3.56%) was significantly decreased (P < 0.01); while that in G1-phase (65.57 ± 4.98%) was significantly increased (P < 0.01) (Figure 4D).
Figure 4.

Overexpression of p53R2-GFP reduced the percentage of cells in S-phase and increased that in G1-phase in both KB (p53 wild-type) and PC3 (p53 truncated) cells. Representative cell cycle profiles were shown as A, KB-GFP and KB-p53R2-GFP cells; B, PC3-GFP and PC3-p53R2-GFP cells. C, quantitative analysis showed that in comparison with KB-GFP cells (28.36±3.12%), the percentage of KB-p53R2-GFP cells in S-phase (14.67 ± 4.49%) was significantly decreased (P < 0.01). Simultaneously, compared with KB-GFP (58.72 ± 5.43%), the percentage of KB-p53R2-GFP cells in G1-phase (74.52 ± 4.22%) was markedly increased (P < 0.01). D, compared with PC3-GFP (S-phase: 23.43± 5.66%, G1-phase: 53.52 ± 3.28 %), the percentage of PC3-p53R2-GFP cells in S-phase (13.57 ± 3.56%) was significantly decreased (P < 0.01), while that in G1-phase (65.57 ± 4.98%) was significantly increased (P < 0.01). The mean and standard errors for the percentage of cells in each phase of the cell cycle were derived from at least three independent experiments, each in duplicate.
Overexpression of p53R2-GFP inhibited proliferation of human cancer cells in vitro and in vivo
The above data strongly suggest that p53R2 may regulate cancer cell proliferation. To further elucidate this question, we compared the growth of p53R2-GFP cancer cells with their control cells. RT-CES traces displayed that proliferation of KB-p53R2-GFP and PC3-p53R2-GFP was significantly slower than their control cells over a period of 96 hours (Figure 5A and 5B). We also observed that transient attenuation of p53R2 by siRNA slightly increased the proliferation of KB and PC3 (data not shown). We further assessed the effect of p53R2-GFP on growth of tumor xenografts in vivo. We found that the growth of KB-p53R2-GFP and PC3-p53R2-GFP xenografts were significantly retarded compared to that of KB-GFP and PC3-GFP xenografts (Figure 5C and 5D, P < 0.05). Mean weight of xenografts was 0.90 ± 0.13/0.29 ± 0.09 g for KB-GFP/PC-GFP cells (n = 8) and 0.56 ± 0.12/0.15 ± 0.07 g for KB-p53R2-GFP/PC3-p53R2-GFP cells (n = 8) respectively, and the decrease was statistically significant (P < 0.05). Western blot analysis also displayed that KB-p53R2-GFP and PC3-p53R2-GFP xenografts expressed more p21 and less cyclin D1 (small pictures in Figure 5C and 5D).
Figure 5.

Overexpression of p53R2-GFP inhibited the growth of KB and PC3 cancer cells in vitro and vivo. A, real time proliferation curves of KB-GFP vs. KB-p53R2-GFP cells and B, PC3-GFP vs. PC3-p53R2-GFP cells. Trace of RT-CES displayed that the growth of KB-p53R2-GFP was significantly slower than KB-GFP cells, and PC3-p53R2-GFP cells grew slightly slower than PC3-GFP over 96 hour. Each trace was an average of 8~12 replicates, *P <0.05 compared with GFP cells after 96 hour culture. C, xenografts curves of KB-GFP vs. KB-p53R2-GFP cells and D, PC3-GFP vs. PC3-p53R2-GFP cells, which described that overexpression of p53R2-GFP significantly inhibited KB-p53R2-GFP and PC3-p53R2-GFP in vivo growth. Small Western blot photo showed the protein level of p21, cyclin D1, p53R2-GFP and p53R2 in one representative tumor xenograft.
Impact of attenuation of p53R2 on DNA damage in KB cells
We further investigated whether p53R2 has a feedback impact on p53, and how p53R2 impacts p21 expression after DNA damage. As shown in Figure 6A, Western blot analysis identified neither the attenuation nor overexpression of p53R2 changed p53 protein level in KB cells. Luciferase report assays also showed that overexpression of p53R2 significantly increased luciferase activity of p21-Luc by 1.6 fold (P < 0.05), but not the luciferase activity of p53 reporter plasmid PG13 (Figure 6B), which consistently indicates the regulation of p53R2 on p21 expression is wild-type p53 independent. We examined the impact p53R2 on p53 and p21 expression under genotoxic stress caused by Adriamycin. Q-RT-PCR analysis and Western blot assays (Figure 6C and 6D) showed that attenuation of p53R2 significantly decreased the p21 mRNA and protein expression, but did not change p53 protein level in KB cells under the genotoxic stress of Adriamycin. DNA damage induced by Adriamycin significantly increased phosphorylation of histone H2AX in KB cells. The level of phosphorylated H2AX was comparable to that of phosphorylated ATM. p53R2-attenuated KB cells had a substantial increase in the phosphorylation of H2AX not only under the stress of Adriamycin but also under normal condition. Consistently, we found that overexpression of p53R2 only marginally decreased the phosphorylation of H2AX and ATM caused by genotoxic stress of adriamycin in KB cells, which indicates that overexpression of p53R2 may slightly increase the resistance of cancer cells against DNA damage reagent (Figure S3).
Figure 6.

Impact of attenuation of p53R2 on DNA damage in KB cells. A, neither attenuation nor overexpression of p53R2 modulated p53 protein level (p53 protein was developed by immunoprecipitation-Western blot; mouse and rabbit anti-p53R2 antibodies were used for pull-down and blot respectively). B, overexpression of p53R2 significantly increased the luciferase activity of p21 promoter reporter: p21-Luc (* P < 0.05), but not the luciferase activity of p53 reporter plasmid PG-13. C, q-RT-PCR and D, Western-blot analysis showed that the attenuation of p53R2 did not change p53 accumulation induced by adramycin, but significantly decreased p21 expression in KB cells. Under both normal and stressed conditions, attenuation of p53R2 substantially enhanced the DNA damage in KB cells, which was indicated by increased phosphorylation of γ-H2AX and phosphorylated ATM proteins.
Discussion
The roles of p53R2 in the biological characteristics of cancer cells and the underlying mechanisms remain largely unclear even controversial. Using GSEA, a more reproducible and more interpretable method that focuses on pathways and processes rather than on high scoring individual gene (20, 23), we surprisingly found that the gene set regulating cell cycle progression was significantly enriched in the p53R2-attenuated KB cells. The finding promoted us to examine the impact of p53R2 on cell cycle progression. Coincidently, transient attenuation of p53R2 by siRNA moderately increased the percentage of cells in S phase in both wild type KB and PC3 cells. To further examine the impact; we constructed two cancer cell lines highly overexpressing p53R2-GFP. Unlike GFP-expressing cells, we observed that the percentage of cells with high-level of p53R2-GFP expression progressively decreased among G418-resistant cells during the process of selection and following culture with G418. The phenotype of p53R2-GFP expressing cells during G418-selection was very similar to cells overexpressing cell cycle inhibitor as reported previously by others (21, 24). Overexpression of GFP may be cytotoxic to cells; however, the phenomena may not be caused by GFP, since the intensity and viability of GFP-expressing cells were stable during G418-selection and culture. We found that overexpression of p53R2-GFP significantly suppressed cancer cells growth regardless of the status of p53. Consistently, early study by Kimura et al revealed that p53R2−/− and p53R2+/− embryonic fibroblasts (MEFs) showed an increase in the rate of proliferation after 7 passages, indicating an inhibitory role of p53R2 in cell proliferation (14). Study by Yamaguchi et al suggested the suppression of cancer growth by inactivation of p53R2-dependent pathway owes to the activation of p53-dependent apoptotic pathway due to the shortage of dNTP for DNA synthesis (25). However, we previously reported that RRM2 may complement p53R2 in response to UV-induced DNA repair in cancer cells with mutant p53 (26). A different study also showed that disruption of the p53R2 mediated DNA repair in ulcerative colitis could initiate carcinogenesis in colon (13).
At the transcriptional level, we found that attenuation of p53R2 in KB cells significantly altered p21 and cyclin D1 levels. Because p21 and cyclin D1 play well-known role in the regulation of cell cycle progression (27, 28), therefore, we further examined the impact of p53R2 on the expression of p21 and cyclin D1 in p53 wild-type cancer cells (KB and MCF-7) and p53 mutated cancer cells (PC3 and MDA-MB-231). A most recent study showed that p53R2 negatively regulates MEK-ERK signal pathway through binging to MEK2 (22). We found that overexpression of p53R2 moderately decreased phosphorylated ERK1/2 in KB cells. Since MEK-ERK signaling pathway regulates diverse cellular functions including cell proliferation, cell cycle progression and cell survival etc, and inhibition of this pathway is a logical therapeutic target for malignancies (29, 30). We further found MEK inhibitor PD98059 partially abolished the impact of p53R2 on expression of cell cycle regulating genes. Therefore, we considered that impact of p53R2 on cancer cells growth may mediate partially through its inhibition on MEK-ERK signaling pathway.
Both p53R2 and p21 are targets and executors of ATM-p53 pathway after DNA damage that provides dNTP for DNA repair and causes cell cycle arrest (21, 31). We further investigated whether p53R2 has a feedback impact on p53, and whether p53R2 still regulates p21 expression after DNA damage. Our data showed that neither attenuation nor overexpression of p53R2 changed wild-type p53 protein in KB cancer cells. Consistent with the observation of study by Devlin et al (12), we also found that p21 protein level was significantly decreased in p53R2-atenuated KB cells under the condition of DNA damage induced by adriamycin, and attenuation of p53R2 impaired DNA damage repair in KB cancer cells as indicated by enhanced phosphorylated γ-H2AX (12, 32). We surprised to find that attenuation of p53R2 in KB cells without genomic stress increased phosphorylated γ-H2AX. We consider that knockdown of p53R2 may increase reactive oxygen species (ROS) from endogenous sources such as oxidative phosphorylation, cytochrome P450 metabolism, peroxisomes etc (33). And ROS may cause DNA damage and induce phosphorylated γ-H2AX. Indeed, the previous study by Kimura et al also found that p53R2−/− MEF cells was much more sensitive to oxidative stress damage that p53R2 wild-type MEF cells (14). Coincidently, a recent study further showed that knockdown of p53 decreased mitochondrial and cellular superoxide levels and increased cellular hydrogen peroxide in human primary fibroblast cells under normal culture conditions, which was accompanied by a reduction of the p53R2 and mtDNA depletion (34).
Several studies found silencing p53R2 enhanced the apoptotic effects of ionizing radiation, UV and genotoxic agent in a wild-type p53R2 dependent manner. p53R2 is a potential therapeutic target for cancer (11, 12, 28). However, both down-regulation and upregulation of p53R2 in human cancer tissues were reported. Studies of ours and others indicated that basal transcription of p53R2 is expressed in almost all of normal human tissues and regulated through the p53-independent mechanism (17, 26). Furthermore, the low constitutive level of p53R2 in mammalian cells is essential for the supply of dNTPs for basal levels of DNA repair and mitochondrial DNA synthesis in G0/G1 cells (35). Studies indicated that p53R2 plays an essential role in maintenance of genomic stability (13, 14). It is well-known that genomic instability also contributes to tumor development, progression and resistance to therapy. Since p53 mutation is frequently observed in human cancers, genomic instability is often seen in cancer tissues without wild-type p53 protein, which may reflect the dysfunction of RNR due to the failure of p53R2 induction (36). Therefore, the novel finding of our study that p53R2 may suppress cancer cells proliferation regardless of p53 status of cells, may be a careful consideration for p53R2-targeted cancer therapy, especially for cancers with mutant p53, since studies indicate that attenuation of p53R2 only enhances the apoptotic effects of ionizing radiation, UV and genotoxic agent in p53 wild-type cancer cells.
Supplementary Material
Acknowledgements
We thank Christina Yen for her excellent revision on the language of the manuscript and Charles Kim for picture edition.
Grant support: This work was partially supported by National Cancer Institute grant CA 127541-01 for Dr. Yun Yen.
Abbreviations list
- RNR
ribonucleotide reductase
- RRM2
R2 subunit of ribonucleotide reductase
- p53R2
the p53-inducible homologue of the R2 subunit of ribonucleotide reductase
- dNDP
deoxyribonucleotides
- GSEA
gene set enrichment analysis
- ERK
extracellular signal-related kinase
- MEK
mitogene-activated protein kinase/ERK kinase
- q-RT-PCR
quantitative reverse transcriptional PCR
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
References
- 1.Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 2.Engstrom Y, Eriksson S, Jildevik I, Skog S, Thelander L, Tribukait B. Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits. J Biol Chem. 1985;260:9114–6. [PubMed] [Google Scholar]
- 3.Zhou B, Yen Y. Characterization of the human ribonucleotide reductase M2 subunit gene; genomic structure and promoter analyses. Cytogenet Cell Genet. 2001;95:52–59. doi: 10.1159/000057017. [DOI] [PubMed] [Google Scholar]
- 4.Tanaka H, Arakawa H, Yamaguchi T, et al. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404:42–9. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 5.Guittet O, Håkansson P, Voevodskaya N, et al. Mammalian p53R2 protein forms an active ribonucleotide reductase in vitro with the R1 protein, which is expressed both in resting cells in response to DNA damage and in proliferating cells. J Biol Chem. 2001;276:40647–51. doi: 10.1074/jbc.M106088200. [DOI] [PubMed] [Google Scholar]
- 6.Fan H, Huang A, Villegas C, Wright JA. The R1 component of mammalian ribonucleotide reductase has malignancy-suppressing activity as demonstrated by gene transfer experiments. Proc Natl Acad Sci USA. 1997;94:13181–6. doi: 10.1073/pnas.94.24.13181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou B, Tsai P, Ker R, et al. Overexpression of transfected human ribonucleotide reductase M2 subunit in human cancer cells enhances their invasive potential. Clin Exp Metastasis. 1998;16:43–9. doi: 10.1023/a:1006559901771. [DOI] [PubMed] [Google Scholar]
- 8.Zhang K, Hu S, Wu J, et al. Overexpression of RRM2 decreases thrombspondin-1 and increases VEGF production in human cancer cells in vitro and in vivo: implication of RRM2 in angiogenesis. Mol Cancer. 2009 doi: 10.1186/1476-4598-8-11. doi:10.1186/1476-4598-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yanamoto S, Iwamoto T, Kawasaki G, Yoshitomi I, Baba N, Mizuno A. Silencing of the p53R2 gene by RNA interference inhibits growth and enhances 5-fluorouracil sensitivity of oral cancer cells. Cancer Lett. 2005;223:67–76. doi: 10.1016/j.canlet.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 10.Okumura H, Natsugoe S, Matsumoto M, et al. The predictive value of p53, p53R2, and p21 for the effect of chemoradiation therapy on oesophageal squamous cell carcinoma. Br J Cancer. 2005;92:284–9. doi: 10.1038/sj.bjc.6602322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yanamoto S, Kawasaki G, Yoshitomi I, Mizuno A. Expression of p53R2, newly p53 target in oral normal epithelium, epithelial dysplasia and squamous cell carcinoma. Cancer Lett. 2003;190:233–43. doi: 10.1016/s0304-3835(02)00588-8. [DOI] [PubMed] [Google Scholar]
- 12.Devlin HL, Mack PC, Burich RA, et al. Impairment of the DNA repair and growth arrest pathways by p53R2 silencing enhances DNA damage-induced apoptosis in a p53-dependent manner in prostate cancer cells. Mol Cancer Res. 2008;6:808–18. doi: 10.1158/1541-7786.MCR-07-2027. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida T, Haga S, Numata Y, et al. Disruption of the p53-p53R2 DNA repair system in ulcerative colitis contributes to colon tumorigenesis. Int J Cancer. 2006;18:1395–1403. doi: 10.1002/ijc.21538. [DOI] [PubMed] [Google Scholar]
- 14.Kimura T, Takeda S, Sagiya Y, Gotoh M, Nakamura Y, Arakawa H. Impaired function of p53R2 in Rrm2b-null mice causes severe renal failure through attenuation of dNTP pools. Nat Genet. 2003;34:440–5. doi: 10.1038/ng1212. [DOI] [PubMed] [Google Scholar]
- 15.Okumura H, Natsugoe S, Yokomakura N, et al. Expression of p53R2 is related to prognosis in patients with esophageal squamous cell carcinoma. Clin Cancer Res. 2006;12:3740–5. doi: 10.1158/1078-0432.CCR-05-2416. [DOI] [PubMed] [Google Scholar]
- 16.Uramoto H, Sugio K, Oyama T, Hanagiri T, Yasumoto K. p53R2, p53 inducible ribonucleotide reductase gene, correlated with tumor progression of non-small cell lung cancer. Anticancer Res. 2006;26:983–8. [PubMed] [Google Scholar]
- 17.Byun DS, Chae KS, Ryu BK, Lee MG, Chi SG. Expression and mutation analyses of p53R2, a newly identified p53 target for DNA repair in human gastric carcinoma. Int J Cancer. 2002;98:718–23. doi: 10.1002/ijc.10253. [DOI] [PubMed] [Google Scholar]
- 18.Liu X, Zhou B, Xue L, et al. Ribonucleotide reductase subunits M2 and p53R2 are potential biomarkers for metastasis of colon cancer. Clin Colorectal Cancer. 2007;6:374–81. doi: 10.3816/CCC.2007.n.007. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Zhou B, Xue L, et al. Metastasis-suppressing potential of ribonucleotide reductase small subunit p53R2 in human cancer cells. Clin Cancer Res. 2006;12:6337–44. doi: 10.1158/1078-0432.CCR-06-0799. [DOI] [PubMed] [Google Scholar]
- 20.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 22.Piao C, Jin M, Kim HB, et al. Ribonucleotide reductase small subunit p53R2 suppresses MEK-ERK activity by binding to ERK kinase 2. Oncogene. 2009;28:2173–84. doi: 10.1038/onc.2009.84. [DOI] [PubMed] [Google Scholar]
- 23.Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Gene. 2003;34:267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 24.Ray A, Shakya A, Kumar D, Ray BK. Overexpression of serum amyloid A-activating factor 1 inhibits cell proliferation by the induction of cyclin-dependent protein kinase inhibitor p21WAF-1/Cip-1/Sdi-1 expression. J Immunol. 2004;172:5006–15. doi: 10.4049/jimmunol.172.8.5006. [DOI] [PubMed] [Google Scholar]
- 25.Yamaguchi T, Matsuda K, Sagiya Y, et al. p53R2-dependent pathway for DNA synthesis in a p53-regulated cell cycle checkpoint. Cancer Res. 2001;1:8256–62. [PubMed] [Google Scholar]
- 26.Zhou B, Liu X, Mo X, et al. The human ribonucleotide reductase subunit hRRM2 complements p53R2 in response to UV-induced DNA repair in cells with mutant p53. Cancer Res. 2003;63:6583–94. [PubMed] [Google Scholar]
- 27.Li Y, Jenkins CW, Nichols MA, Xiong Y. Cell cycle expression and p53 regulation of the cyclin-dependent kinase inhibitor p21. Oncogene. 1994;9:2261–8. [PubMed] [Google Scholar]
- 28.Sherr CJ, Roberts JM. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 29.McCubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–84. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res. 2008;14:342–6. doi: 10.1158/1078-0432.CCR-07-4790. [DOI] [PubMed] [Google Scholar]
- 31.Chang L, Zhou B, Hu S, et al. ATM-mediated serine 72 phosphorylation stabilizes ribonucleotide reductase small subunit p53R2 protein against MDM2 to DNA damage. Proc Natl Acad Sci U S A. 2008;105:18519–24. doi: 10.1073/pnas.0803313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonner WM, Redon CE, Dickey JS, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–67. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annu Rev Pharmacol Toxicol. 2004;44:239–67. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 34.Lebedeva MA, Eaton JS, Shadel GS. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim Biophys Acta. 2009;1787:328–34. doi: 10.1016/j.bbabio.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Håkansson P, Hofer A, Thelander L. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J Biol Chem. 2006;281:7834–41. doi: 10.1074/jbc.M512894200. [DOI] [PubMed] [Google Scholar]
- 36.Hundley JE, Koester SK, Troyer DA, Hilsenbeck SG, Subler MA, Windle JJ. Increased tumor proliferation and genomic instability without decreased apoptosis in MMTV-ras mice deficient in p53. Mol Cell Biol. 1997;17:723–31. doi: 10.1128/mcb.17.2.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.