

Abstract
MicroRNAs (miRNAs or miRs) are short, highly conserved noncoding RNAs that regulate gene expression at the post-transcriptional level by inhibiting translation or promoting the degradation of target mRNA. Even though the field of miRNA biology is relatively young, growing lines of evidence suggest that miRNAs play a key role pathogenesis of heart failure through their ability to regulate genes that govern the process of adaptive and maladaptive cardiac remodeling. Herein, we review the biology of miRNAs in relation to their role in modulating various aspects of the cardiac remodeling process, as well as discuss the potential applications of miRNA biology to the field of heart failure.
Keywords: cardiac remodeling, gene silencing, miRNAs, personalized medicine
The scientific quest for the basic mechanisms that are responsible for the development and progression of heart failure has been exhaustive; nonetheless, no single unifying hypothesis has been unraveled that explains the syndrome of heart failure. Recent studies have identified profound roles for a family of tiny regulatory non-coding RNAs, known as miRNAs in the control of diverse aspects of cardiac function [1-3]. Even though the field of miRNA biology is relatively young, growing lines of evidence suggest that miRNAs play an important role in the pathogenesis of heart failure through their ability to negatively regulate expression levels of genes that govern the process of adaptive and maladaptive cardiac remodeling. Specific miRNAs are differentially regulated in the failing heart, and gain- and loss-of-function experiments in mice have shown that miRNAs modulate various aspects of the heart failure phenotype [1-6]. This article focuses on the biology of miRNAs in relation to their role in the pathogenesis of cardiac remodeling, as well as discusses the potential application of miRNA biology to the field of heart failure.
Biology of miRNAs
miRNAs are short, approximately 22 nucleotides in length, evolutionarily conserved, regulatory RNA molecules that repress gene expression at the post-transcriptional level by targeting mRNA [7]. miRNAs were first discovered as critical regulators of development in Caenorhabditis elegans, however, subsequent studies have shown that miRNA biogenesis and function are conserved in many organisms [8]. Up to 1000 miRNAs are predicted to exist in the human genome, each of which could potentially target hundreds or thousands of mRNAs and regulate as many as 30% of mRNA transcripts in the whole genome [9]. In addition, most 3′ UTRs contain predicted binding sites for a large number of individual miRNAs, allowing cooperative interactions between various unrelated miRNAs. As shown in Figure 1, binding of miRNAs to their cognate target mRNAs commonly leads to decreased expression of target genes through translational repression or mRNA degradation. Increased expression levels of miRNAs can also result in the ‘paradoxical’ upregulation of previously suppressed target genes either directly, by decreasing the expression of inhibitory proteins and/or transcription factors, or indirectly, by inhibiting the expression levels of inhibitory miRNAs. Alternatively, decreased expression levels of inhibitory miRNAs can lead directly to increased target gene expression.
Figure 1. Mechanism for miRNA regulation of target mRNA levels.
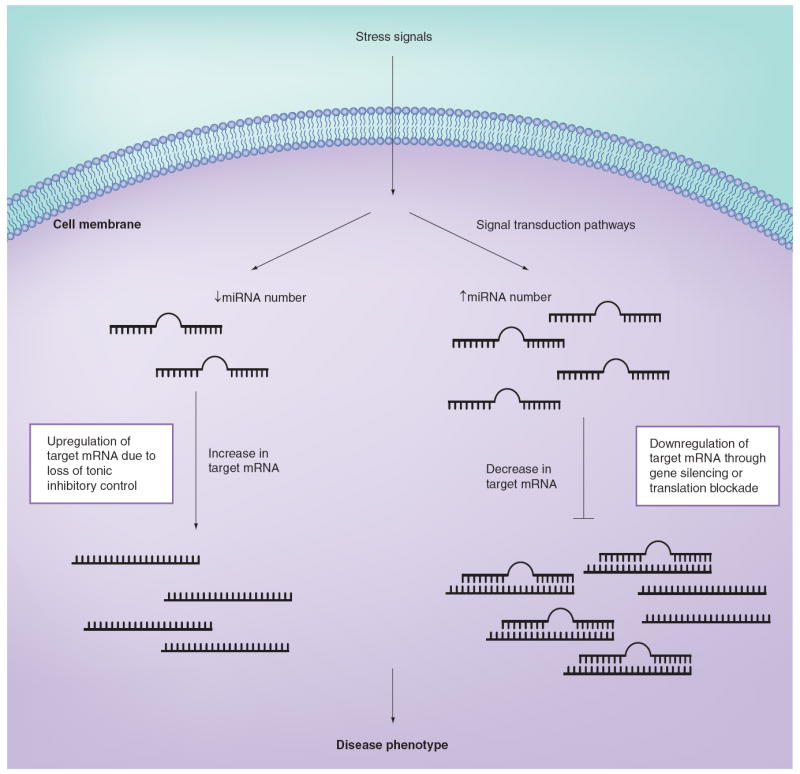
Stress signals (such as hemodynamic overload) activate signal transduction pathways that lead to either the upregulation or downregulation of specific miRNAs. Stress signals that increase the expression levels of miRNAs can result in the downregulation of several target mRNAs through gene silencing or, more commonly, translational blockade of the target mRNA. Alternatively, a stress-induced decrease in the expression levels of inhibitory miRNAs can lead to upregulation of previously suppressed target genes. Ultimately, it is the miRNA-induced pattern of change in gene expression that contributes to the resultant disease phenotype.
Reproduced with permission from [71].
The majority of miRNAs are transcribed by RNA polymerase II from intergenic, intronic or exonic locations generating a long hairpin-shaped structure [10]. These primary miRNAs (pri-miRs) are cleaved in the nucleus by the RNase III-type enzyme, Drosha, into smaller, 70–100 nucleotide-long hairpin shaped precursors, called pre-miRs (Figure 2). Pre-miRs are then transported into the cytoplasm through nuclear pore complexes with the help of exportin-5 and further processed into a 19–25 nucleotide and double stranded miRNA duplex by the RNAse III-type enzyme, dicer [11]. The fully processed miRNA duplex is then incorporated into a multiprotein RNA-induced silencing complex (RISC). During this process, one strand of the miRNA duplex is selected as the ‘mature miRNA’ while the other strand, named as star strand (miRNA*), is rapidly degraded [12,13]. When presented correctly by the RISC, the mature miRNA associates with the 3′ UTR of target mRNA and acts as a negative regulator of gene expression through translational repression or mRNA degradation [14]. Translational repression seems to be the predominant mechanism of regulation in animals, however accumulating evidence suggests that miRNAs not only inhibit translation but also destabilize its target mRNA [15,16].
Figure 2. miRNA biogenesis and function.
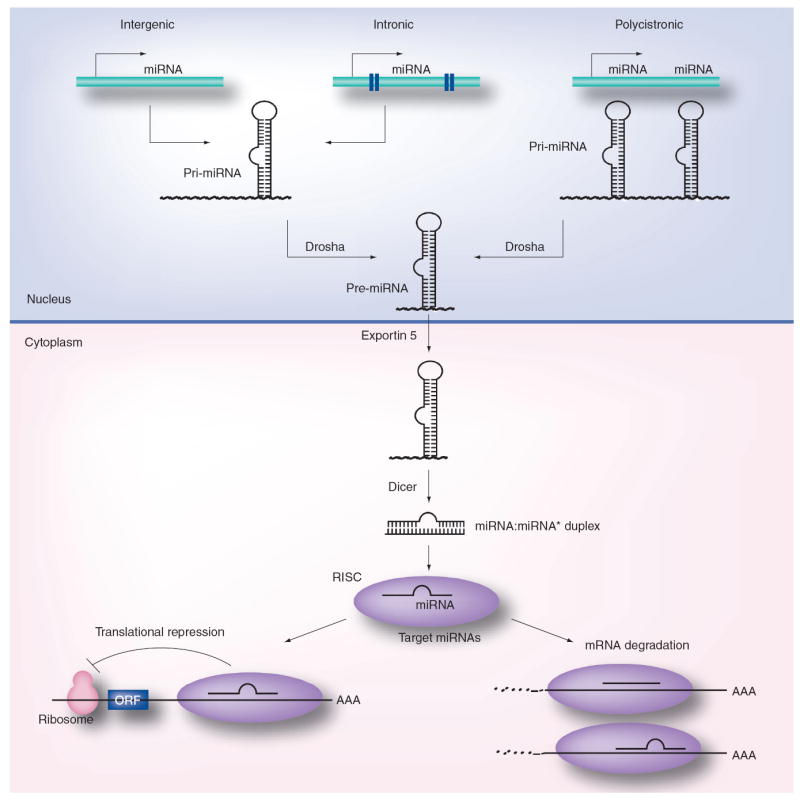
The primary transcripts of miRNAs, called pri-miRNAs, are transcribed as individual miRNA genes, from introns of protein-coding genes or from polycistronic transcripts. The RNase Drosha further processes the pri-miRNA into 70–100 nucleotide-long, hairpin-shaped precursors, called pre-miRNA, which are exported from the nucleus by exportin-5. In the cytoplasm, the pre-miRNA is cleaved by Dicer into a miRNA:miRNA* duplex. Assembled into the RISC, the mature miRNA negatively regulates gene expression by either translational repression or mRNA degradation, which is dependent on sequence complementarity between the miRNA and the target mRNA.
ORF: Open reading frame; RISC: RNA-induced silencing complex.
Reproduced with permission from [72].
Complementarity of the 5′ region of a miRNA, from nucleotide positions 2–7, termed the ‘seed region’ with the 3′ UTR of the corresponding mRNA is thought to confer much of the target recognition specificity [17]. However, since miRNAs are conserved throughout more than the whole length of the mature miRNA, it is likely that the remainder of the miRNA (mismatch tolerant region) may also play a role in target specificity. A number of bioinformatics tools with varying algorithms are available for mammalian target prediction, including TargetScan, miRanda, Pic Tar and DIANA-micro T [18-21]. Potential targets are ranked on the basis of several criteria including seed complementarity, degree of conservation, and multiple binding sites in the 3′ UTR region. However, each miRNA target prediction method is expected to have a sizable rate of both false-positive and false-negative predictions, therefore, experimental validation is of utmost importance for confirmation of target genes.
Expression profiles of miRNAs in experimental & clinical heart failure
The expression profile of the miRNAs failing myocardium have been explored in numerous studies performed by microarray in various well-established murine models of cardiac hypertrophy, including thoracic aortic constriction (TAC) [4,22-24], coronary artery ligation, ischemia-reperfusion [25,26], calcineurin overexpression [4] and Akt overexpression [27], as well as myocardial samples obtained from patients with cardiomyopathy compared with nonfailing controls [4,28-32]. Several points merit consideration before interpretation of the results of these studies. First, it should be noted that the expression analysis has been carried out on an evolving microarray platform and limited by identification of miRNAs with only known sequences. In this regard, next-generation high-throughput miRNA sequencing may allow for discovery and experimental validation of novel or predicted miRNAs with higher sensitivity and specificity, as this technology comes into broader application [33,34]. Second, human heart failure studies of miRNA expression are limited by the lack of standardized protocols, small number of subjects and high degree of variability between groups. In addition, differential expression of particular miRNAs should not always be attributed to cardiomyocytes since other myocardial cell types such as fibroblasts, endothelial cells and inflammatory cells may contribute to the altered levels of miRNAs. This is particularly important given the fact that the majority of these studies performed miRNA profiling on myocardial tissue rather than specific cell types.
The first report of miRNA profiling in human heart failure was performed by Thum and colleagues who compared expression levels of miRNAs from nonfailing, failing and fetal human hearts [32]. They have demonstrated a remarkable overlap of miRNA profiles (85.5% concordance) of the failing and fetal myocardial samples, suggesting that a fetal miRNA profile may also exist and is reactivated in response to myocardial stress and potentially contribute to the reactivation of the fetal gene program during cardiac remodeling. A larger study performed by Ikeda et al. analyzed miRNA expression patterns in myocardial samples from patients with ischemic cardiomyopathy, idiopathic cardiomyopathy and aortic stenosis [31]. Interestingly, using a supervised learning technology, the authors found that miRNA expression profiles correctly grouped subjects by their etiology with an accuracy rate approaching 70%. Sucharov and colleagues have examined myocardial miRNA profiling in patients with ischemic and idiopathic cardiomyopathy compared with nonfailing controls [30]. They demonstrated that the majority of differentially regulated miRNAs (66.7%) were expressed in either ischemic or idiopathic cardiomyopathy patients. Taken together, these results suggest presence of a distinct myocardial miRNA signature in different etiologies of heart failure as a powerful diagnostic tool. More recently, Matkovich et. al. have examined miRNA profiling in patients with heart failure before and after hemodynamic unloading with left ventricular assist devices [28]. Interestingly, they found that 20 of the 28 (71.4%) of the differentially regulated heart failure miRNAs were normalized following left ventricular assist devices support compared with 29 of the 444 (6.5%) differentially regulated mRNAs, suggesting that miRNAs may serve as a marker of myocardial recovery in patients with advanced heart failure.
In an attempt to summarize expression profiles of miRNAs in experimental models of heart failure comparatively with human heart failure, we have generated a 2D graphical map (Figure 3) similar to conventional heat maps generated for gene-expression studies. In this map, each column represents a particular study and each row represents a particular miRNA or miRNA family that is significantly regulated in at least two independent studies. Upregulated miRNAs are depicted in red whereas downregulated miRNAs are depicted in green. miRNAs with no significant changes are uncolored. Unlike a conventional heat map, the color intensity of the miRNAs was not adjusted to reflect the magnitude of change in miRNA expression (i.e., fold change). Evaluation of this heat map reveals a high level of concordance between experimental and human heart failure with 43 out of 62 (69.3%) miRNAs being expressed in both murine and human disease states. Indeed, seven miRNAs or miRNA families (let-7 family, miR-15 family, miR-23a,b, miR-24, miR-125a,b, miR-199a-5p and miR-214) were found to be predominantly upregulated in at least four independent studies (two experimental and two human heart failure studies) suggesting that changes in miRNA expression patterns in experimental models may provide further insight into our understanding of left ventricle (LV) remodeling in human heart failure. Interestingly, the expression profile of miRNAs with decreased expression was far less concordant in experimental models and human heart failure samples. Indeed, there were only two miRNA families (the miR-17 and miR-30 family) that were downregulated in at least four independent studies (two experimental and two human heart failure studies). Analytic approaches such as the depicted heat map may function as a tool for generating hypotheses with respect to exploration of potential roles of specific miRNAs in the development and progression of heart failure.
Figure 3. miRNA expression profiles in experimental models and human heart failure.
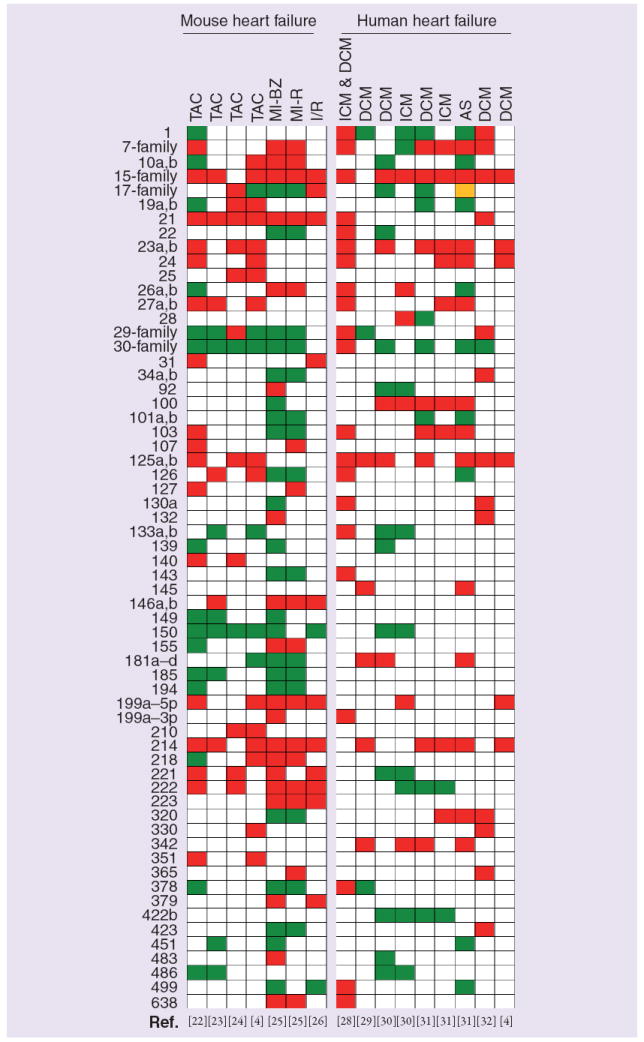
miRNA expression data from 11 relevant studies were represented in 16 columns. miRNAs that were significantly regulated in at least two independent heart failure studies were included and listed on the left of the heat map. miRNAs that shared the same seed region (nucleotides 2–8) were combined into families. The family was labeled as regulated if at least one member was changed. Red color indicates miRs that were significantly upregulated, green color indicates miRs that were significantly downregulated and yellow color indicates miR families that include members that were regulated in opposite directions. The let-7 family includes let-7b to -7j; miR-15 family, miR-15a,b/16/195/424/497; miR-29 family, miR-29a to -29c; miR-30 family, miR-30a to -30e; and miR-17 family, miR-17-5p/20a,b/93/106a.
AS: Aortic stenosis; BZ: Borderzone; DCM: Dilated cardiomyopathy; ICM: Ischemic cardomyopathy; I/R: Ischemia reperfusion; MI: Myocardial infarction; R: Remote; TAC: Thoracic aortic banding.
To better understand how miRNAs may play a role in left ventricular remodeling process, it is useful to recognize that adverse cardiac remodeling represents collective changes in the biology of the cardiac myocyte, the volume of myocyte and non-myocyte components of the myocardium, as well as in the geometry and architecture of the LV chamber (Box 1). Each of these various components of the remodeling process may contribute independently to the progression of heart failure.
Box 1.
Overview of left ventricular remodeling
Alterations in myocyte biology
-
▪
Changes in excitation contraction coupling
-
▪
α-myosin heavy chain (fetal) gene expression
-
▪
β-adrenergic receptor desensitization
-
▪
Hypertrophy with loss of myofilaments
-
▪
Loss of cytoskeletal proteins
Myocardial changes
-
▪
Myocyte loss
-
–
Necrosis
-
–
Apoptosis
-
–
Autophagy
-
–
-
▪
Alterations in extracellular matrix
-
–
Matrix degradation
-
–
Replacement fibrosis
-
–
Impaired angiogenesis
-
–
Alterations in left ventricular chamber geometry
-
▪
Left ventricular dilation
-
▪
Increased left ventricular wall stress
-
▪
Mitral valve incompetence
-
▪
Wall thinning with afterload mismatch
miRNAs involved in cardiac myocyte hypertrophy
In both animal models and the human heart, it is generally held that changes in the biology of the cardiac myocyte are the primary initiating events that lead to cardiac remodeling [35], although it should be noted that cardiac remodeling can occur in the absence of myocyte dysfunction in some experimental models [36,37]. One of the principal changes that occurs in the biology of the failing cardiac myocyte is an increase in cell size (hypertrophy). Based on the extant literature, there is evidence that various miRNAs control and/or modulate key components of the hypertrophic process in cardiac myocytes, and are reviewed below.
miR-1
miR-1 is a muscle specific miRNA that is expressed from bicistronic units with members of the miR-133 family. The vertebrate genome contains two distinct loci for the miR-1/miR-133 cluster producing identical mature miRNA sequences (miR-1–1 and miR-1–2). Cardiac expression of miR-1 is regulated by serum response factor transcription factor [5]. During cardiogenesis, miR-1 levels are suppressed whereas its target, Hand2, a transcription factor that promotes cardiac myocyte proliferation, is expressed at high levels. As the heart develops, miR-1 levels increase and Hand2 protein levels decrease as the cardiac myocytes exit the cell cycle and differentiate into mature cardiac myocytes [5]. Zhao et al. have demonstrated that targeted deletion of a single copy of miR-1 (miR-1–2) resulted in increased late embryonic/postnatal mortality mainly secondary to ventricular septal defects [2]. In addition to its crucial role in cardiac development, a number of studies suggest a potential role of miR-1 in cardiac myocyte hypertrophy. Sayed and colleagues demonstrated that cardiac expression of miR-1 was downregulated as early as 1 day and 7 days after TAC [22]. Care et al. also demonstrated reduced expression of miR-1 (along with miR-133) in three different models of hypertrophy namely TAC, Akt overexpression, and exercise-induced hypertrophy [27]. In a recent study by Ikeda and colleagues, adenoviral overexpression of miR-1 in neonatal rat ventricular cardiomyocytes abolished endothelin-1-induced cardiac myocyte hypertrophy and attenuated endothelin-1-induced expression of fetal genes (NPPA, NPPB and ACTA1), likely via negative regulation of calmodulin, MEF2A and GATA4, key components of calcium signaling pathways necessary for induction of cardiomyocyte hypertrophy [38]. IGF signaling pathway – a key regulator of cardiac myocyte growth and differentiation – was shown to be regulated by miR-1 through translational repression of IGF1 and IGF1R proteins as a potential mechanism of miR-1 for the development of cardiac hypertrophy [39]. The role of miR-1 in the development and progression of human heart failure is still unclear. Some studies report the downregulation of miR-1 levels in the failing heart whereas others report the opposite (Figure 3); therefore, future studies are warranted to explore the role of miR-1 in the development of cardiac hypertrophy.
miR-133
The miR-133 family consists of three members, miR-133a-1, miR-133a-2 and miR-133b, each of which are cotranscribed from bicistronic units with miR-1–2, miR-1–1 and miR-206, respectively. miR-133a-1 and miR-133a-2 are expressed in cardiac and skeletal muscle whereas miR-133b is only expressed in skeletal muscle. Care et al. demonstrated that miR-133 expression levels were downregulated in myocardium in three different models of cardiac hypertrophy namely TAC, Akt overexpression and exercise-induced hypertrophy [27]. In this study, adenoviral overexpression of miR-133 resulted in attenuation of hypertrophic growth and fetal gene expression in phenylephrine or endothelin-1-treated neonatal cardiac myocytes. Moreover, partial in vivo knockdown of miR-133 in mice using specific O-methyl modified, so called, antagomiRs was sufficient to induce hypertrophy at least in part through regulation of RHOA and CDC42 protein levels through translational repression, suggesting an active role for miR-133 in the inhibition of cardiac hypertrophy [27]. By contrast, Liu and colleagues demonstrated that genetic ablation of either miR-133a gene in mice led to a normal cardiac phenotype at baseline and after TAC despite an approximate 50% decrease in miR-133a levels in the heart [40]. It is not clear why genetic deletion of miR-133 and knockdown with antagomiRs provoke different effects; however, transient versus long-term decrease in miR levels may have different effects on the target mRNA regulation. In a study by Matkovich et al. mice overexpressing miR-133a using an α-myosin superimposition of a heavy chain promoter had no effect on cardiac hypertrophy at baseline or following pressure overload. Moreover, as seen in Figure 3, the majority of studies from human heart failure failed to show differential regulation of miR-133a levels [41]. Therefore, further studies are required to determine whether modulation of miR-133 levels would be an interesting therapeutic target in heart failure.
miR-21
miR-21 is one of the few miRNAs which demonstrate a consistent pattern of upregulation in the failing myocardium (Figure 3). miR-21 is also highly expressed in various types of cancer tissues and cell lines, suggesting a common feature of miR-21 in stress response and pathological cell growth [42]. However, the exact role of miR-21 in development of cardiac hypertrophy remains controversial. Knockdown of miR-21 using an antisense oligonucleotide was shown to be sufficient to attenuate phenylyephrine or angiotensin-II induced cell growth and protein synthesis in cultured neonatal cardiac myocytes [23]. By contrast, a different group of investigators demonstrated that knockdown of miR-21 using locked nucleic acid modified antisense oligonuclotide caused increased cardiomyocyte size and induction of hypertrophic gene markers such as ANF and skeletal muscle α-actin [24]. The reasons for this discrepancy remains unclear, however, it may be partially related to differences in the chemical structure of knockdown probes used to modulate miR-21 levels. A carefully designed study by Thum and colleagues revealed that miR-21 is predominantly expressed in cardiac fibroblasts, not cardiomyocytes [43]. Moreover, expression levels of miR-21 in isolated cardiomyocytes did not change in the failing verses nonfailing hearts, suggesting a limited role of miR-21 in cardiomyocytes. Therefore, the change in miR-21 levels in the failing myocardium could be primarily related to an increase in fibroblast cell number or activity rather than changes in cardiomyocyte function. To date, miR-21 has no validated gene targets that are directly related to cardiac myocyte hypertrophy. Accordingly, future studies are needed to investigate effects of miR-21 on cardiac hypertrophy.
miR-23
There are two miR-23 genes (miR-23a and miR-23b) that differ by only one nucleotide in the mature strand. Both miR-23 genes are clustered with miR-24 and miR-27 genes suggesting that these miRNAs may be transcribed together. Lin and colleagues reported that all three members of this cluster (miR-23a, miR-24 and miR-27a) were upregulated in cardiac myocytes upon isoproterenol or aldosterone stimulation; however, only knockdown of miR-23a by a specific antagomiR, not miR-24 or miR-27a, attenuated isoproterenol or aldosterone-induced hypertrophic response [44]. Moreover, in vivo knockdown of miR-23a via an antagomiR was shown to inhibit isoproteronol induced hypertrophy, decrease the HW:BW ratio, and decrease fetal gene expression in a mouse model of heart failure. The authors demonstrated that NFATC3 could directly active miR-23a expression through transcriptional machinery and MURF-1, an antihypertrophic protein, is a direct target of miR-23a as a potential mechanism of prohypertrophic effects of miR-23a [44]. Overexpression of miR-24 under the α-MHC promoter caused embryonal lethality suggesting a role of miR-24 in development [4].
Other candidate miRs involved in cardiac myocyte hypertrophy
Through miRNA microarray profiling of TAC and calcineurin overexpressing models of cardiac hypertrophy, van Rooij et al. identified seven miRNAs that were upregulated and four miRNAs that were downregulated in both models. Six of the upregulated miRNAs (miR-23a, 23b, 24, 195, 199a and 214) were shown to provoke cardiac myocyte hypertrophy when transfected into neonatal cardiac myocytes, whereas two downregulated miRNAs (miR-150 and -181b) caused a reduction in myocyte size [4]. The authors demonstrated that cardiac restricted overexpresion of miR-195 initially induced cardiac growth with presence of severe myocyte disorganization on histologic examination, and followed by progression into a dilated cardiac hypertrophic phenotype by 6 weeks of age. However, the molecular mechanisms for the induction of cardiac hypertrophy in miR-195 overexpressing mice remain unknown. miR-195 is a member of the miR-15 gene family (miR-15a, 15b, 16, 195, 427 and 497) and is clustered with miR-497. As seen in Figure 3, members of miR-15 family are consistently upregulated across mouse models of heart failure, as well as human heart failure, suggesting that members of this family may play an important role in cardiac hypertrophy. In a recent study by Wang et al., upregulation of miR-9 was shown to attenuate cardiac hypertrophy and ameliorate cardiac function in a mouse model of isoproterenol infusion [45]. This effect was mediated at least in part through downregulation of myocardin expression by direct targeting of miR-9. Since miR-9 is not differentially expressed in human heart failure (heat map), additional studies are required to investigate its potential role in cardiac hypertrophy.
miRNAs involved in excitation–contraction coupling
Excitation–contraction coupling refers to the cascade of biological events that begins with cardiac action potential and ends with myocyte contraction and relaxation. Classic studies from explanted failing hearts have shown that patients with end-stage heart failure exhibit decreased contractility and impaired relaxation, which is believed to be secondary to changes in the abundance and/or phosphorylation state of critical calcium (Ca2+) regulatory proteins that are thought to play an important role in crossbridge activation and relaxation.
An additional defect in myocyte function in hypertrophy and heart failure is thought to occur secondary to changes in the actin and myosin myofibrillar crossbridges. Indeed, early studies showed that myofibrillar ATPase was reduced in the hearts of patients who died of heart failure and that these abnormalities in ATPase activity could be explained by an isoform switch from α-MHC, which hydrolyzes ATP rapidly and is expressed in the adult heart, to β-MHC, which hydrolyzes more slowly and is expressed in the fetal heart. Whereas α-MHC accounts for approximately 33% of MHC mRNA in normal human myocardium, the abundance of α-MHC mRNA decreases to approximately 2% in the failing heart [46]. Given the aforementioned link between expression levels of miRNAs and the development of cardiac myocyte hypertrophy, it is logical to consider that miRNAs might also regulate the Ca2+ regulatory proteins involved in excitation – contraction coupling that are downregulated in cardiac hypertrophy.
miR-208
The genome contains of two copies of miR-208 gene (miR-208a and miR-208b), which share the same seed sequence but differ at three nucleotides in their 3′ region. miR-208a and miR-208b are of unique importance to cardiac function as these genes are encoded by introns of α-MHC and β-MHC genes, respectively. The cardiac specific miR-208a, encoded by an intron of the α-MHC gene, is coordinately regulated with the α-MHC gene. van Rooij and colleagues showed that mice deficient for miR-208a(−/−) had a normal cardiac phenotype up to 20 weeks of age; however, these mice had a blunted hypertrophic response, decreased myocardial fibrosis following TAC [3]. In addition, miR-208a−/− mice did not express β-MHC in response to TAC suggesting that miR-208a may regulate the induction of β-MHC expression on cardiac stress. Callis et al. demonstrated that cardiac restricted overexpression of miR-208a was sufficient to induce hypertrophic growth in mice and upregulate expression levels of β-MHC and miR-208b [47]. Interestingly, β-MHC upregulation was restricted to areas of myocardial fibrosis. Regulation of cardiomyocyte growth by miR-208a might be at least in part due to the repression of thyroid hormone receptor-associated protein 1 and myostatin 2, negative regulators of muscle hypertrophy [3,47]. Even though protective in the short-term, long-term depletion of miR-208a led to decreased contractility [3] and abnormalities in cardiac conduction system causing atrial fibrillation, in part through dysregulation of cardiac transcription factors, such as homeodomain-only protein and GATA4, and the gap junction protein connexin 40 [47]. Taken together, these observations suggest that miR-208 is required for the development of cardiac hypertrophy and myocardial fibrosis and that miR-208 is a positive regulator of α-MHC gene expression.
miRNAs involved in regulating the cytoskeleton
The cytoskeleton of cardiac myocytes consists of actin, the intermediate filament desmin, the sarcomeric protein titin, and α- and β-tubulin, which form the microtubules by polymerization. Vinculin, talin, dystrophin and spectrin represent a separate group of membrane-associated cytoskeletal proteins. Disruption of cytoskeletal and/or membrane associated proteins has been implicated in the pathogenesis of heart failure in numerous studies. Indeed, the loss of integrity of the cytoskeleton, with a resultant loss of linkage of the sarcomere to the sarcolemma and extracellular matrix (ECM) would be expected to lead to contractile dysfunction at the myocyte level, as well as at the myocardial level. At present, there is limited evidence that miRNAs are involved in regulating the cytoskeleton; insofar as relatively few studies have examined these targets.
miR-1 & miR-133
As noted above, the decreased expression of miR-1 and miR-133 allows for the increased expression of growth related genes that are responsible for cardiac hypertrophy. Adenoviral-mediated overexpression of miR-1 suppressed sarcomeric α-actin organization that is normally observed in serum-deprived neonatal cardiac myocytes. The effects of miR-1 on cytoskeletal reorganization were also thought to be responsible for the inhibition of endothelin-induced cell spreading in neonatal myocytes [22]. Similarly, adenoviral transfection with miR-133 inhibited the reorganization of the actin myofilaments observed in hypertrophic growth of cardiac myocytes [27]. Two validated gene targets of miR-133 are RHOA and CDC42, a fact that is of interest insofar members of the Rho family of GTP binding proteins are involved in myofibrillar rearrangement and are, therefore, important for cell motility and contractility. miR-1 also targets a number of cytoskeletal-related proteins, including microtubule-related proteins, kinectins, actin binding proteins and cadherins. The significance of the aforementioned miRNA-induced changes in the cytoskeleton vis-a-vis the biology of the failing myocyte remains to be determined.
miRNAs involved in myocardial alterations in the failing heart
The alterations that occur in failing myocardium may be categorized broadly into those that affect the number of cardiac myocytes, as well as those changes that occur in the volume and composition of the ECM (Box 1). With respect to the changes that occur in cardiac myocyte component of the myocardium, there is increasing evidence that suggests that progressive myocyte loss, through necrotic, apoptotic or autophagic cell death pathways, may contribute to progressive cardiac dysfunction and LV remodeling.
miRNAs involved in regulating cell fate
Progressive myocyte loss secondary to apoptosis occurs in failing hearts and contributes to progressive cardiac dysfunction and LV remodeling. Apoptosis requires activation of phylogenetically conserved ensemble of proteins belonging to the extrinsic (death receptor mediated) or intrinsic (mitochondrial) pathways that ultimately lead to activation of executioner caspases and myocyte loss. The extensive synergistic regulation of proand antiapoptotic proteins in response to stress signals raises the possibility that miRs may be involved in regulating programmed cell death.
miR-21
As discussed earlier, miR-21 is upregulated in various types of cancers, and based on its in silico predicted proapoptotic gene targets, has been proposed as a potential antiapoptotic miRNA. Cheng and colleagues showed that miR-21 is upregulated in cultured cardiac myocytes upon exposure to hydrogen peroxide (H2O2), and that modulating miR-21 levels by antisense oligonucleotide knockdown probes led to an increase in H2O2-induced cell-death suggesting a protective role of miR-21 on apoptosis at least in part through targeting PCDC4 protein [48]. As shown in Figure 3, miR-21 is consistently upregulated in heart failure which may represent a prosurvival stress response in response to hemodynamic pressure overload.
miR-1 & miR-133
Even though miR-1 and miR-133 have similar effects on myocyte proliferation, differentiation and cytoskeletal organization; increasing lines of evidence suggest that these miRNAs have opposite roles with respect to regulating cell fate. Xu and colleagues demonstrated that overexpression of miR-1 in H9C2 cell line induced significant apoptotic cell death, which was largely prevented by cotransfection of miR-1 and miR-133 indicating an antiapoptotic effect of miR-133 [49]. Similar effects on oxidative stress induced apoptosis were observed in H2O2-treated H9C2 cells, where miR-1 and miR-133 had promoting and inhibitory effects on cell death, respectively. HSP60 and HSP70 were identified and validated as miR-1 targets whereas caspase 9 was confirmed as a miR-133 target as a potential contributing mechanism of their opposing effects on cell fate.
miR-199a
The miR-199 family has three members, miR-199a-1, miR-199a-2 and miR-199b, which are encoded by the antisense strand of an intron of the dynamin gene (DNM2, DNM3 and DNM1, respectively). Care et al. recently reported that mature miR-199a was rapidly reduced to undetectable levels during cardiac ischemia, and that this reduction was necessary for induction of hypoxia-induced apoptotic gene program at least in part through derepression of HIF1A, an important transcription factor for the induction of gene expression in hypoxic conditions [50]. In addition to targeting HIF1A, downregulation of miR-199a derepressed Sirtuin 1 in cardiac ischemia, which in turn contributed to the accumulation of HIF1A protein, suggesting that miR-199a downregulation has an important role in hypoxia-induced cell death. miR-199a appears to be upregulated in various models of heart failure (Figure 3). Moreover, transfection of miR-199a in cardiac myocytes results in a hypertrophic phenotype [4]. However, the mechanisms for these effects remain unknown at this time.
miR-320
Ren and colleagues demonstrated that miR-320 expression was downregulated in both in vivo and ex vivo models of ischemia-reperfusion (I/R) injury [51]. Overexpression of miR-320 enhanced cell death and apoptosis in cultured adult rat cardiomyocytes on simulated I/R, whereas knockdown was found to be cytoprotective. Transgenic mice with cardiac-specific overexpression of miR-320 revealed increased apoptosis and infarction sizes after in vivo and ex vivo I/R injury compared with the wild-type controls, whereas in vivo treatment with antagomiR-320 reduced infarction size relative to the administration of mutant antagomiR-320 and saline controls, which was in part mediated by regulation of target protein HSP20. Therefore, targeting miR-320 may represent an alternative approach to attenuate myocardial I/R injury.
miRNAs involved in regulating the ECM
Changes within the ECM constitute the second important myocardial adaptation that occurs during cardiac remodeling. The myocardial ECM consists of a basement membrane, a fibrillar collagen network that surrounds myocytes, proteoglycans and glycosaminoglycans, and biologically active signaling molecules. Important changes occur in the ECM during cardiac remodeling, including changes in fibrillar collagen synthesis and degradation, as well as changes in the degree of collagen crosslinking. One of the histological signatures of advancing heart failure is the progressive increase in collagen content of the heart (myocardial fibrosis). The increased fibrous tissue would be expected to lead to increased myocardial stiffness, which would theoretically result in decreased myocardial shortening for a given degree of afterload. In addition, myocardial fibrosis may provide the structural substrate for atrial and ventricular arrhythmias, thus potentially contributing to sudden death.
miR-29
The miR-29 family has three members (miR-29a, -29b and -29c), which are transcribed from two bicistronic miRNA clusters. van Rooij et al. found that members of miR-29 family were downregulated after murine myocardial infarction (MI), particularly in the borderzone. Interestingly, miR-29 family has a high number of targets related to myocardial fibrosis, including collagens, fibrillins and elastin. Knockdown of miR-29 in vivo using an antagomiR approach induced myocardial collagen deposition, whereas overexpression of miR-29 in isolated fibroblasts using a miR-mimic resulted in reduction in collagen gene expression [25]. Similarly, Divakaran and colleagues demonstrated that miR-25 and miR-29a were differentially regulated in SMAD3-/- mice, which demonstrated increased myocardial fibrosis after transaortic constriction compared with wild-type mice [52]. Interestingly, transfection of either miR-29a or miR-25 into primary cardiac fibroblasts resulted in a significant decrease in collagen gene expression, suggesting that collagen genes are targeted by these miRNAs. Taken together, these studies suggest that pharmacological manipulation of the miR-29 family can modulate the myocardial fibrosis that is observed in the failing myocardium.
miR-21
As discussed earlier, miR-21 is predominantly expressed in cardiac fibroblasts and is upregulated in various models of heart failure (Figure 3). Thum et al. demonstrated that miR-21 levels are increased selectively in fibroblasts of the failing heart, augmenting ERK–MAP kinase activity through inhibition of sprouty homologue 1, thereby increasing fibroblast survival and fibrosis [43]. In vivo knockdown of miR-21 with specific antagomiR resulted in attenuation of interstitial fibrosis and cardiac remodeling in mice following TAC. Interestingly, miR-21 was upregulated in the fibroblast enriched borderzone region following MI [25,26] and was shown to increase MMP-2 activation at least in part through targeting PTEN [26].
miR-133a
Liu and colleagues demonstrated that genetic deletion of both copies of miR-133a leads to extensive fibrosis and to a dilated cardiomyopathic phenotype suggesting that miR-133a can be a negative regulator of cardiac fibrosis [40]. Consistent with this finding, Duisters et al. demonstrated that knockdown of miR-133a in cultured cardiac myocytes and fibroblasts caused an increase in target CTGF, and indeed CTGF was validated as a direct target of miR-133a as potential mechanism for its antifibrotic effect on myocardium [53]. Similarly, mice with cardiac overexpression of miR-133a under the α-MHC promoter were protected from myocardial fibrosis after transaortic constriction [41]. Interestingly, CTGF mRNA levels were increased more than twofold in miR-133a transgenic hearts, suggesting presence of an alternative mechanism for the antifibrotic effects of miR-133a overexpression in myocardium.
miR-208a
As noted earlier, van Rooij and colleagues have shown that miR-208, a cardiac-specific miRNA encoded within an intron of the α-MHC gene, is required for cardiac fibrosis in response to hemodynamic pressure overload. Indeed, miR-208 deficient mice were resistant to developing fibrosis following TAC [3]. Similarly, myocardial fibrosis was significantly attenuated when heart failure mice with cardiac restricted overexpression of calcineurin was cross bred with miR-208 deficient mice [3]. Thus, miR-208 appears to be required for the development of cardiac fibrosis; however, the mechanisms for this effect remain currently unknown.
miRNAs involved in electrophysiological alterations
In addition to cardiac remodeling and neurohormonal activation, there are important changes in ion channel function and expression in the failing heart that lead to alterations in the electric phenotype of both atrial and ventricular myocytes. This ‘electrophysiological remodeling’ renders the heart more vulnerable to ventricular arrhythmias that underlie sudden cardiac death. Although the exact role of miRNAs in the development of atrial and ventricular arrhythmias in the setting of clinical heart failure is unknown, the experimental literature suggests that cardiac specific miRNAs are sufficient to provoke cardiac arrhythmias.
miR-1 & miR-133
It has been suggested that the pacemaker current (If) carried by hyperpolarization activated channels encoded by HCN2 and HCN4 genes plays an important role in cardiac automaticity, and that increased activity of these channels contributes to the risk of arrhythmias under pathological conditions such as cardiac hypertrophy [54]. Interestingly, HCN2 was experimentally validated as a target for both miR-1 and miR-133, and HCN4 was established as a target for miR-1 only. Moreover, co-transfection of miR-1 and miR-133 was shown to abrogate angiotensin II-induced upregulation of HCN2 and HCN4 protein levels in cultured ventricular myocytes, suggesting that downregulation of miR-1 and miR-133 can induce arrhythmias in myocardium [55]. Zhao and colleagues reported that mice with genetic deletion of miR-1–2 exhibited abnormal cardiac electrical activity, including bradycardia, shortened PR interval, widened QRS complexes associated with increased mortality. The ECG abnormalities were in partially attributed to upregulation of iroquois family transcription factor 5, which is a confirmed target for miR-1 and a negative transcriptional regulator of KCND2 gene (Kv 4.2 channel) and important for cardiac repolarization [2]. By contrast, Yang et al. demonstrated that overexpression of miR-1 exacerbated arythmogenesis in both normal and post-MI myocardium at least in part by post-transcriptionally repressing KCNJ2 (which encodes the K+ channel subunit Kir2.1) and GJA1 (which encodes connexin 43), suggesting a proarrhythmic effect of miR-1 in the post-MI setting [1]. In addition to modulating cardiac conduction and repolarization current, adenoviral overexpression of miR-1 in cardiac myocytes was shown to induce spontaneous arrhythmogenic oscillations of intracellular Ca2+ in presence of isoproterenol [56]. This was mediated in part through a decrease in expression of the protein phosphatase, PP2A regulatory subunit B56α, a miR-1 target leading to hyperphosphorylation of l-type Ca2+ channels as well as RYR2. A recent study by Matkovich and colleagues showed mice with cardiac restricted overexpression of miR-133a had a prolonged QT interval on ECG surface and prolonged action potential in isolated ventricular myocytes compared with wild-type controls [41]. Interestingly, miR-133a overexpression induced K+ channel remodeling in the myocardium, and was associated with a reduction in TAC associated decrease in Ito,f current partially through downregulation of KCNIP2 gene that encodes the Ito,f channel accessory subunit KChIP2 [41].
Role of miRNAs in personalized heart failure therapy
As reviewed in the preceding discussion, growing lines of evidence suggest that miRNAs contribute to adverse/pathological cardiac remodeling through regulating expression of genes or gene networks that drive the so-called ‘heart failure phenotype’ (Figure 4). Therefore, it is tempting to speculate that miRNAs, acting singly or in combination, may be responsible for modulating the transition from adaptive to pathological cardiac remodeling. Although predicting the success of future therapeutic strategies in the field of heart failure is fraught with difficulty, one can envision that miRNA biology may contribute to management of heart failure through multiple different applications.
Figure 4. Role of miRNAs in cardiac remodeling process.
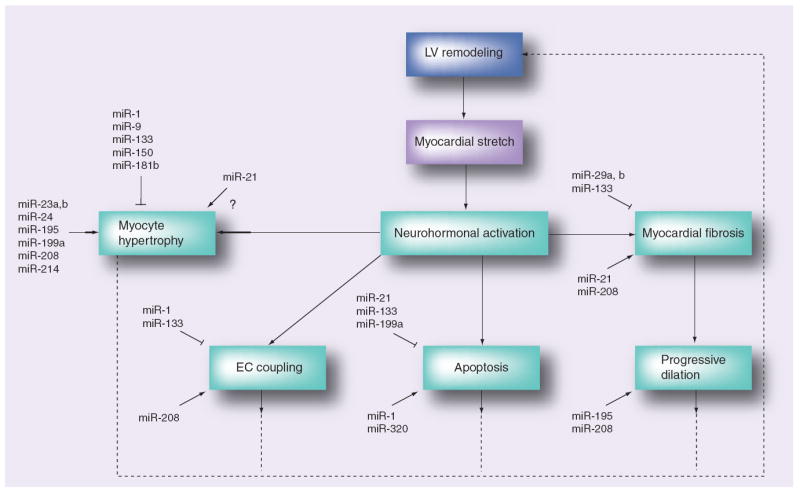
Candidate miRNAs with suggested roles in the cardiac remodeling process depicted with respect to their target LV remodeling component.
ECM: Extracellular matrix; LV: Left ventricle.
Potential of miRs as diagnostic & prognostic markers in heart failure
Ikeda and colleagues have first reported that myocardial miRNA expression profiles segregated the patients by the etiology of heart failure (ischemic vs idiopathic vs aortic stenosis) with 100% accuracy, using a discriminant analyis technique, suggesting that each form of heart failure is characterized by a distinct miRNA expression profile [32]. A major limitation to clinical application of this approach is the need for an invasive procedure to obtain myocardial samples. Recent evidence suggest that endogenous circulating miRs, which are remarkably stable and are protected from endogenous RNAse activity may serve as biomarkers of cardiovascular disease [57,58]. Indeed, Tijsen et al. demonstrated that miR-423–5p was enriched in serum of patients with heart failure and significantly correlated with circulating brain natriuretic peptide levels as well as left ventricular ejection fraction; however, the source and mechanism of increased circulating miR-423–5p levels in patients with heart failure is unknown [58].
Two studies have examined miR expression in patients with mechanical circulatory support. Through microarray profiling, Matkovich and colleagues showed that 28 different miRs that were significantly altered in heart failure group was near normalized in the post-treatment left ventricular assist device group [28]. Another study using RT-PCR, demonstrated a decrease in miR-1, miR-133a, and miR-133b levels following mechanical support in patients with dilated cardiomyopathy, whereas an opposite trend was noted in patients with ischemic cardiomyopathy [59]. Taken together, these studies suggest that unique patterns of miRNA expression may provide fingerprints that may serve as molecular biomarkers for the diagnosis, prognosis of disease, and prediction of therapeutic response in patients with heart failure.
Potential of miRs as therapeutic targets in heart failure
In addition to their utility as markers of diagnosis and disease progression, miRNAs are emerging as potential therapeutic targets in the treatment of heart failure. Modulation of cardiac miRNA expression can be achieved through a number of pharmacological interventions, including antisense oligonucleotide mediated (anti-miR) knock-down and miR-mimic mediated overexpression techniques. One of the potential advantages of miRNA based therapies over conventional therapeutics used in the treatment of heart failure would be the ability to modulate multiple target genes or networks related to cardiac remodeling process. In this regard, it is important to note that since miRNAs typically act as negative regulators of gene expression, overexpressing a miRNA through miR-mimics would downregulate its target genes, whereas inhibiting a miRNA through antimiR knockdown approach would relieve the inhibition of genes that are normally targeted by the miRNA.
AntimiRs are antisense oligonucleotides with the reverse complementarity sequence of the mature target miRNA. Upon cellular uptake, antimiRs bind to target miR and reduce its activity. Since native nucleic acids are rapidly degraded by the enzymatic activity in biological environments, various chemical modifications have been proposed to enhance their stability, cellular uptake, and inhibitory action on the target miRNA. Krutzfeld and colleagues reported on the first mammalian miRNA knockdown using cholesterol conjugated, ‘antagomiRs’, to inhibit a liver specific miRNA (miR-122), which caused upregulation of genes in cholesterol biosynthesis and a reduction of serum cholesterol levels [60]. The utility of antagomiRs in preventing and/or reversing various components of adverse/pathological cardiac remodeling has been demonstrated in a number of animal studies [25,27,43,44]. Alternative modification techniques such as 2′-O-methoxyethyl phosphorothioate substitution and use of locked nucleic acid chemistry were shown to effectively knockdown target miRNA levels, and the use of locked nucleic acid technology is currently under investigation in the first clinical human trial [61,62].
In situations in which an increase in the activity of a specific miRNA is desired, the use of miR-mimics has been used to achieve elevated cellular concentrations of given miRNA. A miR-mimic is a double stranded oligonucleotide that involves the mature miRNA sequence (guide strand), as well as a complementary passenger strand, which is required for the efficient recognition and loading of the guide strand into the RNA-induced silencing complex. Whereas miR-mimics have been used in vitro to decrease the expression of target mRNAs, their efficacy has not yet been tested in vivo. Since miR-mimics function as mature miRNAs, they cannot be modified or stabilized like anti-miRs, requiring a much higher dosing, which would in turn increase their potential risk for side effects. However, the feasibility of the RNA interference technique through the use of cardiotropic adeno-associated virus carrying short hairpin RNAs has been demonstrated, suggesting that a strategy that targets select mRNAs may be effective in vivo. For example, Suckau and colleagues showed that delivery of short hairpin RNAs targeting phospholamban reduced phosholamban levels and significantly improved cardiac function without apparent toxicity in rats undergoing transaortic constriction [63]. Therefore, the use of miR-mimics may represent an attractive approach of enhancing myocardial miRNA levels for those that are downregulated in the failing heart.
Even though a number of studies discussed in this article demonstrate that miRNA-based therapies hold promise for the treatment of heart failure, there are several limitations to overcome before this technology can be safely and successfully applied in clinical medicine. First, given that a single miRNA may regulate expression of hundreds or thousands of genes, modulation of a miRNA of interest can lead to unintended side effects through regulation of the remaining as well as unknown target mRNAs with pathological consequences. Second, pharmacokinetics, biodistribution and tissue penetration of miRNA based therapies needs to be improved through molecular modifications to optimize tissue specificity. Alternatively, catheter-based miRNA-based delivery techniques including intramyocardial injection as well as transcoronary infusion may serve as an alternative attractive to intravenous application in patients with heart failure.
miRNA polymorphisms & potential implications for heart failure pharmogenomics
Given the pivotal role of miRNAs in the regulation of gene expression, it is tempting to speculate that naturally occurring polymorphisms or mutations in miRNA regulatory genes may have profound effects on mechanisms of disease progression and response to medical and/or device therapy in patients with heart failure. Polymorphisms in miRNAs encoding genes may lead to sequence variations in the pri-, pre- or mature miRNAs and profoundly influence processing and/or target specificity of the effected miRNAs. A recent study identified a polymorphism in the seed region of miR-125a that significantly inhibited processing of pri-miRNA to pre-miRNA and attenuated miR-125a mediated translational suppression of a known target mRNA [64]. In addition to sequence variations in miRNA encoding genes, polymorphisms effecting 3′ UTR of the target mRNAs may also interfere with the regulatory function of the miRNA on the target mRNA expression. Using computer alignment, Martin and colleagues have demonstrated that +1166A/C polymorphism in the human AT1R, which was previously shown to be associated with cardiovascular disease, was indeed recognized by a specific miRNA, miR-155 [65]. In the presence of the +1166 C allele, these authors demonstrated that the ability of miR-155 to interact with 3′ UTR of target AT1R gene was disrupted, resulting in increased expression of AT1R gene and activation of ERK1/2, as a potential mechanism leading to cardiovascular disease. A bioinformatics survey of human SNP database by Saunders and colleagues revealed a relatively low level of variation in functional regions of miRNAs, but an appreciable level of variation at target sites [66]. Interestingly, the likelihood of a SNP occurring in a miRNA seed region was less than 1%, suggesting that seed regions are highly conserved from sequence variation from an evolutionary standpoint. Last, polymorphisms in genes encoding proteins that are involved in various steps of miRNA transcription, processing, export and targeting such as RNA polymerase II, Drosha/Pasha, exportin-5, Dicer and the RISC complex, may have effects on the miRNA function (Figure 5) [67,68]. In this regard, it is interesting to note that cardiomyocyte specific deletions of Dicer or Dgcr8 cause development of heart failure in mice [69,70].
Figure 5. miRNA polymorphisms effecting cardiac remodeling.
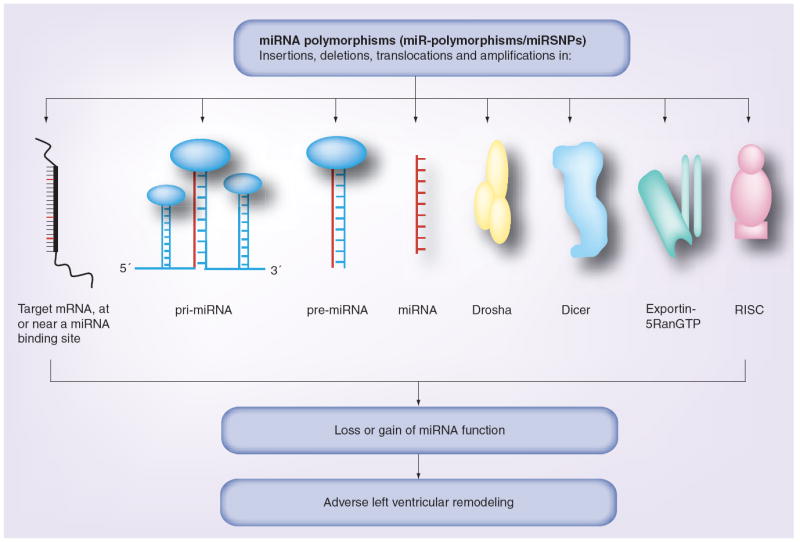
Polymorphisms present in the target mRNA, pri-miRNA, pre-miRNA, mature miRNA, Drosha, Dicer, exportin-5RanGTP and in the RISC complex may affect miRNA-mediated regulation in the cell. The miR-polymorphisms can be present in the form of insertions, deletions, amplifications or chromosomal translocations, leading to loss or gain of a miRNA function. Since miRNAs are predicted to regulate target genes that are involved in the cardiac remodeling process, genetic polymorphisms in miRNA pathways may lead to adverse left ventricular remodeling.
RISC: RNA-induced silencing complex.
Modified with permission from [68].
Even though our understanding of genetic variations in the miRNA pathways and their effects on adverse cardiac remodeling processes is very limited, miR polymorphisms have tremendous potential for clinical application in the diagnosis, prognosis, and treatment of heart failure patients. For example, certain miR-SNPs may serve as a powerful clinical tool to assess disease severity and prognosis in patients with heart failure. In addition, miR-SNPs can be used as predictors of response to medical and/or device therapy based on patient-specific genetic make-up, which would allow physicians to tailor personalized therapies for these patients. Therefore, understanding the role and function of miRNA polymorphisms has a promising future in pharmacogenomics and personalized heart failure therapies.
Future perspective
Since the initial discovery of miRNAs as key regulators of gene expression in mammalian cells, research in this field has been growing at an exponential rate. Given that a number of distinct miRNAs and/or miRNA families are differentially regulated in the failing heart, and an individual miRNA may target thousands of different mRNAs, we are only beginning to understand the extent of miRNA function in the pathogenesis of heart failure. Over the next 5–10 years, we anticipate that an increasing number of miRNA–mRNA interactions will be discovered and significantly contribute to our understanding of basic mechanisms of the disease process.
From a clinical standpoint, it is expected that serum and/or myocardial miRNA profiling will serve as biomarkers of disease severity and therapeutic response in patients with heart failure. In addition, screening for genetic polymorphisms in miRNA–mRNA regulatory pathways will predict response to medical and device therapies and enable physicians to tailor personalized therapies for heart failure patients. Advancements in pharmacology and RNA silencing technology will eventually lead to discovery of cardiac specific miR-based therapeutics (miR-mimics, antagomiRs and so on) and allow us to modulate cardiac miRNA levels while minimizing their off-target effects. These agents are expected to be in clinical use and may further improve outcomes in patients with heart failure in the foreseeable future.
Footnotes
Financial & competing interests disclosure
This research was supported by research funds from the NIH (RO1 HL58081, HL-73017–0, HL089543–01 and T32HL007081). DL Mann is a consultant for Miragen (Boulder, CO, USA). The authors have no other relevant affiliations or financial involvement in any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Bibliography
Papers of special note have been highlighted as:
-
▪
of interest
-
▪▪
of considerable interest
- 1.Yang B, Lin H, Xiao J, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13(4):486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 2.Zhao Y, Ransom JF, Li A, et al. Dysregulation of cardiogenesis, cardiac conduction and cell cycle in mice lacking miRNA-1–2. Cell. 2007;129(2):303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 3.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 4.van Rooij E, Sutherland LB, Liu N, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA. 2006;103(48):18255–18260. doi: 10.1073/pnas.0608791103.. ▪▪ First study to demonstrate role of miRNAs in cardiac hypertrophy and heart failure.
- 5.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436(7048):214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 6.Xiao J, Luo X, Lin H, et al. MicroRNA miR-133 represses HERG K+ channel expression contributing to QT prolongation in diabetic hearts. J Biol Chem. 2007;282(17):12363–12367. doi: 10.1074/jbc.C700015200. [DOI] [PubMed] [Google Scholar]
- 7.Bartel DP. MicroRNAs, genomics, biogenesis, mechanism and function. Cell. 2008;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 8.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 9.Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell. 2005;120(1):21–24. doi: 10.1016/j.cell.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 10.Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004;10(12):1957–1966. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17(24):3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115(2):199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 13.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115(2):209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 14.Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the microprocessor complex. Nature. 2004;432(7014):231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- 15.Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 16.Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006;20(5):515–524. doi: 10.1101/gad.1399806. [DOI] [PubMed] [Google Scholar]
- 17.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 18.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115(7):787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 19.Griffiths-Jones S, Grocock RJ, van DS, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34(Database issue):D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krek A, Grun D, Poy MN, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37(5):495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 21.Kiriakidou M, Nelson PT, Kouranov A, et al. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004;18(10):1165–1178. doi: 10.1101/gad.1184704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100(3):416–424. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 23.Cheng YH, Ji RR, Yue JM, et al. MicroRNAs are aberrantly expressed in hypertrophic heart – do they play a role in cardiac hypertrophy? Am J Pathol. 2007;170(6):1831–1840. doi: 10.2353/ajpath.2007.061170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tatsuguchi M, Seok HY, Callis TE, et al. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2007;42(6):1137–1141. doi: 10.1016/j.yjmcc.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Rooij E, Sutherland LB, Thatcher JE, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roy S, Khanna S, Hussain SR, et al. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res. 2009;82(1):21–29. doi: 10.1093/cvr/cvp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Care A, Catalucci D, Felicetti F, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13(5):613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 28.Matkovich SJ, van Booven DJ, Youker KA, et al. Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of the microRNA signature by biomechanical support. Circulation. 2009;119(9):1263–1271. doi: 10.1161/CIRCULATIONAHA.108.813576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naga Prasad SV, Duan ZH, Gupta MK, et al. A unique microRNA profile in end-stage heart failure indicates alterations in specific cardiovascular signaling networks. J Biol Chem. 2009;284:27487–27499. doi: 10.1074/jbc.M109.036541. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Sucharov C, Bristow MR, Port JD. miRNA expression in the failing human heart: functional correlates. J Mol Cell Cardiol. 2008;45(2):185–192. doi: 10.1016/j.yjmcc.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikeda S, Kong SW, Lu J, et al. Altered microRNA expression in human heart disease. Physiol Genomics. 2007;31(3):367–373. doi: 10.1152/physiolgenomics.00144.2007. [DOI] [PubMed] [Google Scholar]
- 32.Thum T, Galuppo P, Wolf C, et al. MicroRNAs in the human heart – a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116(3):258–267. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 33.Morin RD, O’Connor MD, Griffith M, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18(4):610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willenbrock H, Salomon J, Sokilde R, et al. Quantitative miRNA expression analysis: comparing microarrays with next-generation sequencing. RNA. 2009;15(11):2028–2034. doi: 10.1261/rna.1699809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dash R, Kadambi V, Schmidt AG, et al. Interactions between phospholamban and β-adrenergic drive may lead to cardiomyopathy and early mortality. Circulation. 2001;103(6):889–896. doi: 10.1161/01.cir.103.6.889. [DOI] [PubMed] [Google Scholar]
- 36.Urabe Y, Hamada Y, Spinale FG, et al. Cardiocyte contractile performance in experimental biventricular volume-overload hypertrophy. Am J Physiol. 1993;264:H1615–H1623. doi: 10.1152/ajpheart.1993.264.5.H1615. [DOI] [PubMed] [Google Scholar]
- 37.Anand IS, Liu D, Chugh SS, et al. Isolated myocyte contractile function is normal in postinfarct remodeled rat heart with systolic dysfunction. Circulation. 1997;96(11):3974–3984. doi: 10.1161/01.cir.96.11.3974. [DOI] [PubMed] [Google Scholar]
- 38.Ikeda S, He A, Kong SW, et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol. 2009;29(8):2193–2204. doi: 10.1128/MCB.01222-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elia L, Contu R, Quintavalle M, et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation. 2009;120(23):2377–2385. doi: 10.1161/CIRCULATIONAHA.109.879429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu N, Bezprozvannaya S, Williams AH, et al. MicroRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22(23):3242–3254. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matkovich SJ, Wang W, Tu Y, et al. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res. 2010;106(1):166–175. doi: 10.1161/CIRCRESAHA.109.202176.. ▪ Demonstrates distinct patterns of miRNA expression before and after left ventricular assist device support.
- 42.Krichevsky AM, Gabriely G. MiR-21: a small multi-faceted RNA. J Cell Mol Med. 2009;13(1):39–53. doi: 10.1111/j.1582-4934.2008.00556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thum T, Gross C, Fiedler J, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456(7224):980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 44.Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF. MiR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci USA. 2009;106(29):12103–12108. doi: 10.1073/pnas.0811371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang K, Long B, Zhou J, Li PF. MiR-9 and NFATc3 regulate myocardin in cardiac hypertrophy. J Biol Chem. 2010;285(16):11903–11912. doi: 10.1074/jbc.M109.098004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakao K, Minobe W, Roden R, Bristow MR, Leinwand LA. Myosin heavy chain gene expression in human heart failure. J Clin Invest. 1997;100:2362–2370. doi: 10.1172/JCI119776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Callis TE, Pandya K, Seok HY, et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest. 2009;119(9):2772–2786. doi: 10.1172/JCI36154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng Y, Liu X, Zhang S, Lin Y, Yang J, Zhang C. MicroRNA-21 protects against the H2O2-induced injury on cardiac myocytes via its target gene PDCD4. J Mol Cell Cardiol. 2009;47(1):5–14. doi: 10.1016/j.yjmcc.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu C, Lu Y, Pan Z, et al. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J Cell Sci. 2007;120(17):3045–3052. doi: 10.1242/jcs.010728. [DOI] [PubMed] [Google Scholar]
- 50.Rane S, He M, Sayed D, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1a and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res. 2009;104(4):879–886. doi: 10.1161/CIRCRESAHA.108.193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ren XP, Wu J, Wang X, et al. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation. 2009;119(17):2357–2366. doi: 10.1161/CIRCULATIONAHA.108.814145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Divakaran V, Adrogue J, Ishiyama M, et al. Adaptive and maladptive effects of SMAD3 signaling in the adult heart after hemodynamic pressure overloading. Circ Heart Fail. 2009;2(6):633–642. doi: 10.1161/CIRCHEARTFAILURE.108.823070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duisters RF, Tijsen AJ, Schroen B, et al. MiR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009;104(2):170–178. doi: 10.1161/CIRCRESAHA.108.182535. [DOI] [PubMed] [Google Scholar]
- 54.Fernandez-Velasco M, Goren N, Benito G, Blanco-Rivero J, Bosca L, Delgado C. Regional distribution of hyperpolarization-activated current (If) and hyperpolarization-activated cyclic nucleotide-gated channel mRNA expression in ventricular cells from control and hypertrophied rat hearts. J Physiol. 2003;553(2):395–405. doi: 10.1113/jphysiol.2003.041954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luo X, Lin H, Pan Z, et al. Downregulation of miRNA-1/miRNA-133 contributes to re-expression of pacemaker channel genes HCN2 and HCN4 in hypertrophic heart. J Biol Chem. 2008;283:20045–20052. doi: 10.1074/jbc.M801035200. [DOI] [PubMed] [Google Scholar]
- 56.Terentyev D, Belevych AE, Terentyeva R, et al. MiR-1 overexpression enhances Ca(2+) release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56α and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ Res. 2009;104(4):514–521. doi: 10.1161/CIRCRESAHA.108.181651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang GK, Zhu JQ, Zhang JT, et al. Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur Heart J. 2010;31(6):659–666. doi: 10.1093/eurheartj/ehq013. [DOI] [PubMed] [Google Scholar]
- 58.Tijsen AJ, Creemers EE, Moerland PD, et al. MiR423–5p as a circulating biomarker for heart failure. Circ Res. 2010;106(6):1035–1039. doi: 10.1161/CIRCRESAHA.110.218297.. ▪ Shows that circulating miRNAs can serve as disease biomarkers in patients with heart failure.
- 59.Schipper ME, van Kuik J, de Jonge N, Dullens HF, de Weger RA. Changes in regulatory microRNA expression in myocardium of heart failure patients on left ventricular assist device support. J Heart Lung Transplant. 2008;27(12):1282–1285. doi: 10.1016/j.healun.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 60.Krutzfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438(7068):685–689. doi: 10.1038/nature04303.. ▪▪ First demonstration of mammalian miRNA knockdown using antagomir approach.
- 61.Esau C, Davis S, Murray SF, et al. MiR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3(2):87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 62.Elmen J, Lindow M, Schutz S, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452(7189):896–899. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 63.Suckau L, Fechner H, Chemaly E, et al. Long-term cardiac-targeted RNA interference for the treatment of heart failure restores cardiac function and reduces pathological hypertrophy. Circulation. 2009;119(9):1241–1252. doi: 10.1161/CIRCULATIONAHA.108.783852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duan R, Pak C, Jin P. Single nucleotide polymorphism associated with mature miR-125a alters the processing of pri-miRNA. Hum Mol Genet. 2007;16(9):1124–1131. doi: 10.1093/hmg/ddm062. [DOI] [PubMed] [Google Scholar]
- 65.Martin MM, Buckenberger JA, Jiang J, et al. The human angiotensin II type 1 receptor +1166 A/C polymorphism attenuates microRNA-155 binding. J Biol Chem. 2007;282(33):24262–24269. doi: 10.1074/jbc.M701050200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.Saunders MA, Liang H, Li WH. Human polymorphism at microRNAs and microRNA target sites. Proc Natl Acad Sci USA. 2007;104(9):3300–3305. doi: 10.1073/pnas.0611347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mishra PJ, Mishra PJ, Banerjee D, Bertino JR. MiRSNPs or MiR-polymorphisms, new players in microRNA mediated regulation of the cell: introducing microRNA pharmacogenomics. Cell Cycle. 2008;7(7):853–858. doi: 10.4161/cc.7.7.5666. [DOI] [PubMed] [Google Scholar]
- 68.Mishra PJ, Bertino JR. MicroRNA polymorphisms: the future of pharmacogenomics, molecular epidemiology and individualized medicine. Pharmacogenomics. 2009;10(3):399–416. doi: 10.2217/14622416.10.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.da Costa Martins PA, Bourajjaj M, Gladka M, et al. Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation. 2008;118(15):1567–1576. doi: 10.1161/CIRCULATIONAHA.108.769984. [DOI] [PubMed] [Google Scholar]
- 70.Rao PK, Toyama Y, Chiang HR, et al. Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ Res. 2009;105(6):585–594. doi: 10.1161/CIRCRESAHA.109.200451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mann DL. MicroRNAs and the failing heart. N Engl J Med. 2007;356(25):2644–2645. doi: 10.1056/NEJMcibr072068. [DOI] [PubMed] [Google Scholar]
- 72.van Rooij E, Olson EN. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest. 2007;117(9):2369–2376. doi: 10.1172/JCI33099. [DOI] [PMC free article] [PubMed] [Google Scholar]