

Abstract
Coffin–Lowry syndrome (CLS) is a rare form of X-linked mental retardation caused by mutations of the RSK2 gene, associated with cognitive impairment and skeletal malformations. We conducted the first morphometric study of CLS brain morphology by comparing brain volumes from two CLS families with healthy controls. Individuals with CLS consistently showed markedly reduced total brain volume. Cerebellum and hippocampus volumes were particularly impacted by CLS and may be associated with specific interfamilial RSK2 mutations. We provide preliminary evidence that the magnitude of hippocampus volume deviation from that of controls may predict general cognitive outcome in CLS.
Keywords: Coffin–Lowry syndrome, RSK2, MRI, Hippocampus, X-linked mental retardation
Introduction
Coffin–Lowry syndrome (CLS) is a rare form of X-linked mental retardation (XLMR). The syndrome is characterized by skeletal malformations, particularly involving the face and hands, growth retardation, hearing deficit, and increased risk for paroxysmal movement disorders as well as cognitive impairment in affected males and some carrier females. CLS is caused by mutations in a gene located at Xp22.2 that encodes ribosomal S6 kinase-2 (RSK2) [1-5]. RSK2 is expressed in various brain structures, including cortex, cerebellum, and hippocampus [6, 7]. Previous qualitative studies of gross neuropathology in CLS indicate that hydrocephalus, white matter lesions, abnormal gyrification, and dysgenesis of the corpus callosum can occur in affected individuals [8-10]. To date, no quantitative studies of brain morphology in CLS have been conducted.
In this report, we examined brain morphology associated with CLS in an effort to better understand the associations among genes, brain, and cognition. We obtained whole brain and regional volumes, including tissue specific, gray, and white matter measurements, from magnetic resonance brain images (MRI) acquired from two families with CLS. We then compared these volumes descriptively with those of age-and gender-matched typically developing controls.
Materials and methods
Participants
Six individuals from two families with CLS were recruited for this study through Greenwood Genetics Center in South Carolina. Family 1 consisted of a 31.6-year-old carrier woman and her two affected sons, ages 8.9 and 10.7 years. The mutation in family 1 is located in the N-terminal kinase catalytic domain in a conserved region of RSK2, 12 amino acids downstream of the first adenosine triphosphate (ATP) binding site. Family 2 consisted of fraternal-twin female carriers age 7.3 years and their affected brother, age 4.4 years. The mutation in Family 2 is located in the 3′end of the gene in close proximity to the externally regulated kinase (ERK) docking domain. Intellectual functioning (IQ) was assessed for all CLS participants using the Stanford Binet Intelligence Scale, fourth edition [11], with the exception of the 4-year-old child whose IQ was measured using the Developmental Profile II IQ Equivalent [12].
MRI acquisition
MRI scans for Family 1 were acquired at Self Regional Healthcare in Greenwood, South Carolina, and scans for Family 2 were acquired at Stanford University. Recent studies suggest that anatomical MRI data collected across sites using comparable acquisition procedures and equipment can be compatible [13]. All MRI scans used in this study were obtained with whole-body GE 1.5T Signa scanners (GE medical systems, Milwaukee) at Stanford and Self Regional Healthcare. Additionally, MRI scans of 58 age- and gender-matched typically developing controls were obtained from the structural neuroimaging database at Stanford University School of Medicine. Participants were excluded for MRI contraindications (e.g., orthodontia). Controls were selected after exclusion for any history of neurological, cognitive, or psychiatric disorders. Institutional Review Boards at both sites approved this study. Coronal brain images were acquired with an identical 3D volumetric radio frequency spoiled gradient echo pulse sequence using the following scan parameters: TR=35 ms, TE=6 ms, flip angle=45°, NEX=1, matrix size=256×192, field of view=24 cm, slice thickness=1.5 mm, and 124 contiguous slices.
MRI analysis
All image processing was completed at Stanford University. Scans conducted at Self Regional Healthcare were transferred to Stanford via optical disks. MRI scans were imported into BrainImageJava (cibsr.stanford.edu/tools) for semi-automated whole brain segmentation and quantification in the coronal plane. Data processing steps included removal of non-brain tissues from the images, correction of equipment-related image artifacts, segmentation of tissue components [gray, white, and cerebrospinal fluid (CSF)], normalization of image position, and parcellation of the cerebral cortex into lobe and subcortical regions based on a stereotaxic atlas template, as previously described and validated [14, 15]. The hippocampus was manually delineated for each participant in the coronal plane according to previously described methods [16], using gray-scale volume stacks derived from the whole brain analysis. However, only a subset of the typically developing controls had available hippocampus measurements for comparison (n=27). Interrater reliability obtained by interclass correlation exceeded 0.90 for all variables reported in this study.
Typically developing controls were grouped by age and gender based on the following conventions: young pediatric males (mean age=4.9±2.6, range=1.8–8.5, n=16); older pediatric males (mean age=9.5±1.1, range=8–10.8, n=12); pediatric females (mean age=7.8±0.8, range=5.7–8.7, n=17); and adult females (mean age=30.9±1.8, range=26.6–33.4, n=11). Means and standard deviations were calculated for each MRI volume for the control groups. Corrected, or proportional, volumes also were computed using the ratio of regional volumes to total brain volume (multiplied by 100) to determine differences that were disproportionate to overall brain volume. MRI volume results for the CLS families were compared to the appropriate control group by calculating z scores for each participant in the CLS families. Z scores that were at least ±1.5 were considered “significant”.
Results
Individuals with CLS, including carrier females and affected males, consistently demonstrated significantly reduced total brain volumes. This tended to include both gray and white matter as well as the cerebral lobes, particularly the temporal lobe (Fig. 1; Tables 1 and 2). Temporal lobe, cerebellum, and hippocampus volumes were particularly impacted by CLS. There was variation in the profile of these regions in CLS, with individuals showing disproportionately enlarged or reduced volumes. The hippocampus demonstrated a relatively consistent trend within a particular family with volumes being enlarged in Family 1 and reduced in Family 2 (Tables 1 and 2; Fig. 2).
Fig. 1.
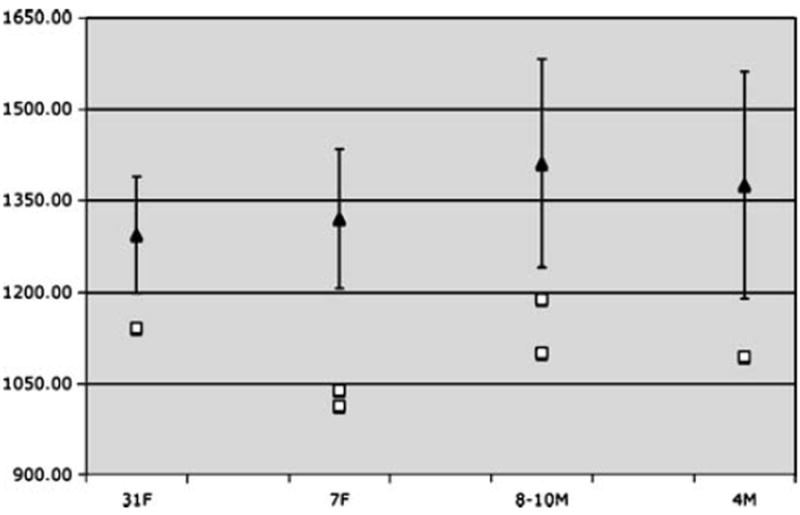
Mean typically developing control (black triangles with SD bars) and individual CLS (white squares) total brain volumes (shown in cm3) showing marked reduction in the CLS subjects compared to age- and gender-matched controls. 31F 31-year-old carrier woman; 7F 7-year-old carrier female children; 8–10M 8- and 10-year-old affected male children; 4M 4-year-old affected male child
Table 1.
Absolute brain volumes for female individuals with CLS and controls
| Adult female controls | 31F | z score | Pediatric female controls | 7F (T1) | z score | 7F (T2) | z score | |
|---|---|---|---|---|---|---|---|---|
| Age | 30.88 (1.76) | 31.59 | 7.76 (0.80) | 7.28 | 7.28 | |||
| TBV | 1,293.53 (95.0) | 1,140.81a | −1.61 | 1,319.57 (113.5) | 1,012.84a | −2.70 | 1,038.73a | −2.47 |
| Cerebral gray | 556.36 (54.6) | 513.16 | −0.79 | 651.68 (57.5) | 485.66a | −2.89 | 494.80a | −2.73 |
| Cerebral white | 447.78 (40.3) | 403.68 | −1.10 | 398.93 (47.7) | 263.02a | −2.85 | 280.23a | −2.49 |
| Cerebral CSF | 117.28 (19.1) | 70.82a, b | −2.43 | 99.47 (26.6) | 124.41 | 0.94 | 94.95 | −0.17 |
| Subcortical gray | 39.35 (4.8) | 36.01 | −0.70 | 44.08 (3.2) | 35.53a | −2.68 | 30.98a | −4.11 |
| Subcortical white | 46.20 (4.4) | 37.80a, b | −1.91 | 42.38 (5.6) | 28.30a | −2.54 | 29.60a, b | −2.30 |
| Ventricular CSF | 9.22 (2.0) | 6.53 | −1.34 | 7.32 (3.6) | 14.91a | 2.12 | 13.79a | 1.81 |
| Cerebellum | 119.29 (12.2) | 105.16 | −1.16 | 119.89 (11.3) | 99.77a | −1.78 | 118.40c | −0.13 |
| Frontal | 365.52 (27.9) | 339.60c | −0.93 | 381.72 (37.0) | 284.90a | −2.61 | 281.20a | −2.71 |
| Parietal | 252.17 (14.8) | 233.70 | −1.25 | 266.86 (24.7) | 205.90a | −2.47 | 231.70a, c | −1.42 |
| Temporal | 189.54 (13.9) | 163.70a, b | −1.86 | 199.93 (18.5) | 120.40a | −4.31 | 124.50a, b | −4.09 |
| Occipital | 111.39 (14.2) | 106.10 | −0.37 | 115.65 (17.7) | 73.70a | −2.37 | 77.10a, b | −2.18 |
| Hippocampus | 7.62 (0.61) | 7.15c | −0.77 | 6.59 (0.30) | 4.93a | −5.56 | 5.30a | −4.32 |
Control data shown as mean (standard deviation)
31F 31-year-old carrier woman; 7F 7-year-old carrier female children; family 1 relatives marked in bold; family 2 relatives marked in italics
Significantly different from controls (z score of absolute volume=±1.5)
Disproportionately reduced compared to controls (z score of proportional volume=−1.5)
Disproportionately enlarged (z score of proportional volume=+1.5)
Table 2.
Absolute brain volumes for male individuals with CLS and controls
| Older male controls | 8M | z score | 10M | z score | Younger male controls | 4M | z score | |
|---|---|---|---|---|---|---|---|---|
| Age | 9.47 (1.1) | 8.85 | 10.65 | 4.97 (2.7) | 4.39 | |||
| TBV | 714.66 (46.4) | 1,188.06a | −2.91 | 1,099.83a | −3.77 | 1,374.87 (185.8) | 1,093.53a | −1.51 |
| Cerebral gray | 468.54 (41.3) | 563.47a | −3.26 | 538.04a | −3.81 | 686.16 (83.8) | 564.24a | −1.46 |
| Cerebral white | 116.46 (35.8) | 352.01a | −2.82 | 331.57a | −3.32 | 400.48 (83.6) | 285.93 | −1.37 |
| Cerebral CSF | 47.90 (4.7) | 106.08 | −0.29 | 87.56 | −0.81 | 113.83 (25.0) | 112.90 | −0.04 |
| Subcortical gray | 45.24 (6.6) | 38.79a | −1.96 | 34.00a | −2.99 | 45.44 (7.0) | 33.35a, b | −1.73 |
| Subcortical white | 9.34 (3.2) | 33.50a | −1.79 | 34.00a | −1.72 | 38.66 (8.5) | 28.20 | −1.23 |
| Ventricular CSF | 130.88 (15.5) | 10.98c | 0.51 | 6.88 | −0.76 | 10.12 (6.7) | 12.99 | 0.43 |
| Cerebellum | 1,487.60 (102.8) | 118.94c | −0.77 | 102.91a | −1.81 | 120.40 (20.3) | 79.71a, b | −2.01 |
| Frontal | 422.22 (36.3) | 313.90a, b | −2.98 | 317.10a | −2.89 | 390.30 (66.1) | 327.30 | −0.95 |
| Parietal | 294.43 (28.7) | 224.40a | −2.44 | 220.80a | −2.57 | 274.83 (38.9) | 213.50a | −1.58 |
| Temporal | 231.13 (15.7) | 182.10a | −3.12 | 165.10a | −4.21 | 210.86 (30.5) | 151.90a, b | −1.93 |
| Occipital | 142.28 (12.8) | 122.80a | −1.53 | 98.70a | −3.42 | 126.57 (20.0) | 96.00a | −1.53 |
| Hippocampus | 7.30 (0.66) | 6.64c | −1.01 | 6.74c | −0.86 | 6.81 (0.87) | 4.39a, b | −2.76 |
Control data shown as mean (standard deviation)
8–10M 8- and 10-year-old affected male children; 4M 4-year-old affected male child; family 1 relatives marked in bold; family 2 relatives marked in italics
Significantly different from controls (z score of absolute volume=±1.5)
Disproportionately reduced compared to controls (z score of proportional volume=−1.5)
Disproportionately enlarged (z score of proportional volume=+1.5)
Fig. 2.
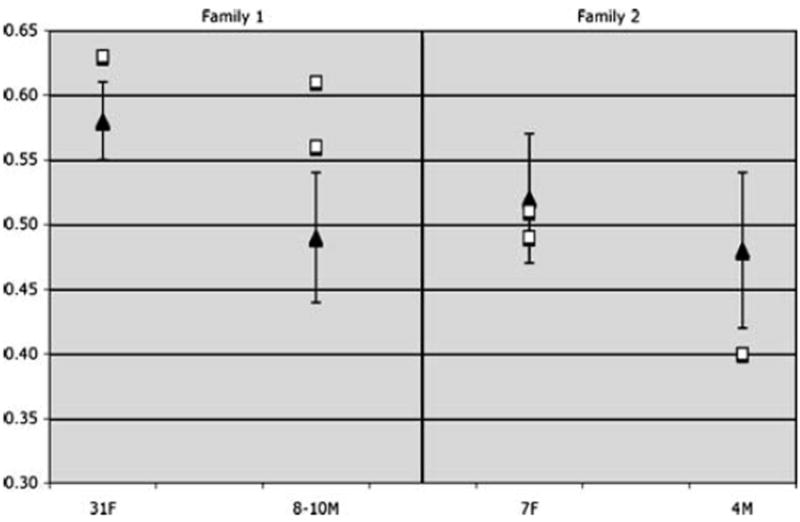
Mean typically developing (black triangles) and individual CLS (white squares) proportional hippocampus volumes demonstrating abnormal hippocampus morphology associated with CLS. Additionally, similar interfamilial pattern of hippocampus deviation from control means is noted with individuals in Family 1 showing enlarged hippocampi and individuals in Family 2 showing reduced hippocampi. 31F 31-year-old carrier woman; 7F 7-year-old carrier female children; 8–10M 8-and 10-year-old affected male children; 4M 4-year-old affected male child
Discussion
We report in this paper the first volumetric study of the neurodevelopment associated with Coffin–Lowry syndrome (CLS). These are very preliminary data based on case studies. However, as CLS and many other XLMR disorders occur infrequently, case studies offer a novel opportunity to investigate the potential impact of particular X-linked genes on neurodevelopment. We compared brain morphological findings from individuals with CLS with those from age- and gender-matched typically developing controls.
The most consistent finding across individuals with CLS was the reduced total brain volume compared to controls (Fig. 1). Decreased brain volume in CLS is likely to stem from disruption of core RSK2 protein functions associated with mutations of this gene. The RSK2 protein activates the transcription factor cyclic AMP response element binding protein (CREB) [17, 18], which in turn, regulates neuronal survival factors and is required for axonal growth, neuro-protection, and synaptic plasticity associated with long-term memory. Abnormalities in CREB function may result in increased apoptosis [19, 20]. Accordingly, our present results suggest that diffuse reduction in brain volume may be a neuroanatomical marker for decreased neuronal survival within the developing CLS brain.
Our findings further suggest that development of the temporal cortex, cerebellum, and hippocampus may be particularly affected by RSK2 mutations. RSK2 is known to be highly expressed in the human cerebellum and hippocampus [6]. The present study offers the first evidence, albeit preliminary, of morphological abnormalities of these structures in association with CLS. In Family 1 of the present study, the 8-year-old affected male child had disproportionately enlarged cerebellum, whereas his affected brother did not. The brothers’ cognitive abilities were similarly impaired, but the 8-year-old child demonstrated a 33% lower finger-tapping score, a measure of fine motor function [21]. Additionally, in family 2, the female carrier twin 2 (T2; Tables 1 and 2) appeared to have more serious morphological abnormalities compared to her sister, who also showed disproportionately enlarged cerebellum. Although finger-tapping scores were not available for this individual, she did demonstrate much lower IQ than her sister who did not demonstrate disproportionately large cerebellum volume. Therefore, abnormalities of cerebellum morphology in CLS may be associated with greater risk for certain cognitive deficits. However, replication of these findings with larger samples and more specific cognitive testing are required.
The impact of RSK2 on the temporal lobe, particularly the hippocampus, may involve signaling pathways responsible for learning and memory. Persistent neuronal stimulation activates a cascade of molecular components that results in transcription and translation of new proteins, which strengthen and increase the number of synapses [18, 22]. The RSK2 protein is directly involved in these signaling pathways [23, 24]. Thus, alteration of RSK2 protein function resulting from mutations in CLS may disrupt the mechanisms necessary for the development and maintenance of new synapses in the hippocampus, a structure characterized by high synaptic activity, which is crucial for cognitive function.
However, variation from normative hippocampus morphology was not consistent across the two families with CLS, with family 1 members manifesting enlarged volume (absolute and proportional) and family 2 relatives showing reduced absolute volume (Fig. 2; reduction in proportional hippocampus volume in the 4-year-old affected male child in family 2 approached our definition of significance: z=−1.39). This interfamilial resemblance of hippocampus morphology may initially suggest some genetic influence common to each family (unrelated to RSK2), although previous studies have shown that hippocampus volume is less heritable than other brain volumes [25] and is likely to be influenced by factors such as environmental stimulation and cortisol function [26]. It is possible that these divergent morphological findings may also be due, in part, to different RSK2 mutations giving rise to variations of RSK2 protein function in the two families. Family 1 relatives demonstrated a mutation of the N-terminal kinase catalytic domain of RSK2, while the mutation in family 2 was in close proximity to the ERK docking domain. CREB and ERK pathway function have both been directly implicated in hippocampus development and function [27-31]. While a hypothesis linking differences in RSK2 mutations to neuroanatomical variation is speculative at this point, further genotype–brain phenotype studies in CLS may provide more definitive data in the future.
Although hippocampus morphology was different between the two families, it is interesting to note that the correlation of the absolute value of hippocampus z scores (i.e., the magnitude of deviation from the control group mean) with IQ approached significance when examined across both families (r=−0.64, p=0.06, Supplementary Fig. 1). This finding suggests that in CLS, the degree of hippocampus abnormality, rather than the direction (i.e., increased or decreased volume), most accurately predicts functional outcome. Although the small sample size of the combined CLS group greatly limits the interpretability of these results, they are broadly consistent with the occurrence of hippocampus volume abnormalities in multiple populations with cognitive deficits, including Alzheimer’s disease, schizophrenia, and Down’s syndrome [32]. These findings are very preliminary and require replication with larger samples but give direction for future studies involving the brain morphology and cognitive outcome of CLS as well as other XLMR syndromes.
RSK2 mutations may modulate the risk for cognitive impairment in CLS through altered early neurodevelopment (i.e., reduced or enlarged volumes) in combination with disruptions in ongoing neural organization and plasticity via the learning and memory signaling pathways. It will be essential to study the morphology and function of specific brain regions involved in learning and memory (e.g., hippocampus, prefrontal cortex) to determine the potential impact of disrupted neuronal plasticity in CLS. Additionally, studies involving assessment of neuroanatomy, mutation type, and cognitive-behavioral outcome in CLS are required to elucidate essential links among gene, brain, and cognition. Continued studies combining genetic analysis, neuroimaging, and cognitive outcome with larger CLS samples hold promise for improving our understanding of the influence of X-linked genes on brain development and function.
Supplementary Material
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10048-007-0080-6) contains supplementary material, which is available to authorized users.
Contributor Information
Shelli R. Kesler, Center for Interdisciplinary Brain Sciences Research, Stanford University School of Medicine, 401 Quarry Road, MC5795, Stanford, CA 94305-5795, USA, skesler@stanford.edu
Richard J. Simensen, Greenwood Genetic Center, Greenwood, SC, USA
Kytja Voeller, Greenwood Genetic Center, Greenwood, SC, USA.
Fatima Abidi, Greenwood Genetic Center, Greenwood, SC, USA.
Roger E. Stevenson, Greenwood Genetic Center, Greenwood, SC, USA
Charles E. Schwartz, Greenwood Genetic Center, Greenwood, SC, USA
Allan L. Reiss, Center for Interdisciplinary Brain Sciences Research, Stanford University School of Medicine, 401 Quarry Road, MC5795, Stanford, CA 94305-5795, USA
References
- 1.Ahuja SR, et al. Coffin–Lowry syndrome. Indian J Pediatr. 2003;70(12):1001–1002. doi: 10.1007/BF02723830. [DOI] [PubMed] [Google Scholar]
- 2.Hanauer A, Young ID. Coffin–Lowry syndrome: clinical and molecular features. J Med Genet. 2002;39(10):705–713. doi: 10.1136/jmg.39.10.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simensen RJ, et al. Cognitive function in Coffin–Lowry syndrome. Clin Genet. 2002;61(4):299–304. doi: 10.1034/j.1399-0004.2002.610410.x. [DOI] [PubMed] [Google Scholar]
- 4.Stephenson JB, et al. The movement disorders of Coffin–Lowry syndrome. Brain Dev. 2005;27(2):108–113. doi: 10.1016/j.braindev.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 5.Touraine RL, Zeniou M, Hanauer A. A syndromic form of X-linked mental retardation: the Coffin–Lowry syndrome. Eur J Pediatr. 2002;161(4):179–187. doi: 10.1007/s00431-001-0904-6. [DOI] [PubMed] [Google Scholar]
- 6.Guimiot F, et al. Expression of the RSK2 gene during early human development. Gene Expr Patterns. 2004;4(1):111–114. doi: 10.1016/j.modgep.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Zeniou M, et al. Expression analysis of RSK gene family members: the RSK2 gene, mutated in Coffin–Lowry syndrome, is prominently expressed in brain structures essential for cognitive function and learning. Hum Mol Genet. 2002;11(23):2929–2940. doi: 10.1093/hmg/11.23.2929. [DOI] [PubMed] [Google Scholar]
- 8.Ozden A, et al. Callosal dysgenesis in a patient with Coffin–Lowry syndrome. Indian J Pediatr. 1994;61(1):101–103. doi: 10.1007/BF02753570. [DOI] [PubMed] [Google Scholar]
- 9.Coffin GS. Postmortem findings in the Coffin–Lowry syndrome. Genet Med. 2003;5(3):187–193. doi: 10.1097/01.GIM.0000067991.73943.4F. [DOI] [PubMed] [Google Scholar]
- 10.Kondoh T, et al. New radiological finding by magnetic resonance imaging examination of the brain in Coffin–Lowry syndrome. J Hum Genet. 1998;43(1):59–61. doi: 10.1007/s100380050038. [DOI] [PubMed] [Google Scholar]
- 11.Thorndike R, Hagen E, Sattler J. The Stanford–Binet intelligence scale. 4. Riverside Publishing; Chicago, IL: 1986. [Google Scholar]
- 12.Alpern G, Boll T, Shearer M. Developmental profile II manual. Western Psychological Services; Los Angeles, CA: 2000. [Google Scholar]
- 13.Ewers M, et al. Multicenter assessment of reliability of cranial MRI. Neurobiol Aging. 2006;27:1051–1059. doi: 10.1016/j.neurobiolaging.2005.05.032. [DOI] [PubMed] [Google Scholar]
- 14.Kates WR, et al. Automated Talairach atlas-based parcellation and measurement of cerebral lobes in children. Psychiatry Res. 1999;91(1):11–30. doi: 10.1016/s0925-4927(99)00011-6. [DOI] [PubMed] [Google Scholar]
- 15.Reiss AL, et al. Reliability and validity of an algorithm for fuzzy tissue segmentation of MRI. J Comput Assist Tomogr. 1998;22(3):471–479. doi: 10.1097/00004728-199805000-00021. [DOI] [PubMed] [Google Scholar]
- 16.Kates WR, et al. Reliability and validity of MRI measurement of the amygdala and hippocampus in children with fragile X syndrome. Psychiatry Res. 1997;75(1):31–48. doi: 10.1016/s0925-4927(97)00019-x. [DOI] [PubMed] [Google Scholar]
- 17.Weeber EJ, Levenson JM, Sweatt JD. Molecular genetics of human cognition. Mol Interv. 2002;2(6):376–391. 339. doi: 10.1124/mi.2.6.376. [DOI] [PubMed] [Google Scholar]
- 18.Johnston MV, Alemi L, Harum KH. Learning, memory, and transcription factors. Pediatr Res. 2003;53(3):369–374. doi: 10.1203/01.PDR.0000049517.47493.E9. [DOI] [PubMed] [Google Scholar]
- 19.Lonze BE, et al. Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron. 2002;34(3):371–385. doi: 10.1016/s0896-6273(02)00686-4. [DOI] [PubMed] [Google Scholar]
- 20.Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35(4):605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 21.Lezak M. Neuropsychological assessment. 3. Oxford University Press; New York, NY: 1995. [Google Scholar]
- 22.Sweatt JD, Weeber EJ, Lombroso PJ. Genetics of childhood disorders: LI. Learning and memory, part 4. Human cognitive disorders and the ras/ERK/CREB pathway. J Am Acad Child Adolesc Psychiatry. 2003;42(6):741–744. doi: 10.1097/01.CHI.0000046859.56865.A8. [DOI] [PubMed] [Google Scholar]
- 23.Harum KH, Alemi L, Johnston MV. Cognitive impairment in Coffin–Lowry syndrome correlates with reduced RSK2 activation. Neurology. 2001;56(2):207–214. doi: 10.1212/wnl.56.2.207. [DOI] [PubMed] [Google Scholar]
- 24.Woo MS, et al. Ribosomal S6 kinase (RSK) regulates phosphorylation of filamin A on an important regulatory site. Mol Cell Biol. 2004;24(7):3025–3035. doi: 10.1128/MCB.24.7.3025-3035.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sullivan EV, et al. Heritability of hippocampal size in elderly twin men: equivalent influence from genes and environment. Hippocampus. 2001;11(6):754–762. doi: 10.1002/hipo.1091. [DOI] [PubMed] [Google Scholar]
- 26.Toga AW, Thompson PM. Genetics of brain structure and intelligence. Annu Rev Neurosci. 2005;28:1–23. doi: 10.1146/annurev.neuro.28.061604.135655. [DOI] [PubMed] [Google Scholar]
- 27.Countryman RA, et al. CREB phosphorylation and c-Fos expression in the hippocampus of rats during acquisition and recall of a socially transmitted food preference. Hippocampus. 2005;15(1):56–67. doi: 10.1002/hipo.20030. [DOI] [PubMed] [Google Scholar]
- 28.Hao Y, et al. Mood stabilizer valproate promotes ERK pathway-dependent cortical neuronal growth and neurogenesis. J Neurosci. 2004;24(29):6590–6599. doi: 10.1523/JNEUROSCI.5747-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu B, et al. Changes in trkB-ERK1/2-CREB/Elk-1 pathways in hippocampal mossy fiber organization after traumatic brain injury. J Cereb Blood Flow Metab. 2004;24(8):934–943. doi: 10.1097/01.WCB.0000125888.56462.A1. [DOI] [PubMed] [Google Scholar]
- 30.Mantamadiotis T, et al. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet. 2002;31(1):47–54. doi: 10.1038/ng882. [DOI] [PubMed] [Google Scholar]
- 31.Ying SW, et al. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci. 2002;22(5):1532–1540. doi: 10.1523/JNEUROSCI.22-05-01532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geuze E, Vermetten E, Bremner JD. MR-based in vivo hippocampal volumetrics: 2. Findings in neuropsychiatric disorders. Mol Psychiatry. 2005;10(2):160–184. doi: 10.1038/sj.mp.4001579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.