

Abstract
As part of an approach to the synthesis of the antitubercular agent elisapterosin B, we prepared two different chiral, non-racemic olefinic substrates and examined their diastereoselective ring closure using mercury salts. The effort yielded potential precurors to elisapterosin B in good yield with good to excellent diastereocontrol.
Keywords: benzothiazine, elisapterosin B, Friedel-Crafts, mercuric acetate, tuberculosis
1. Introduction
Tuberculosis is a major worldwide health problem, with as many as one third of the planet’s population infected with the causative organism. Approximately 10% of those infected proceed to active disease and between one and two million people die annually as a result. Current chemotherapies against TB are often effective, but require months of treatment, during which time compliance with dosing regimens can be compromised. This and other factors have given rise to the emergence of multidrug resistant (MDR) and extremely (extensively) drug resistant (XDR) strains of tuberculosis.1 These require new, more aggressive therapies to effect cures. There is thus a need for new chemotherapeutic agents that are more potent and efficacious that those currently in clinical use, both to combat resistant strains of TB and to shorten the time necessary for treatment of all forms of TB, so that treatment can be effected in a conveniently timely fashion, mitigating problems of noncompliance with long-term chemotherapies.
There is therefore much current interest in exploring new leads for the treatment of TB. One approach involves the discovery of natural products that have antitubercular activity.2 For example, elisapterosin B (1), was isolated from the gorgonian coral Pseudopterogorgia elisabethae and found to have antitubercular activity, inhibiting the growth of M. tuberculosis H37Rv to the extent of 79% at 1.5 μg/mL.3 This is but one of a family of related terpenes that also showed various degrees of activity against tuberculosis. Another example is the amphilectane diterpene, pseudopteroxazole.4
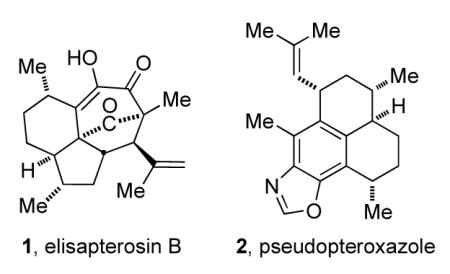
Our interest in this area was stimulated both by the serious nature of the medical problem and the fact that we had developed benzothiazine chemistry that we thought applicable to the synthetic challenges of compounds like 1 and 2. In fact, we reported that our completely stereoselective approach to benzothiazines5 was a powerful approach to the synthesis of pseudopteroxazole.6 Further, we used the chemistry to synthesize related molecules like curcumene, curcuphenol and erogorgiaene in enantiomerically pure form;7,8 and used it in an approach to seco-pseudopteroxazole.9
Our approach to elisapterosin B is presently centered on a formal total synthesis that intersects the Rychnovsky route10,11 to 1 at 3, which can be converted to 1 via a Lewis acid-mediated (5+2)-cycloaddition (Scheme 1). It is anticipated that 3 can be obtained from 4 and that the stereoselective formation of 4 can be accomplished by an intramolecular Friedel-Crafts alkylation (IFCA) of 5. Finally, 5 would be available using our benzothiazine synthesis, leading retrosynthetically to 6 and 7 (Scheme 1).
Scheme 1.
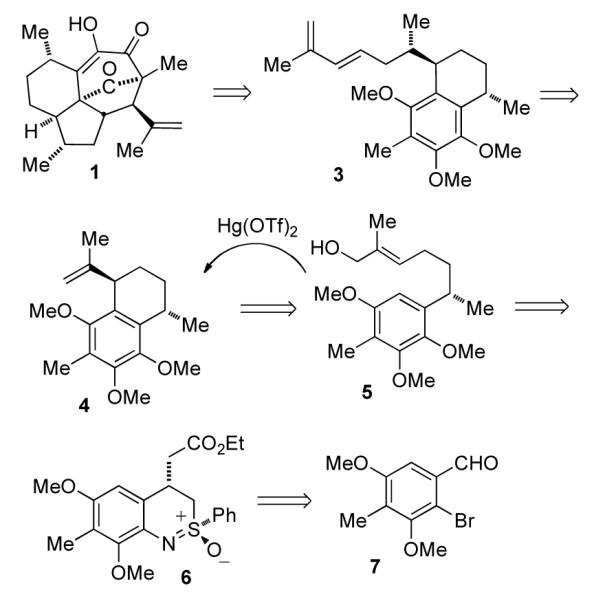
Retrosynthesis for elisapterosin B.
The intramolecular cyclization, the subject of this paper, was anticipated to be successful based on a report by the Nishizawa group, who reported a mercury-catalyzed arylene cyclization (Eq. 1).12 Allylic alcohol 8 was cyclized to 9 in good yield. However, the critical factor for us was the diastereocontrol available in such a process.
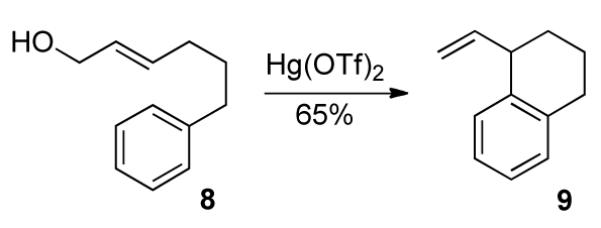 |
(1) |
2. Preparation of benzothiazine 6
The synthesis of benzothiazine began with the known aldehyde 7.13 A Horner-Wadsworth-Emmons (HWE) reaction afforded the ortho-bromocinnamate 10 in quantitative yield. A Buchwald-Hartwig coupling reaction14 between 10 and enantiomerically pure (+)-sulfoximine (S)-11 gave an N-arylated sulfoximine intermediate, which was converted with complete diastereocontrol into benzothiazine 6 via intramolecular Michael addition8 by using LiHMDS as base.5
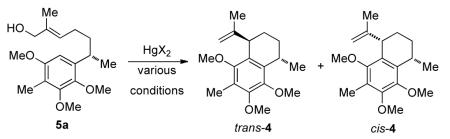
3. Preparation of 5a and 5b
In considering a study of the cyclization process and its diastereoselectivity, we chose to synthesize both 5a and its desmethyl analogue 5b. Thus, benzothiazine 6 was reduced with LAH and desulfurized with Na/Hg to give aniline 12 in excellent yield. Oxidation of 12 with CAN afforded quinone 13 in 76% yield,15 which was reduced and methylated to give alcohol 14 in one pot. Compound 14 was oxidized to the aldehyde, which was homologated to 15. A HWE reaction using Roush conditions16 and reduction with DIBAL afforded 5a and 5b in good yield.
4. The mercury-catalyzed cyclization of 5a
In an effort to optimize the preparation of 4, particularly in the context of stereocontrol, conditions were sought for the mercury-catalyzed cyclization of 5a that would be optimal. The results of the study are shown in Table 1. Using catalytic amounts of Hg(OTf)2, the reaction was performed at different temperatures in toluene. Beginning the process at a relatively high temperature was better than beginning at ambient temperature followed by warming (entries 1-4). However, when the initial temperature as increased to 110 °C, both the conversion and diastereoselectivity decreased (entry 5). Under these reaction conditions, the reaction was not clean. Side reactions such as double bond migration may have occurred due to presence of triflic acid produced in the reaction. In order to suppress these side reactions, 2,6-tert-butylpyridine was added; however, no reaction was observed even at 110 °C (entry 6). Further, solvent effects were studied (entries 7-10). The reaction did not proceed in acetonitrile. Interestingly, when dichloromethane was the solvent, the diastereoselectivity was high (entry 8); and with 1,2-dichloroethane was the solvent, the diastereoselectivity was even higher, up to 15:1 (entry 9). However, both reactions gave low yields due to unwanted side reactions. When Hg(SO3C4F9)2 was the catalyst, the reaction was very poor (entry 10).
Table 1.
Mercury-catalyzed IFCA reaction of 5a
| Entry | HgX2, (equiv) | Conditions | Yield %,a (trans:cis)b |
|---|---|---|---|
| 1 | Hg(OTf)2, (0.005) | PhMe, rt, 1.5 h; 48 °C, 25 h | 40%,d 12.5:1 |
| 2 | Hg(OTf)2, (0.005) | PhMe, rt, 20 min; 60 °C, 25 h | 54%,d 8:1, |
| 3 | Hg(OTf)2, (0.005) | PhMe, 45 °C, 4 h | 54%, 9:1 |
| 4 | Hg(OTf)2, (0.005) | PhMe, 55 °C, 4 h | 50%, 9:1 |
| 5 | Hg(OTf)2, (0.005) | PhMe, 110 °C, 10 min | 34%, <9:1 |
| 6 | Hg(OTf)2,(0.01)c | PhMe, 60 °C, 30 min; 110 °C, 12 hc | 0 |
| 7 | Hg(OTf)2, (0.005) | MeCN, rt, 20 min; 70-80 °C, 19 h | 0 |
| 8 | Hg(OTf)2, (0.005) | CH2Cl2, rt, 35 min; 90 °C, 5 h | 13%,d 12.5:1 |
| 9 | Hg(OTf)2, (0.005) | ClCH2CH2Cl, rt, 35 min; 90 °C, 5 h | 27%,d 15:1 |
| 10 | Hg(SO3C4F9)2, (0.005) | PhMe, rt, 24 h; 80 °C, 15 min | e |
Yields are for isolated products unless otherwise stated.
The diastereomeric ratio was determined by analysis of the 1H NMR spectrum of the crude reaction mixture.
2,6-di-tert-butylpyridine was added.
Crude yield.
A complex mixture was formed.
The structural assignment of the products was carried out using proton and carbon NMR. The stereochemical assignment was based on the fact that the alkene methylene protons of the major trans isomer were upfield from those of the minor isomer, which was assigned as cis.17 A similar phenomenon was observed for the isomers of cyclization products derived from 5b, and the stereochemistry of the major product was established by X-ray analysis in that case. Furthermore, a report by Schmalz on the cyclization of a compound related to 5a (and which did not work with 5a) indicated that same basic pattern in chemical shifts, the methylene protons of the trans cyclization products resonated upfield from those of the corresponding cis isomer.18
5. The mercury-catalyzed cyclization of 5b
The mercury-catalyzed IFCA reaction of the allylic alcohol 5b was also investigated. The results of this reaction are tabulated in Table 2. Using benzene as solvent and Hg(SO3C4F9)2 as catalyst, at room temperature, the cyclized product 16 was obtained in 50% isolated yield with good selectivity (dr = trans:cis > 10:1, entries 1 and 2, Table 2), but the reaction was slow. At higher temperatures, the yield improved but the selectivity dropped somewhat and remained relatively constant over a temperature range of 70-110 °C. This suggests kinetic control in the cyclization process, as one would expect an increase in the amount of trans-16 at higher temperatures if the reaction were reversible.19 Overall, the best reaction conditions appeared to be heating at 70 °C in benzene or toluene for 15 min (Table 2, entries 3, 6).
Table 2.
IFCA reaction of allylic alcohol 5b
| Entry | HgX2, (equiv) | Conditions | Yield % (dr = trans:cis) |
|---|---|---|---|
| 1 | 0.02 equiv Hg(SO3C4F9)2 | benzene, rt, 2d | trace |
| 2 | 0.02 equiv Hg(SO3C4F9)2 | benzene, rt, 4d | 50%, dr > 10:1 |
| 3 | 0.02 equiv Hg(OTf)2 | benzene, rt, 1h, 70 °C, 30 min | 73%, dr = 6.3:1 |
| 4 | 0.02 equiv Hg(OTf)2 | benzene, 70 °C, 15 min | 73%, dr = 6.3:1 |
| 5 | 0.005 equiv Hg(OTf)2 | benzene, rt, 35 min, 80 °C, 1.5 h | 73%, dr = 5.9:1 |
| 6 | 0.02 equiv Hg(OTf)2 | PhMe, 70 °C, 15 min | 72%, dr = 6.3:1 |
| 7 | 0.005 equiv Hg(OTf)2 | PhMe, 110 °C, 5 min | 73%, dr = 5.6:1 |
| 8 | 0.005 equiv Hg(OTf)2 | PhMe, 110 °C, 20 min | 58%, dr = 5.6:1 |
Yields are for isolated products.
The diastereomeric ratio was determined by analysis of the 1H NMR spectrum of the crude reaction mixture
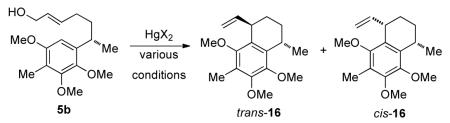
In order to verify the structure of 16, an attempt was made to produce a crystalline derivative. Hydroboration of alkene 16 with BH3·THF, followed by oxidation gave 17 in 71% isolated yield. Reaction of 17 with TsCl furnished 18 in 84% isolated yield. Treatment of 18 with thiophenol and potassium carbonate in DMSO, followed by oxidation of intermediate sulfide with m-CPBA, furnished the sulfone 19 in 63% yield over two steps (Scheme 4). The major isomer of 19 was separable by recrystallization from MeOH/CH2Cl2. The X-ray crystal structure20 confirmed the stereochemistry of 16. It should be noted that the terminal vinyl proton trans to the internal vinyl proton resonated at 4.62 ppm for trans-16 but appeared at 4.89 ppm in cis-16. This helped in assigning the stereochemistry of the isomers of 4, as mentioned above.
Scheme 4.
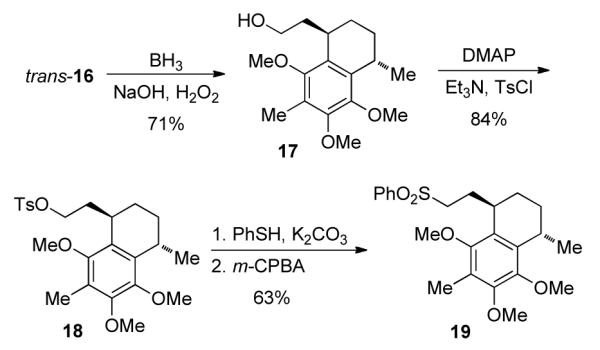
Synthesis of a crystalline derivative of trans-16.
The basis for the trans selectivity in these cyclization presumbably arises from a chairlike six-membered transition state in which the substituents are disposed in a pseudoequatorial orientation. How substitution patterns on the aromatic ring and the putative allylic cation intermediate affect this selectivity must be the subject of future studies.
6. Conclusion
In summary, using benzothiazine chemistry and Hg(OTf)2-catalyzed diastereoselective IFCA, we have synthesized the potential precursors (4 and 16) to elisapterosin B. We are now targeting the synthesis of diene 3 to finish this total synthesis. The results of these studies will be reported in due course.
Supplementary Material
Scheme 2.
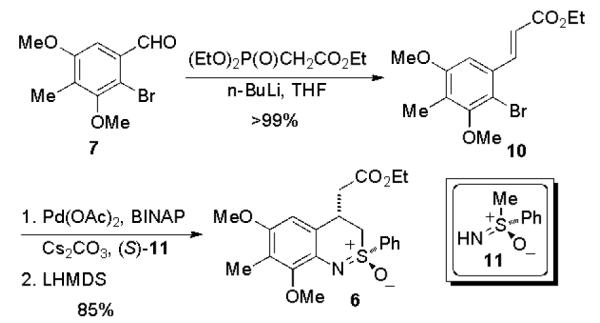
Synthesis of benzothiazine 6.
Scheme 3.

Synthesis of alcohols 5a and 5b.
Acknowledgments
This work was supported by the NIH (1R01-AI59000-01A1) to whom we are grateful.
Footnotes
Supplementary data Supplementary data (experimental procedures, charaterization data; 1H and 13C spectra for new compounds) associated with this letter can be found in the online versions at doi:XXXXXXXX.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Haydel SE. Pharmaceuticals. 2010;3:2268. doi: 10.3390/ph3072268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gutierrez-Lugo M-T, Bewley CA. J. Med. Chem. 2008;51:2606. doi: 10.1021/jm070719i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodriguez AD, Ramirez C, Rodriguez II, Barnes CL. J. Org. Chem. 2000;65:1390–1398. doi: 10.1021/jo9914869. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez AD, Ramirez C, Rodriguez II, Gonzalez E. Org. Lett. 1999;1:527–530. doi: 10.1021/ol9907116. [DOI] [PubMed] [Google Scholar]
- 5.Harmata M, Hong X. J. Am. Chem. Soc. 2003;125:5754. doi: 10.1021/ja034744z. [DOI] [PubMed] [Google Scholar]
- 6(a).Harmata M, Hong X. Org. Lett. 2005;7:3581–3583. doi: 10.1021/ol0515412. [DOI] [PubMed] [Google Scholar]; (b) Harmata M, Cai Z, Chen Y. J. Org. Chem. 2009;74:5559. doi: 10.1021/jo9009112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harmata M, Hong X, Barnes CL. Tetrahedron Lett. 2003;44:7261. [Google Scholar]
- 8.Harmata M, Hong X, Schreiner PR. J. Org. Chem. 2008;73:1290. doi: 10.1021/jo701935s. [DOI] [PubMed] [Google Scholar]
- 9.Harmata M, Zheng P. Heterocycles. 2009;77:279. [Google Scholar]
- 10.Kim AI, Rychnovsky SD. Angew. Chem., Int. Ed. 2003;42:1267. doi: 10.1002/anie.200390325. [DOI] [PubMed] [Google Scholar]
- 11(a).For other syntheses of elisapterosin B, see: Waizumi N, Stankovic AR, Rawal VH. J. Am. Chem. Soc. 2003;125:13022. doi: 10.1021/ja035898h. Jarvo ER, Lawrence BM, Jacobsen EN. Angew. Chem. Int. Ed. 2005;44:6043. doi: 10.1002/anie.200502176. Boezio AA, Jarvo ER, Lawrence BM, Jacobsen EN. Angew. Chem. Int. Ed. 2005;44:6046. doi: 10.1002/anie.200502178. Davies HML, Dai Xing, Long MS. J. Am. Chem. Soc. 2006;128:2485. doi: 10.1021/ja056877l.
- 12.Namba K, Yamamoto H, Sasaki I, Mori K, Imagawa H, Nishizawa M. Org. Lett. 2008;10:1767. doi: 10.1021/ol800450x. [DOI] [PubMed] [Google Scholar]
- 13.Harmata M, Ying W, Barnes CL. Tetrahedron Lett. 2009;50:2326. doi: 10.1016/j.tetlet.2010.10.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harmata M, Pavri N. Angew. Chem., Int. Ed. 1999;38:2419. doi: 10.1002/(sici)1521-3773(19990816)38:16<2419::aid-anie2419>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 15.Further details of this oxidation will be reported elsewhere.
- 16.Blanchette MA, Choy W, Davis JT, Essenfeld AP, Masamune S, Roush WR, Sakai T. Tetrahedron Lett. 1984;25:2183. [Google Scholar]
- 17.4.79 and 3.94 ppm for trans-4 and 4.81 and 4.43 ppm for cis-4.
- 18.Werle S, Fey T, Neudörfl J. r. M., Schmalz H-G. Org. Lett. 2007;9:3555–3558. doi: 10.1021/ol071228v. [DOI] [PubMed] [Google Scholar]
- 19.Molecular mechanics calculations that the most stable conformation of trans-16 was 5.9 kcal/mol more stable that the most stable conformation of cis-16.
- 20.Crystallographic data (excluding structure factors) for 19 has been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 734229. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [Fax: +44(1223)336033; deposit@ccdc.ac.uk].
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.