

Abstract
In histidine and tryptophan biosynthesis, two related isomerization reactions are generally catalyzed by two specific single-substrate enzymes (HisA and TrpF), sharing a similar (β/α)8-barrel scaffold. However, in some actinobacteria, one of the two encoding genes (trpF) is missing and the two reactions are instead catalyzed by one bisubstrate enzyme (PriA). To unravel the unknown mechanism of bisubstrate specificity, we used the Mycobacterium tuberculosis PriA enzyme as a model. Comparative structural analysis of the active site of the enzyme showed that PriA undergoes a reaction-specific and substrate-induced metamorphosis of the active site architecture, demonstrating its unique ability to essentially form two different substrate-specific actives sites. Furthermore, we found that one of the two catalytic residues in PriA, which are identical in both isomerization reactions, is recruited by a substrate-dependent mechanism into the active site to allow its involvement in catalysis. Comparison of the structural data from PriA with one of the two single-substrate enzymes (TrpF) revealed substantial differences in the active site architecture, suggesting independent evolution. To support these observations, we identified six small molecule compounds that inhibited both PriA-catalyzed isomerization reactions but had no effect on TrpF activity. Our data demonstrate an opportunity for organism-specific inhibition of enzymatic catalysis by taking advantage of the distinct ability for bisubstrate catalysis in the M. tuberculosis enzyme.
Keywords: amino acid biosynthesis, enzyme evolution, mycobacteria, active site substrate adaptability, protein structure symmetry
Enzymes that fold into (β/α)8 barrels provide the most widely found prototype scaffold to study the molecular mechanisms underlying enzymatic diversity (1, 2). Among those investigated, many catalyze specific reactions, but there are also examples of enzymes with broader substrate profiles (2, 3). Generally the level of functional promiscuity seems to be correlated with active site conformational plasticity (2, 4), conceptually described as “metamorphic proteins” (5). There is, however, at least one documented case in which two alternative solutions have evolved to catalyze two distinct isomerization reactions involved in histidine (his) and tryptophan (trp) biosynthesis, either by two single-substrate enzymes or by one bisubstrate enzyme, all folded as (β/α)8 barrels. The mechanism of bisubstrate specificity in the latter has remained unknown to date.
In most bacteria with these biosynthesis pathways extant, two different genes, hisA and trpF (6), encode two distinct single-substrate enzymes (HisA, TrpF) that catalyze the isomerization of distinct metabolites from two amino acid biosynthesis pathways, N′-[(5′-phosphoribosyl)-formimino]-5-aminoimidazole-4-carboxamide ribonucleotide (ProFAR, his biosynthesis) and phosphoribosyl anthranilate (PRA, trp biosynthesis). Biochemical data indicate that both isomerization reactions are catalyzed by an acid/base-assisted Amadori rearrangement (7). In structural terms, both single-substrate enzymes are folded into (β/α)8 barrels (8, 9). However, because the two substrates differ by about a factor of two in molecular weight, the active sites of HisA and TrpF substantially differ in overall size. Whereas the larger HisA substrate ProFAR is bound via two terminal phosphate groups in symmetric positions across the active site of the HisA (β/α)8-barrel fold, the smaller TrpF substrate PRA is bound via only one conserved phosphate group-binding site into the TrpF active site (9, 10).
However, in most of the known genomes of the actinobacteria phylum, the trpF gene is missing from the trp operon. A hisA-like gene, referred to as priA, was shown to encode an enzyme with bisubstrate specificity in Streptomyces coelicolor, rendering a bifunctional his/trp biosynthesis isomerase (11). Although some active site residues from the C-terminal (β/α)8-barrel face in PriA have been shown to be involved in PriA catalysis (12–14), it was not known which molecular mechanism renders PriA a bisubstrate enzyme, in the absence of structural insight into the positioning of any of the two substrates within the active site. Here, we have investigated the PriA enzyme from Mycobacterium tuberculosis to resolve this question. Because this pathogen, like S. coelicolor, does not have a trpF gene, we expected bisubstrate activity in the corresponding PriA enzyme as well. Based on three separate structures—presenting the apo conformation and distinct substrate-induced conformations of each of the two isomerization reactions—we have unraveled an unexpected ability of the enzyme to form two different active site structures that adapt to the respective his and trp biosynthesis substrates. We furthermore demonstrate that one of two activities (PRA isomerization) involves active site residues that are distinct from the analogous single-substrate enzyme TrpF, and we show that these differences can be exploited with PriA-specific inhibitors.
Results
Structural Basis of the Substrate-Dependent Active Site Properties of PriA.
To determine the molecular basis of bisubstrate specificity, we crystallized PriA from M. tuberculosis in the presence of two reaction ligands involved in HisA-like ProFAR isomerization and TrpF-like PRA isomerization (Figs. 1 and 2 and Table S1). Crystals of the catalytically impaired PriA(D11N) variant, grown in the presence of the substrate ProFAR, diffracted to ultrahigh resolution (1.33 Å). The electron density map revealed the presence of the product N′-[(5′-phosphoribulosyl)formimino]-5-aminoimidazole-4-carboxamide ribonucleotide (PrFAR), with an opened phosphoribulosyl moiety, indicating residual substrate turnover under crystallization conditions. The structure of wild-type PriA, in the presence of the reduced product analogue 1-(o-carboxyphenylamino)-1-deoxyribulose 5-phosphate (rCdRP), was determined as well. The crystallization conditions and resulting crystal forms were unrelated to each other (Table S1).
Fig. 1.

Ligand-induced active site changes of PriA. (A) Active site loop structure of the PriA complexes with sulfate (Left), rCdRP (Center), and PrFAR (Right). The eight active site loops 1–8 are colored in yellow (1,5), green (2,6), cyan (3,7), and magenta (4,8), emphasizing the twofold repeated (β/α)4 half-barrel elements (9); those loop segments that are disordered are indicated by dashed lines. The remaining structures are shown in surface presentation (β-strands, light gray; α-helices, dark gray). Each ligand is shown in stick presentation. (B) Ligand-specific Asp175 recruitment: by Arg143, in the presence of rCdRP (trp biosynthesis, Left); and by Arg19, in the presence of PrFAR (his biosynthesis, Right). Loop 5 changes from a β-hairpin conformation (rCdRP) to a knot-like conformation (PrFAR), allowing Arg143 and Trp145 to switch positions. Hydrogen bonds are shown with dashed lines. The areas shown in B approximately correspond to the red boxes in A.
Fig. 2.

Active site structures of the PriA-rCdRP and PriA-PrFAR complexes. Each ligand (rCdRP, A; PrFAR, B) is shown in 2Fo-Fc electron density at a 1,5 σ contour level. Hydrogen bonds between each ligand and PriA residues, in part mediated by ordered solvent molecules (red spheres), are represented with dashed lines. Highlighted residues and ligands are shown in stick presentation, using atom-specific colors (oxygen, red; nitrogen, blue; sulfur, yellow; phosphorus, magenta). The carbon atoms of PriA residues are colored according to the scheme used in Fig. 1; carbon atoms of ligands are in gray. Residues involved in side chain-specific interactions with ligands are labeled. The labels of those residues that have been identified in catalysis or ligand-induced active site recruitment are colored red. The remaining part of each PriA structure is shown in ribbon representation.
As reference, we also solved the PriA crystal structure in the presence of sulfate ions, which mimic the phosphate groups of the reaction ligands (apo form) (Fig. S1 A and B and Table S1). Comparison of this structure with those of the same enzyme from S. coelicolor in the presence of sulfate (12, 13) reveals no significant changes of the overall fold and active site loop structure, indicating that the conformational changes observed in the two PriA-ligand complexes are caused by the presence of the reaction ligands.
The overall structure of M. tuberculosis PriA is a (β/α)8 barrel with a twofold repeated array of active site loops, 1–4 and 5–8 (Fig. 1A and Fig. S1A). Comparison of the two PriA-ligand complexes reveals that the overall orientation of the two reaction compounds, PrFAR and rCdRP, is similar (Figs. 1A and 2 and Fig. S1C), despite substantial differences in the active site environment (see below). The phosphoribulosyl moiety of both reaction ligands is anchored by the same conserved phosphate-binding site, which is created by loops 7 and 8, and superimposes well in the PriA active site (Fig. 2 and Figs. S1C and S2). In contrast, the 5-aminoimidazole-4-carboxamide ribonucleotide moiety of PrFAR exceeds the rCdRP structure and, therefore, requires a larger PriA active site binding area. One of the sulfate ions of the apo-structure superimposes with the common terminal phosphate group of the two reaction compounds (Fig. 1A and Fig. S1C). The second phosphate of PrFAR is bound by the symmetry-related loops 3 and 4 and superimposes with the second sulfate ion of the apo structure.
In contrast to the rigid PriA (β/α)8-barrel core, which is virtually identical in all three structures, the active site loops 1, 5, and 6 from the C-terminal (β/α)8-barrel face show reaction-specific conformational changes with movements up to 20 Å and, in part, order/disorder transitions (Fig. 1, Fig. S3, and Movies S1 and S2). Loop 1 wraps over the PriA active site only in the presence of PrFAR (his biosynthesis), whereas in the other two structures it is either partly unfolded or tilted away from the active site. The symmetry-related loop 5 folds over the active site in the presence of both reaction compounds (PrFAR, rCdRP), while being partly disordered in the apo conformation. Strikingly, depending on the type of bound ligand, loop 5 either adopts a β-sheet-like hairpin structure (rCdRP) or twists into a knot-like conformation (PrFAR) (Fig. 1B). In these two arrangements, Arg143 and Trp145 exchange positions, which we refer to as “Arg143/Trp145 switch” hereafter. Loop 6 wraps similarly over the active site in the presence of both reaction compounds, whereas it is turned away from the active site in the apo structure. Collectively, these loop movements change the PriA active site from an open conformation (apo), to a half-closed arrangement in the presence of rCdRP, and to an entirely closed active site cavity in the presence of PrFAR (Fig. S4A and Movies S1 and S2). The structural data of the PriA-PrFAR complex suggest that ProFAR isomerization by PriA is entirely sequestered from the external solvent.
The structural details of the two bound reaction compounds PrFAR and rCdRP allow the categorization of residues involved in ProFAR (his biosynthesis) and PRA (trp biosynthesis) isomerization: (i) catalysis of the isomerization of the aminoaldose ring, (ii) binding of the functional groups that differ in the two substrates (anthranilate in the case of PRA, formimino-5-aminoimidazole-4-carboxamide ribonucleotide in the case of ProFAR), and (iii) anchoring of the terminal phosphate groups (Fig. 2 and Fig. S2). The structures of both PriA-ligand complexes (PrFAR, rCdRP) reveal the same pair of residues, Asp11 and Asp175, as candidates to catalyze the isomerization of the respective his and trp biosynthesis substrates, ProFAR and PRA. Asp175 interacts with the 4′-hydroxyl group of the ribulosyl moiety in both complexes, whereas Asp11 interacts with the 2′-keto group (PrFAR) or 2′-hydroxyl group (rCdRP) of the ribulosyl moiety. The position and orientation of Asp11, which is situated at the C terminus of strand β1, is nearly identical in all three PriA structures. In contrast, Asp175, which is located on the flexible loop 6, is tilted away from the PriA active site in the apo conformation. By contrast, in both PriA-ligand (rCdRP, PrFAR) structures the side chain of the residue is oriented toward the center of the PriA active site, associated with a movement of more than 10 Å with respect to the apo conformation (Fig. 1A, Fig. S3, and Movies S1 and S2). However, whereas Asp175 is bound to Arg19 from loop 1 in the PriA-PrFAR complex (his biosynthesis), such an interaction is not possible in the rCdRP complex (trp biosynthesis) as loop 1 does not fold over the active site. Instead, the β-hairpin conformation of loop 5, only observed in the presence of rCdRP, allows recruitment of Asp175 via a charged interaction with Arg143 from loop 5, thus assigning this loop a specific function in active site recruitment for PRA isomerization (trp biosynthesis) (Fig. 1B).
In contrast to the central, chemically active ribulose segment, the residual moieties of the two reaction compounds, the anthranilate carboxylate group of rCdRP and the formimino group of PrFAR, are in different orientations and thus determine binding specificity by interacting with different PriA residues (Fig. 2 and Figs. S1C and S2). Because of the larger size of PrFAR, the found specific ligand interactions with PriA residues exceed those of rCdRP. In addition, some of the interactions with PrFAR require major active site loop movements, using the PriA apo conformation as reference. Notably, in the structure of the PriA-rCdRP complex, Asp130 is shielded away from the anthranilate carboxylate group of the ligand by Arg143, which inserts its guanidinium group like a finger in between Asp175, Thr170, Asp130, and the rCdRP molecule (Fig. 1B). The interaction is enabled because of the Arg143/Trp145 switch, exchanging Trp145 and Arg143 in the PrFAR (his biosynthesis) and rCdRP (trp biosynthesis) complexes with contributions to the ligand-specific active site pockets. In the single-substrate HisA enzyme, the residue equivalent to Asp130 in PriA was originally considered to be the second catalytic residue for ProFAR isomerization (7, 9, 15, 16), whereas the contribution of the equivalent of Asp175 was not tested.
Requirements for Bisubstrate Catalysis in PriA.
To further investigate our findings, we measured the catalytic activities for both isomerization reactions of the wild-type enzyme by steady-state kinetics (Table 1). The catalytic efficiencies (kcat/KM) are 1.2 × 104 s-1 M-1 for ProFAR isomerization and 1.7 × 105 s-1 M-1 for PRA isomerization. The approximately 14-fold difference in catalytic efficiency almost entirely originates from differences in substrate turnover (kcat(ProFAR isomerization) = 0.23 s-1, kcat(PRA isomerization) = 3.6 s-1), whereas the Michaelis constants are almost identical for both reactions (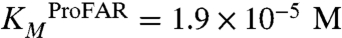 ,
, 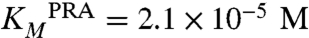 ). Similar trends for the ratio of ProFAR/PRA catalysis were also observed for the equivalent single-substrate enzymes HisA and TrpF from Thermotoga maritima (7).
). Similar trends for the ratio of ProFAR/PRA catalysis were also observed for the equivalent single-substrate enzymes HisA and TrpF from Thermotoga maritima (7).
Table 1.
Comparison of structural and functional properties of the bisubstrate enzyme PriA and single-substrate enzymes TrpF and HisA
| mtbPriA | tmHisA* | tmTrpF* | |
| ProFAR isomerization (his biosynthesis) | |||
| kcat [s-1] | 2.3 × 10-1 | 6.7 × 10-1 | |
| KM [M] | 1.9 × 10-5 | 6.0 × 10-7 | |
| kcat/KM [M-1 s-1] | 1.2 × 104 | 1.1 × 106 | |
| Catalytic residues | D11/D175 | D8/D169 | |
| Active site recruiter |
R19 |
none |
|
| PRA isomerization (trp biosynthesis) | |||
| kcat [s-1] | 3.6 | 3.7 | |
| KM [M] | 2.1 × 10-5 | 2.8 × 10-7 | |
| kcat/KM [M-1 s-1] | 1.7 × 105 | 1.3 × 107 | |
| Catalytic residues | D11/D175 | C7/D126 | |
| Active site recruiter | R143 | none | |
*Kinetic data taken from Henn-Sax et al. (7).
In a series of subsequent experiments, we removed the side chain-specific functions of several active site residues via site-directed mutagenesis, and we biochemically characterized their activities toward the two PriA substrates, ProFAR and PRA (Fig. 3 and Table S2). Two PriA variants, D11A and D175A, did not show detectable activity for either of the two catalyzed reactions, thus supporting our structural data that suggested that the two residues act as acid/base pair catalysts during isomerization of both substrates ProFAR and PRA. We were particularly interested in the functional roles of three key residues (Arg19, Arg143, and Trp145) that are located on flexible active site loops and are thus expected to play important roles in the substrate-specific formation of the PriA active site (Fig. 1B). ProFAR isomerization is largely diminished or even abolished when any of the three residues is mutated, whereas PRA isomerization is substantially affected in the R143A mutant only. The data thus support a role for Arg19 (loop 1) as a substrate-specific recruiter of Asp175 into the PriA active site for ProFAR isomerization (his biosynthesis), as suggested by the structure of the PriA-PrFAR complex. The structure of the PriA-rCdRP complex, by contrast, suggests a role for Arg143 (loop 5) as an Asp175 recruiter for PRA isomerization (Figs. 1B and 2A and Figs. S2B and S4B). The structure of the PriA-PrFAR complex does not support such an assignment for Arg143 in the catalysis of ProFAR isomerization, because the residue does not participate in the PriA active site conformation, adapted for ProFAR isomerization. However, in this conformation, Arg143 forms a salt bridge with Glu109, which is apparently critical to form the observed knot-like conformation of loop 5, providing the basis of the observed Arg143/Trp145 switch (Fig. 1B).
Fig. 3.

Biochemical analysis of PriA ProFAR/PRA isomerization. Relative catalytic efficiencies of several PriA variants (wild-type PriA = 100%), in which active site residues were mutated into alanines: ProFAR isomerization, black bars; PRA isomerization, gray bars (A). The complete data are listed in Table S2. The effects of the PriA mutations on ProFAR isomerization (B) and PRA isomerization (C) are graphically mapped onto the respective PriA-ligand complexes. The color codes of the mutated residues, shown in ball-and-stick presentation, are red, < 1% wt activity; orange, < 5% wt activity; green, < 50% wt activity; blue, > 50% wt activity. The orientation of PriA is as in Fig. 1.
Remarkably, the PriA variant, in which the other loop 5 switch residue was mutated (W145A), shows little effect on PRA isomerization, but completely abolishes the ability of the enzyme to isomerize ProFAR (Fig. 3 and Table S2). The data thus indicate that the knot-like conformation of loop 5, observed only in the PriA-PrFAR complex (Fig. 1B), is essential to properly position Trp145 into the PriA active site conformation adapted for ProFAR isomerization. In contrast, in the PriA-rCdRP complex, the same residue does not participate in the active site, due to the unrelated β-turn conformation of loop 5, thus explaining the lack of any significant effects of this PriA variant on PRA isomerization. Similarly, all remaining PriA variants (T105A, D130A, and T170A) largely diminish or even abolish ProFAR isomerization, while showing only minor effects on PRA isomerization (Fig. 3). All these effects are supported by our structural data and demonstrate the complexity of structural active site requirements for ProFAR isomerization, exceeding those for PRA isomerization.
Relationship of the Bisubstrate PriA Isomerase to Corresponding Single-Substrate Enzymes.
In the next step of our analysis, we used available structural data of several (β/α)8-barrel enzymes from the his and trp biosynthesis pathways (17) to elucidate possible relationships between the bisubstrate enzyme PriA and the equivalent single-substrate enzymes HisA and TrpF (Figs. S5 and S6). The structure-based sequence identity of PriA and HisA is 24% and reveals identical or conserved residues along the entire sequences. At the level of active site topography, the overall similarity of HisA/PriA is also reflected by structural matches of most of the conserved active site residues in both enzymes (Fig. S5A). The observations point to a close evolutionary relationship between PriA and HisA. However, in contrast to available apo structures of PriA from M. tuberculosis (this contribution) and S. coelicolor (12, 13), in which Asp175 is either remote from the active site or invisible, requiring recruitment into the active site upon substrate binding, in the available apo structure of HisA from T. maritima (9), the equivalent residue (Asp169) is oriented into the active site (Fig. S6A).
In contrast, superposition of the PriA structure onto TrpF does not reveal significant structural similarity of the active site, except a common phosphate-binding site at the C terminus of strand β7 (10) (Fig. S5B). In addition, none of the two catalytic residues in TrpF (Cys7, Asp126) structurally matches the two PriA catalytic residues (Asp11, Asp175). Moreover, in contrast to Asp175 in PriA, which needs to be recruited into the active site of the bisubstrate enzyme, both catalytic residues in TrpF are in rigid β-strand positions (Fig. S6B), thus discarding a hypothetical activation mechanism in TrpF by conformational changes of the active site. Moreover, almost no conformational changes could be detected in TrpF upon binding of the PRA isomerization product analogue rCdRP (7, 8).
To experimentally validate these findings, we established an approach to detect possible differences between the bisubstrate enzyme PriA and the corresponding single-substrate enzymes from T. maritima with two separate genes (7). We identified and characterized six compounds that inhibited both PriA-catalyzed reactions, ProFAR and PRA isomerization, by screening a library of 20,000 small molecule compounds. The six compounds were subsequently subjected to complete Michaelis–Menten inhibition analysis (Table 2). All selected compounds were competitive inhibitors for PRA isomerization (Fig. S7), demonstrating binding into the PriA active site. Several of them showed Ki values below 10 μM for PRA isomerization. When we then tested these compounds against the single-substrate T. maritima enzymes, all of them also inhibited ProFAR isomerization by HisA, although with diminished affinities, confirming our findings of a closely related ProFAR isomerization mechanism in the two enzymes. In contrast, none of these compounds showed any inhibition of the TrpF-catalyzed PRA isomerization, indicating a level of active site structural differences in TrpF and PriA that hinders cross-inhibition of the same reaction in the two enzymes.
Table 2.
Effects of selected inhibitors on PriA (ProFAR, PRA), HisA (ProFAR), and TrpF (PRA) activity
| ID | MW |
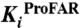 [M] [M] |
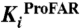 [M] [M] |
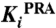 [M] [M] |
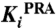 [M] [M] |
| PriA | HisA | PriA | TrpF | ||
| #16827 | 296 | 1.3 ± 0.2 × 10-5 | 2.2 ± 0.4 × 10-4 | 3.6 ± 0.5 × 10-6 | none |
| #17146 | 275 | 2.9 ± 0.3 × 10-5 | 3.0 ± 0.6 × 10-4 | 8.8 ± 2.2 × 10-6 | none |
| #19746 | 297 | 7.2 ± 1.7 × 10-5 | 1.4 ± 0.2 × 10-4 | 2.4 ± 0.8 × 10-4 | none |
| #20022 | 176 | 1.8 ± 0.1 × 10-5 | 7.0 ± 1.4 × 10-4 | 3.7 ± 0.7 × 10-5 | none |
| #20138 | 539 | 1.3 ± 0.2 × 10-4 | 1.7 ± 0.5 × 10-4 | 4.1 ± 1.1 × 10-5 | none |
| #20169 | 676 | 5.7 ± 1.0 × 10-5 | 3.2 ± 0.1 × 10-4 | 1.7 ± 0.5 × 10-5 | none |
Discussion
Our structural and biochemical data reveal the molecular mechanism of PriA catalysis of both ProFAR and PRA isomerization with identical catalytic residues (Asp11, Asp175) and with a substrate-specific recruitment mechanism for Asp175. Our data, however, do not support previous hypotheses on either the potential use of the twofold repeated half-barrel symmetry or general loop flexibility considerations, which present features largely shared by the bisubstrate PriA enzyme and the single-substrate HisA enzyme (12, 14). These hypotheses were raised based on available apo PriA structures only, without experiment-based knowledge of the type of conformational changes associated with substrate binding.
We next wondered what kind of structural constraints restrict HisA and TrpF acting as single-substrate isomerases, in contrast to PriA. Whereas for TrpF the answer is obvious—the respective active site is too small to bind the HisA substrate ProFAR—the reasons for single-substrate catalysis by HisA are less conceivable. However, some rationalization is possible by a comparative analysis of a conserved HisA key active site residue for specific ProFAR isomerization: Asp127 (7). The equivalent position in TrpF is replaced by other noncharged amino acids in all known trpF sequences, suggesting that removal of the aspartate side chain may be essential for PRA isomerization activity. Therefore, it has remained an open question how PriA could tolerate an aspartate in the same position (Asp130), despite PRA isomerization activity. The structure of the PriA-rCdRP complex reveals how the rCdRP molecule is shielded from potential repulsive interactions with Asp130 by the insertion of the positively charged side chain of Arg143 (Fig. 1B). The structural observations thus fit our biochemical data indicating that Arg143 is the only residue, in addition to the catalytic residue pair Asp11/Asp175, that is highly sensitive when mutated to both PRA and ProFAR isomerization activity (Fig. 3). The structure-based sequence alignment of HisA/PriA shows that whereas in known PriA sequences this residue is highly conserved as an arginine, it is variable in available HisA sequences (Fig. S5A), suggesting that it could be a key contributor to the evolved bisubstrate specificity in PriA.
A comparison of the measured catalytic efficiencies both for ProFAR and PRA isomerization in PriA and data from single-substrate HisA and TrpF enzymes, however, reveals a negative trade-off for bisubstrate specificity in PriA (Table 1). Most of the reduction in activity is because of a substantial decrease in substrate affinity. Similar trends were previously observed for the equivalent enzyme from S. coelicolor (12, 13). A comparison of the PriA-rCdRP and TrpF-rCdRP (7) product analogue structure complexes may provide further hints for different substrate binding affinities in PRA isomerization that we observed in PriA and TrpF. In the two structures, although the conserved phosphate group-binding site for rCdRP is identical, the orientation of the anthranilate group is opposite, leading to unrelated specific interactions (Fig. S6C). Whereas in PriA the anthranilate carboxylate group is deeply buried and hydrogen-bound by two conserved active site residues (His50, Ser81) (Fig. 2A and Fig. S2B), in the equivalent TrpF complex the same group is only loosely bound to residues from loop 1.
Does this negative trade-off in PriA bisubstrate specificity reflect evolutionary ancestry, and possibly even primitive enzyme features, or evolutionary advantages such as adaptation to specific living conditions? Established evolutionary hypotheses, which suggest that broad specificity could be precursors of related more specific enzymes (18), may point to PriA as a potential ancestral enzyme. This idea may be further supported by ancestral properties of the actinobacteria phylum, however, with largely undetermined relations to other bacteria phyla (19). Therefore, ultimate proof of these observations will require further studies of bacterial species without a trpF operon gene under native living conditions. Nevertheless, irrespective of the precise evolutionary relationships among PriA, TrpF, and HisA enzymes, our data allow a mechanistic comparison with recent laboratory-based evolution experiments to convert the scaffold of the single-substrate substrate enzyme HisA from T. maritima into an enzyme with TrpF-like properties catalyzing the isomerization of PRA—albeit at the expense of its native ProFAR isomerization activity (15, 20).
A comparison of the structure of PriA and that of the directed evolution version of HisA (20) shows that two of the four mutated HisA residues (D127V, D169V) have equivalents (Asp130, Asp175) in the PriA active site with key roles for PriA catalysis, as evidenced by our structural and biochemical data. As mutation of Asp175 in PriA abolishes activity for both ProFAR and PRA isomerization while being required for PRA isomerization activity in HisA, the suggested role of the residue as an acid/base catalyst inevitably needs to be carried out by a different active site residue in the engineered HisA variant with TrpF-like activity, assuming a similar biochemical isomerization mechanism. The additional requirement for replacing Asp127 in the same HisA variant to generate PRA isomerization activity fits all known TrpF sequences without an aspartate in the equivalent position and may be necessary because the local active site environment in HisA is not sufficient to shield Asp127 from unwanted ligand interactions in PRA catalysis. This is in contrast to the bisubstrate enzyme PriA, in which the presence of an aspartate in the same position is tolerated. Our structural data on the PriA-PrFAR complex, by indicating direct involvement of Asp130 in catalysis, also explain why this replacement in the HisA variant with TrpF-like activity loses the original ProFAR isomerization activity (20).
We conclude that the solutions for generating PRA isomerization activity either on the HisA scaffold by laboratory-based evolution or on PriA, presenting a HisA-like scaffold, are different. Whereas the directed evolution experiment has led to an active site conformation with different functionality, with parallel loss of the original activity, by replacing key active site residues, we have found a flexible active site architecture in PriA that involves identical residues for both ProFAR and PRA isomerization. Comparison of our structural data suggests that an extraordinary level of active site loop flexibility coupled with an active site recruitment mechanism for Asp175 are key parameters for its bisubstrate specificity properties and make it a paradigm for substrate-induced active site metamorphosis of the same enzyme scaffold. To the best of our knowledge, substrate-induced recruitment of active site residues to generate distinct bisubstrate specificity is without precedence. The failure to capture a bisubstrate profile by directed evolution experiments on the corresponding single-substrate enzymes to date demonstrates the underlying complexity of bisubstrate specificity.
Finally, comparison of our structural and functional data on PriA from M. tuberculosis and those from the two single-substrate enzymes HisA and TrpF indicate substantial differences in the active site architecture for PRA isomerization by PriA and TrpF, whereas the structural requirements for ProFAR isomerization in PriA and HisA are highly conserved. These findings are supported by a lack of any measurable effect of six selected PriA inhibitors for PRA isomerization in TrpF (Table 2). Although the aim of this study was not drug discovery, our findings demonstrate the potential of unraveling the structural and functional details of molecular mechanisms in enzymes with suspected altered specificity profiles, in this particular case genetically indicated by the loss of the trpF gene in M. tuberculosis and related organisms from the actinobacteria phylum. As shown by our PriA inhibitor study, the identification of specific enzyme activity mechanisms in potential protein targets that may be different from other organisms, such as the pathogen host, may provide unprecedented opportunities for the development of organism-specific inhibitors and ultimately drugs.
Materials and Methods
PriA from M. tuberculosis, HisA from T. maritima, and TrpF from T. maritima were expressed in Escherichia coli and purified by immobilized metal affinity chromatography. For crystallization of the PriA-ligand complexes, drops containing 300 μM (8 mg/mL) PriA were mixed with either 5 mM ProFAR or 5 mM rCdRP and mixed 1∶1 with the reservoir solution. PriA crystals in the presence of ProFAR were obtained from 1.4 M tri-sodium citrate dihydrate in 0.1 M Hepes (pH 7.5) buffer. PriA crystals in the presence of rCdRP were grown from 32% (wt/vol) PEG 6000 in 0.1 M sodium acetate buffer (pH 5.7). The resulting X-ray structures were solved and refined to a resolution of 2.5 Å (apo conformation), 2.4 Å (PriA-rCdRP), and 1.33 Å (PriA-PrFAR). All PriA variants were biochemically investigated by steady-state kinetics by either monitoring changes of the absorbance (ProFAR isomerization) (21) or fluorescence (PRA isomerization) (22). A search for potential PriA inhibitors was performed using a library of 20,000 small molecule compounds (FMP). The library was designed to represent structural diversity and predicted high solubility in water. Primary screening was performed with compounds at a concentration of 50 μM each, using the ProFAR isomerization assay, based on quantification of absorption decrease at 300 nm.
Supplementary Material
Acknowledgments.
We thank Reinhard Sterner, University of Regensburg, Regensburg, Germany, for technical assistance, for the provision of plasmids and genetically manipulated E. coli strains, and for comments on the manuscript. Felix List provided us with Movies S1 and S2. The work has been supported by Grants DFG-1058/6-1, DFG-1058/6-2, DFG-1058/6-3, and the grant “SystemTB” by the European Commission (HEALTH-F4-2010-241587) (to M.W.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 2Y89 (PriA, apo conformation), 2Y85 (PriA-rCdRP complex), and 2Y88 (PriA-PrFAR complex)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1015996108/-/DCSupplemental.
References
- 1.Gerlt JA, Raushel FM. Evolution of function in (β/α)8-barrel enzymes. Curr Opin Chem Biol. 2003;7:252–264. doi: 10.1016/s1367-5931(03)00019-x. [DOI] [PubMed] [Google Scholar]
- 2.Glasner ME, Gerlt JA, Babbitt PC. Mechanisms of protein evolution and their application to protein engineering. Adv Enzymol Relat Areas Mol Biol. 2007;75:193–239. doi: 10.1002/9780471224464.ch3. [DOI] [PubMed] [Google Scholar]
- 3.Khersonsky O, Roodveldt C, Tawfik DS. Enzyme promiscuity: Evolutionary and mechanistic aspects. Curr Opin Chem Biol. 2006;10:498–508. doi: 10.1016/j.cbpa.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 4.Tokuriki N, Tawfik DS. Protein dynamism and evolvability. Science. 2009;324:203–207. doi: 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- 5.Murzin AG. Biochemistry. Metamorphic proteins. Science. 2008;320:1725–1726. doi: 10.1126/science.1158868. [DOI] [PubMed] [Google Scholar]
- 6.Xie G, Keyhani NO, Bonner CA, Jensen RA. Ancient origin of the tryptophan operon and the dynamics of evolutionary change. Microbiol Mol Biol Rev. 2003;67:303–342. doi: 10.1128/MMBR.67.3.303-342.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henn-Sax M, et al. Two (β/α)8-barrel enzymes of histidine and tryptophan biosynthesis have similar reaction mechanisms and common strategies for protecting their labile substrates. Biochemistry. 2002;41:12032–12042. doi: 10.1021/bi026092h. [DOI] [PubMed] [Google Scholar]
- 8.Hennig M, Sterner R, Kirschner K, Jansonius JN. Crystal structure at 2.0 A resolution of phosphoribosyl anthranilate isomerase from the hyperthermophile Thermotoga maritima: Possible determinants of protein stability. Biochemistry. 1997;36:6009–6016. doi: 10.1021/bi962718q. [DOI] [PubMed] [Google Scholar]
- 9.Lang D, Thoma R, Henn-Sax M, Sterner R, Wilmanns M. Structural evidence for evolution of the β/α barrel scaffold by gene duplication and fusion. Science. 2000;289:1546–1550. doi: 10.1126/science.289.5484.1546. [DOI] [PubMed] [Google Scholar]
- 10.Wilmanns M, Hyde CC, Davies DR, Kirschner K, Jansonius JN. Structural conservation in parallel β/α-barrel enzymes that catalyze three sequential reactions in the pathway of tryptophan biosynthesis. Biochemistry. 1991;30:9161–9169. doi: 10.1021/bi00102a006. [DOI] [PubMed] [Google Scholar]
- 11.Barona-Gomez F, Hodgson DA. Occurrence of a putative ancient-like isomerase involved in histidine and tryptophan biosynthesis. EMBO Rep. 2003;4:296–300. doi: 10.1038/sj.embor.embor771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuper J, Doenges C, Wilmanns M. Two-fold repeated (β/α)4 half-barrels may provide a molecular tool for dual substrate specificity. EMBO Rep. 2005;6:134–139. doi: 10.1038/sj.embor.7400330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright H, et al. The structure/function relationship of a dual-substrate (β/α)8-isomerase. Biochem Biophys Res Commun. 2008;365:16–21. doi: 10.1016/j.bbrc.2007.10.101. [DOI] [PubMed] [Google Scholar]
- 14.Noda-Garcia L, et al. Identification and analysis of residues contained on β → α loops of the dual-substrate (βα)8 phosphoribosyl isomerase A specific for its phosphoribosyl anthranilate isomerase activity. Protein Sci. 2010;19:535–543. doi: 10.1002/pro.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jurgens C, et al. Directed evolution of a (β/α)8-barrel enzyme to catalyze related reactions in two different metabolic pathways. Proc Natl Acad Sci USA. 2000;97:9925–9930. doi: 10.1073/pnas.160255397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leopoldseder S, Claren J, Jurgens C, Sterner R. Interconverting the catalytic activities of (β/α)8-barrel enzymes from different metabolic pathways: sequence requirements and molecular analysis. J Mol Biol. 2004;337:871–879. doi: 10.1016/j.jmb.2004.01.062. [DOI] [PubMed] [Google Scholar]
- 17.Vega MC, Lorentzen E, Linden A, Wilmanns M. Evolutionary markers in the (β/α)8-barrel fold. Curr Opin Chem Biol. 2003;7:694–701. doi: 10.1016/j.cbpa.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 18.Jensen RA. Enzyme recruitment in evolution of new function. Annu Rev Microbiol. 1976;30:409–425. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- 19.Ventura M, et al. Genomics of Actinobacteria: Tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev. 2007;71:495–548. doi: 10.1128/MMBR.00005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Claren J, Malisi C, Hocker B, Sterner R. Establishing wild-type levels of catalytic activity on natural and artificial (β/α)8-barrel protein scaffolds. Proc Natl Acad Sci USA. 2009;106:3704–3709. doi: 10.1073/pnas.0810342106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klem TJ, Davisson VJ. Imidazole glycerol phosphate synthase: the glutamine amidotransferase in histidine biosynthesis. Biochemistry. 1993;32:5177–5186. doi: 10.1021/bi00070a029. [DOI] [PubMed] [Google Scholar]
- 22.Hommel U, Eberhard M, Kirschner K. Phosphoribosyl anthranilate isomerase catalyzes a reversible amadori reaction. Biochemistry. 1995;34:5429–5439. doi: 10.1021/bi00016a014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.