

Abstract
The identification of several families of innate pattern recognition receptors has greatly enhanced our understanding of the host innate immune response against a variety of pathogens. One such family of innate receptors is the NOD-like receptors (NLRs). NOD2, a member of the NLRs, was characterized as a cytosolic sensor of bacteria peptidoglycan (PGN). For almost 10 years, NOD2 was assigned with the function of mediating the RICK- and NF-κB induced pro-inflammatory response triggered by PGN. Recent studies have extended the biological activity of NOD2 to include the induction of autophagy and antiviral responses, as well as mediating direct T cell activation. Here, we highlight and discuss these new findings in the context of immune activation and pathogen detection.
Pathogen sensing
Multicellular organisms are exposed to a variety of microbes, most are innocuous but some are potentially detrimental to the well being of the host. The innate immune system, the first line of defense against deleterious microorganisms, is delegated with the duty of detecting and removing these potentially pathogenic microbes. Once triggered, the innate immune response is fully activated within minutes; unlike the adaptive immune response, which requires days to reach maturity. The immediacy of the innate immune system can be attributed to rapid pathogen detection mediated by the host germline-encoded pathogen recognition receptors (PRRs). PRRs recognize conserved microbial structures often referred to as pathogen-associated molecular patterns (PAMPs). Currently, there are several described families of PRRs. Perhaps the best-known are the Toll-like receptors (TLRs). TLRs, which are localized at the cell surface, or within endosomes, recognize microbial structures from Gram-positive and -negative bacteria, RNA and DNA viruses, fungi, and protozoans [1]. Several additional families of innate receptors have been identified, including the intracellular nucleotide-binding domain and leucine rich repeat containing receptors (NLRs) and the retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs). To date, NLRs are primarily activated in response to bacterial products, while RLRs are helicases that typically sense viruses [2, 3]. The existence of multiple PRRs capable of recognizing a single microorganism ensures the maximum induction of immune response and provides redundancy even when a sensor or its signaling pathway is a target of inhibition by the pathogen. The activation of PRRs results in the activation of multiple signaling pathways, including nuclear factor kappa B (NF-κB), mitogen-activated protein kinases (MAPKs), and the type I interferon (IFN) response, which lead to the induction of inflammatory and immune-specific gene expression [1–3]. This review focuses on the NLR family member NOD2 and recent findings describing novel NOD2 functions, such as viral recognition, autophagy and T cell activation, in the context of immune activation and bacteria sensing.
NOD2 in sensing bacteria
NOD2 was first identified almost 10 years ago [4], and is one of the most well studied members of the NLR family. Initially, NOD2 expression was reported to be relatively tissue-specific and confined to antigen-presenting cells (APCs) such as monocytes, macrophages, and dendritic cells (DCs) and certain epithelial cells [2]. More recently, in some other cell types, it appears that basal levels of NOD2 expression are typically quite low but can be induced by a variety of inflammatory signals such as LPS, TNF-α, and IFN-γ [5, 6]. This type of crosstalk with other PRR and cytokine signaling pathways represents an additional regulatory layer by which to control NOD2 signaling. Indeed, several recent reports have shown that NOD2 expression can be induced by inflammatory signals in other cell types including dental pulp tissue [7] and a variety of epithelial cell types such as ophthalmic [8], kidney [9], liver [10], and lung [11].
Initial biochemical characterization of NOD2 revealed that it induced NF-κB activation in a TLR-independent manner [12, 13]. Further studies showed that NOD2 is activated by molecules produced during the synthesis and/or degradation of bacterial peptidoglycan (PGN). NOD2 is activated by muramyl dipeptide (MDP), a PGN motif present in all Gram-positive and –negative bacteria [14, 15]. Based on the molecular mechanism of other NLR receptors, it is generally accepted that NOD2 undergoes conformational changes during ligand-induced activation resulting in self-oligomerization. This is followed by the recruitment and activation of the serine threonine kinase RICK (also called RIP2), which is essential for activation of the NF-κB and MAPK signaling pathways, leading to the transcription of immune response genes [16]. The inability of RICK null cells or mice to activate NF-κB signaling downstream of NOD2 indicates that RICK is essential for the signaling and the induction of inflammatory responses [12, 17]. Therefore MDP and/or bacterial stimulation activates the ‘classical’ RICK-dependent NOD2 signaling pathway.
Based on our current understanding of the NOD2 signaling pathway (Figure 1), one would predict that a variety of bacterial pathogens are capable of activating RICK via NOD2. Consistent with this idea, in vitro studies have shown NOD2 activation by pathogens including Streptococcus pneumoniae [18], Staphyrococcus aureus [14], Salmonella enterica serovar Typhimurium [19], Shigella flexneri [20], and Mycobacterium tuberculosis [21]. Moreover, several reports have shown that NOD2-deficient mice are more susceptible to bacterial infection in vivo. For example, Nod2−/− mice had decreased production of type I cytokines and reduced recruitment of CD4+ and CD8+ T cells during Mycobacterium bovis bacillus Calmette-Guerin infection [22]. In addition, NOD2- and RICK-null mice infected with Chlamydophila pneumoniae exhibited impaired NO and CXCL1 production and delayed neutrophil recruitment to the lungs [23]. Lastly, NOD2 has been shown to be important for bacterial recognition and host defense against L. monocytogenes when TLR signaling is inhibited after prolonged exposure to TLR ligands and E. coli in vivo [17].
Figure 1.
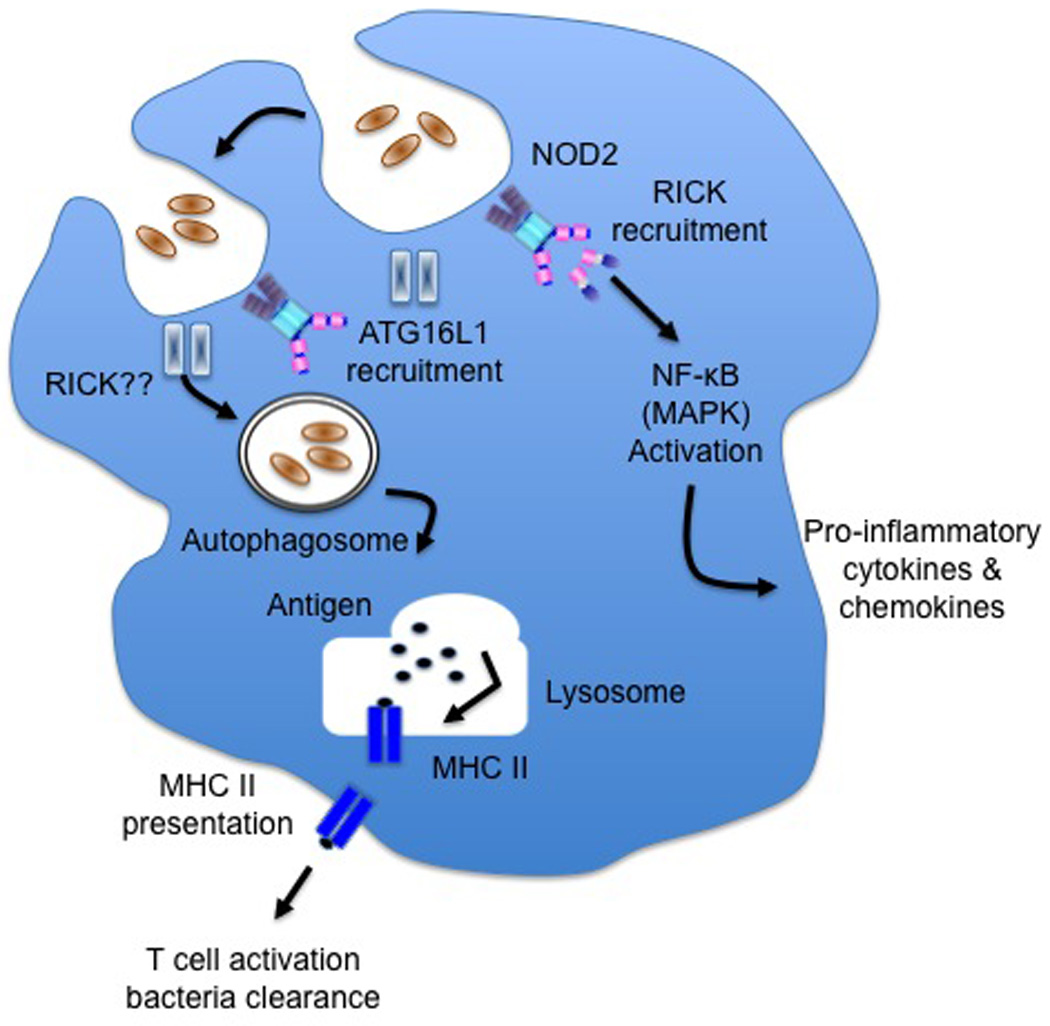
NOD2 activates the autophagy pathway. Bacteria is engaged by NOD2 and functions to recruit ATG16L1 to the plasma membrane. The interaction of NOD2 and ATG16L1 leads to the formation of autophagosome. The fusion of the autophagosome with the lysosome promotes the processing/loading of bacterial peptide onto MHC II molecule. The peptide-MHC II complex leads to appropriate T cell activation and bacteria clearance. Consistent with the role of NOD2 induction of autophagy is the finding that NOD2 mutation fails to induce autophagy.
NOD2-mediated recognition of commensal microbiota impacts host immunity
In addition to detecting bacterial pathogens, recent studies have suggested that NOD2 signaling is required for the regulation of commensal microbiota. Deficiencies in NOD2 or RICK lead to increased bacterial load of certain strains of commensal bacteria in the terminal ileum [24]. This phenomenon might be associated with impaired bacteria killing activity due to blunted α-defensin production by Paneth cells within the ileal crypts of the NOD2-deficient host [24, 25]. Although NOD2 signaling appears to control the number and composition of commensal microbiota, the microbiota itself can regulate the expression of NOD2. The expression of NOD2 and RICK, but not MyD88 or Tirap – adaptor proteins that are important for TLR signaling – was lower in terminal ileum of germ-free mice compared with specific pathogen free, SPF, mice [24]. In addition, NOD2 expression in the intestine of germ-free mice was restored following reconstitution with commensal bacteria, such as Lactobacillus plantarum or non-pathogenic E. coli, suggesting that commensal microbiota positively regulate NOD2-RICK signaling [24]. These observations indicate that a mutual balance exists between NOD2 and commensal bacteria, creating a negative feedback loop in which gut-residing bacteria positively regulate NOD2 and its associated signaling molecules, which in turn negatively regulate commensal flora. These findings raise more questions then they answer. It is unclear whether the in vivo increase in bacteria load observed in the Nod2−/− mice is truly a consequence of impaired microbicidal activity of the Paneth cells, because the bacterial load in germ-free Nod2−/− mice after mono-colonization with a relevant commensal bacterial has yet-to-be examined. It would also be of interest to determine whether the composition of the microbiota of patients with NOD2 mutations is skewed towards one or more specific bacterial species. Nevertheless, it is tempting to speculate that mutations observed in the NOD2 gene in Crohn’s disease (CD) are associated with alterations in the composition and bacteria load in the intestine that may facilitate disease pathogenesis.
The significance of the intestinal microbial flora for the pathogenesis of acute graft-versus-host disease (GVHD) was first discovered in experimental mouse models and confirmed in human trials in the 1970s [26, 27]. GVHD is a potentially lethal complication in patients undergoing allogeneic hematopoietic stem cell transplantation (HSCT). It is characterized by damage of epithelial surfaces in target organs, such as the liver, skin and intestine, which is caused by alloreactive donor T cells that recognize recipient tissue antigens. A common feature of the primary GVHD target tissues is their exposure to microbes and microbial products through the epidermis, intestinal mucosa, and portal circulation. Recently, single nucleotide polymorphisms (SNPs) in the NOD2 gene have been associated with a higher incidence of GVHD [28, 29]. How does NOD2 regulate the development of GVHD? It was observed that in Nod2−/− allogeneic bone marrow (allo-BM) transplant recipients there was an increase in both the severity and incidence of GVHD in both MHC-mismatched and MHC-matched models [30]. It was further demonstrated that NOD2 deficiency in the hematopoietic system aggravates both experimental GVHD as well as experimental trinitrobenzene sulfonate colitis [30]. Finally, the DCs of Nod2−/− allo-BM transplant recipients exhibited increased activation and function leading to enhanced proliferation and activation of allogeneic donor T cells, resulting in target organ damage. These data support the hypothesis that recognition of commensal microbiota through NOD2 in antigen-presenting cells, such as DCs, contributes to the prevention of GVHD. However, the contribution of NOD2 in GVHD remains controversial given that the association of SNPs in NOD2 with GVHD was not found by all [28, 29, 31, 32]. The conflicting results between the different clinical studies are most probably explained by differences between the study cohorts including the NOD2 SNP frequency, intestinal decontamination with antibiotics prior to and after transplant, and environmental factors such as diet and variations in the exposure level and the type of microbes. Despite the discrepancies in data on the role of NOD2 in mediating GVHD, the importance of commensal bacteria for disease pathogenesis is underscored by the reduced severity of GVHD in germ-free mice or when intestine decontamination was performed to minimize the level of bacteria stimulation [33, 34]. Taken together these observations support an emerging role for NOD2 as a key sensor of commensal microbiota in the gut and a regulator of host intestinal homeostasis and inflammation.
NOD2 controls bacterial infection via induction of autophagy
Recent findings provide evidence that NOD2-mediated bacterial sensing is linked to the induction of autophagy. Autophagy is a highly conserved self-degradation system that plays an important role in maintaining cellular homeostasis through the elimination of misfolded proteins and damaged organelles [35]. The core pathway of mammalian autophagy begins with the formation of an isolation membrane (also called phagophore), followed by the transport and maturation of the autophagosome, which ultimately fuses with the lysosome to degrade their cargo and recycle biomolecules to the cytoplasm [36]. In addition to these housekeeping duties, autophagy is crucial for host defense against bacterial, viral, and parasitic pathogens [37]. MDP induces autophagy in non-myeloid cells, such as fibroblasts and epithelial cells, or myeloid monocytic cells, such as macrophages and DCs, in a NOD2-dependent manner [38–40]. Consistent with ligand-induced autophagy, infection with invasive bacteria sensed by NOD2 also triggers autophagy, thus contributing to NOD2-mediated host defense [38–40]. The role of RICK in NOD2-dependent autophagy is currently unclear. Two independent studies suggest a crucial role for RICK [38, 39], while another study demonstrated that induction of autophagy is unimpaired in RICK-deficient cells [40]. Nevertheless, all three studies demonstrate that NOD2-induced autophagy is not dependent on NF-κB signaling that is ‘classically’ associated with NOD2 activation [38–40]. NOD2 autophagy induction requires interaction between NOD2 and a known autophagy protein, ATG16L1 [40]. Like NOD2, polymorphisms in ATG16L1 are also associated with increased risk for CD [38, 39]; NOD2 promotes the recruitment of ATG16L1 to the bacterial entry site in human epithelial cell lines or murine fibroblasts and macrophages [40]. It has also been reported that autophagy in turn enhances the MDP-induced NOD2 signaling thus creating a positive feedback loop during intracellular bacterial infection [39]. Consistent with this idea, NOD2-dependent NF-κB activation in human colonic epithelial cells is decreased by the inhibition of the early autophagic response or lysosomal fusion, suggesting that autophagy positively regulates NOD2 signaling. This implicates autophagy in the intracellular trafficking of MDP for NOD2 signaling. However, this is inconsistent with normal NOD2-induced cytokine secretion that is observed by Atg16l1 null macrophages after MDP stimulation. This raises the interesting question of whether the mechanism of MDP uptake and subsequent intracellular trafficking is cell-type specific? The finding that NOD2 plays a regulatory role in the induction of autophagy is certainly novel. However, these initial characterizations of NOD2 were performed in vitro utilizing either human cell lines or cytokine and/or growth factor-derived DCs and macrophages; therefore, care must be taken to extrapolate these data to an in vivo setting. Is NOD2-mediated autophagy more relevant in cells such as DCs and macrophages than other cell types? Recently, it has been demonstrated that autophagy-associated genes are crucial for Paneth cell function, as ATG16L1-deficient Paneth cells exhibit granule abnormalities [41]. It will be interesting to investigate how NOD2-autophagy interaction affects Paneth cell function. In addition, it has been shown that autophagy dampens inflammasome activation, and ATG16L1-deficient mice produce higher amounts of IL-1β and IL-18 following LPS stimulation [42]. Could CD patients with defective NOD2-mediated autophagy lack this key modulatory function? Despite raising more questions, these studies have narrowed the gaps in our knowledge regarding the pathogenesis of CD.
Alternative role of NOD2 in host defense
NOD2 is ‘classically’ described as a MDP ‘sensor’ with RICK acting as a critical signaling intermediate. No data, however, have demonstrated direct NOD2-MDP binding. So does NOD2 have biological functions other than sensing bacterial-derived PGN? A recent study challenged the classic function of NOD2 by highlighting that single-stranded RNA (ssRNA) viruses, which do not contain PGN, also activate NOD2, leading to IRF3-dependent type I IFN secretion and antiviral immunity [43]. It was demonstrated in vitro that NOD2 facilitated type I IFN production after infection with viruses containing a ssRNA genome such as, respiratory syncytial virus (RSV), vesicular stomatitis virus (VSV), or influenza virus [43]. Furthermore, it was observed, in a cell-free assay, that NOD2 specifically interacts with RSV ssRNA but not with control mRNA. The biological significance of this NOD2-mediated IRF3 activation is underscored by the enhanced susceptibility of NOD2-deficient animals following infection with RSV and influenza virus [43]; however, it remains to be determined whether humans with NOD2 mutations exhibit increased susceptibility to certain viral pathogens. RICK might not be important in type I IFN induction by NOD2, as silencing of endogenous RICK expression does not alter NOD2-mediated production of IFN-β in RSV-infected cells [43]. Interestingly it was found that viral activation of NOD2 requires MAVS, the mitochondrial antiviral signaling protein that mediates RIG-I- and MDA5-induced IRF3 activation [43]. During viral infection an increased fraction of NOD2 relocalizes to the mitochondria, where it associated with MAVS [43]. The interaction between NOD2 and MAVS appears to be the essential event linking recognition of ssRNA to IRF3 activation, because the loss of MAVS impaired NOD2-dependent interferon production following infection with influenza virus [43]. This mode of NOD2 interacting with MAVS is reminiscent of the binding of NLRX1, a mitochondrial NLR protein that inhibits MAVS and diminishes IRF3 activation [44]. Taken together, these recent findings demonstrate the existence of an ‘alternative’ pathway (Figure 2) for triggering a NOD2-mediated immune response that is RICK-independent, but MAVS-dependent.
Figure 2.
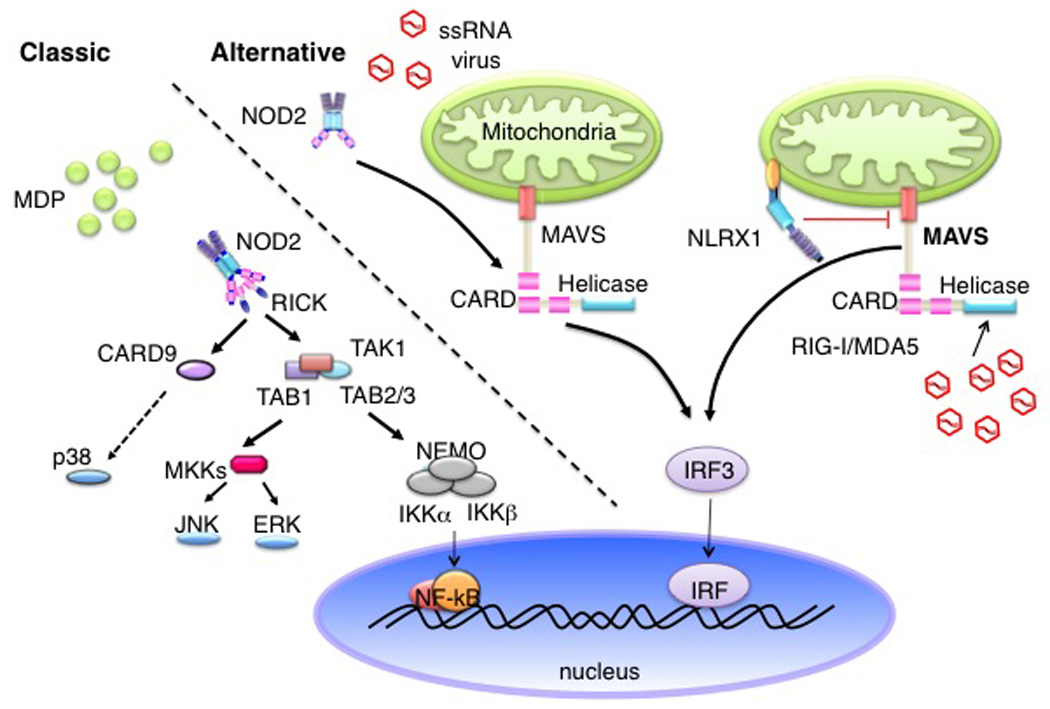
‘Classic’ versus ‘alternative’ pathways activated by NOD2. The classical pathway is identified by the engagement of NOD2 by MDP, which involves the kinase, RICK. As a consequence this leads to the activation of NF-κB, leading to inflammatory gene expression. The ‘alternative’ pathway involves ssRNA virus, where ssRNA interacts with NOD2, which binds to MAVS present on the mitochondria; leading to IRF3 activation and IFN gene expression that is RICK-independent. NLRX1 is an NLR family member that inhibits MAVS at the mitochondrial membrane.
Role of NOD2 in adaptive immune responses
A key role for NOD2 in innate immune defense has been well established. Only a few studies, however, have examined the contribution of NOD2 to the induction adaptive immune response, despite the fact that MDP is the ‘minimum’ effective component of complete Freund’s adjuvant that promotes adaptive immune responses [45–47].
It has been demonstrated that MDP-induces the production of IL-17 from human memory CD4+ T cells, but not naïve CD4+ T cells, in a NOD2-dependent manner. The target of MDP in this model was shown to be DCs. MDP stimulation of DCs alone was not sufficient to induce IL-17 production by T cells. In contrast, MDP in combination with other TLR agonists enhanced the capacity of DCs to specifically promote IL-17 secretion by memory CD4+ T cells [48]. Mechanistically, MDP and TLR co-stimulation of DCs induces the production of IL-23p19 and IL-1α/β, cytokines that are important for promoting IL-17 production by activated/memory T cells [48]. Therefore, the role of MDP and TLR co-stimulation may not be to promote the differentiation of naïve CD4+ T cells into IL-17-producing cells per se, but rather to expand the memory pool of IL-17 producing CD4+ cells through mediators such as IL-23 and IL-1.
In mice, the role of MDP-stimulated NOD2 activation in driving antigen (Ag)-specific responses was explored by two independent studies, which resulted in distinct findings and conclusions. In one study, NOD2 was demonstrated to trigger a potent Ag-specific immune response that was biased for Th2 polarization, characterized by the induction of IL-4 and IL-5 by T cells and an IgG1 antibody response [49]. In this same study, NOD2 was also critical for the induction of both Th1 and Th2-type responses following costimulation with TLR agonists [49]. In a separate study, the adjuvant properties of the MDP-NOD2 pathway were systematically tested under various immunization regimes [50]. This study concluded that MDP by itself, compared to the TLR agonist, LPS, has a marginal adjuvant effect for IgG1 and by itself cannot stimulate IgG2b or IgG2c production [50]. Interestingly, it was observed in the same study that NOD2 was required for an optimal IgG1 and IgG2c response in the absence of exogenous TLRs or NLR agonists, suggesting a MDP-independent pathway of NOD2 activation.
As alluded to above, activation of the MDP-NOD2 pathway in DCs can stimulate the development of an adaptive immune response. But how does MDP-NOD2 signaling in DCs promote the generation of an adaptive immune response? One possibility is that NOD2 regulates the antigen presentation capacity of DCs through autophagy [38–40]. Autophagy is responsible for the delivery of cytosolic proteins for MHC class II presentation in DCs [38]. It was demonstrated that NOD2 autophagy induction influences NOD2-mediated MHC class II, but not MHC class I, trafficking, surface expression and MHC class II antigen presentation in DCs [38]. Consistent with this observation, the antigen-specific proliferation of autologous CD4+ T cells was reduced after siRNA-mediated silencing of NOD2 or ATG16L1 in DCs. More recently, Nod signaling was shown to enhance Ag cross-presentation, or cross priming, by DCs [51]. Ag cross-presentation denotes the ability of DCs to endocytose, process and present extracellular antigens with MHC class I molecules to CD8+ T cells to generate cytotoxic CD8+ T cell (CTL) immunity to tumors and viral infections that do not infect DCs. In this study, DCs stimulated with MDP exhibited increased expression of co-stimulatory and cross-presentation-associated molecules [51]. In addition, injection of Nod agonist into wild type mice significantly enhanced the ability of DCs cells to cross-prime Ag-specific CD8+ T cells as indicated by the increased CTL activity against peptide-pulsed target cells, as well as in vivo anti-tumor CTL activity [51]. Taken together, there is accumulating evidence suggesting the NOD2 signaling in antigen-presenting cells, particularly DCs, can positively influence the maturation of the host adaptive immune response. One question that remains is whether or not NOD2 can function in other cells types to regulate adaptive immune responses?
Recently it was demonstrated that NOD2 functions in a T cell autonomous manner to regulate adaptive immunity [52]. The initial observation that Nod2−/− mice exhibit enhanced susceptibility following Toxoplasma gondii (T. gondii) infection suggested that NOD2 may serve as a ‘sensor’ for this intracellular parasite that lacks PGN. In contrast to the role of NOD2 in bacterial infection, in vitro and in vivo APC functions, such as cytokine production and antigen presentation, were normal in the absence of NOD2 following T. gondii infection. Thus it was hypothesized that NOD2 was intrinsically required for T cell function (Figure 3). In vitro and in vivo experiments revealed this to be the case, with a specific defect in the ability of NOD2-deficient T cells to produce IFN-γ following infection with this parasite [52]. In addition to impaired resistance against T. gondii infection, NOD2 was found also required for lymphopenia-induced homeostatic T cell proliferation and in a T cell mediated colitis model [52]. The impaired T cell function in the absence of NOD2 was also associated with impaired production of IL-2, which plays a critical role in T cell priming for IFN-γ production [53]. Given that NOD2-deficient T cells are phenotypically similar to T cells lacking the co-stimulatory molecule, CD28, could NOD2 regulate T cell activity by acting downstream of CD28 signaling pathway? In vitro biochemical and functional studies suggest a scaffolding role for NOD2 in the pathway of CD28 stimulation for c-Rel-mediated IL-2 production, thereby positively regulating T cell activation and differentiation [52]. The finding that NOD2 directly regulates T cell function might seem to contradict with previous studies demonstrating normal proliferative potential of NOD2-deficient T cells in a GVHD model [30], normal Th2 induction after immunization with NOD1 agonist [49] and antibody production after treatment with TLR7 agonist [54] in the absence of NOD2. In the GVHD study [30], it is likely that any effect CD28 signaling might have on cellular proliferation could have been ‘masked’ by the proliferative signals generated through the T-cell receptor. This idea is consistent with the observation that CD28−/− T cells exhibit normal proliferation and are able to reconstitute irradiated recipients [55]. This is also in agreement with the finding that NOD2-mediated effects on T cell expansion is only revealed utilizing competitive adoptive transfer experiments [52]. The discrepancy in the requirements for NOD2 in generating effective adaptive immunity in these studies might be due to the use of different adjuvants, which could differentially influence the expression of other B7-related costimulatory molecules [56] and/or tumor necrosis factor receptor superfamily molecules [57] on antigen-presenting cells. Therefore, the influence of NOD2 on adaptive immune responses in vivo might be determined by whether or not CD28 signaling is ‘preferentially’ required. The finding that NOD2 functions in a T cell autonomous manner raises several issues. First, does NOD2 mediate signals exclusively downstream of CD28, or other families of co-stimulatory molecules, expressed on T cells? Next, studies have demonstrated that c-Rel that is activated in response to TCR and CD28 triggering binds to regulatory element present in the Foxp3 promoter region to facilitate Foxp3 expression and to promote T-regulatory (Treg) cell differentiation [58–63]. These findings lead one to speculate that the dysregulated immune response observed in the absence of NOD2 is the result of impaired Treg differentiation due to reduced CD28-mediated c-Rel activity. Lastly, do other NLR members, in addition to NOD2, directly regulate T cell function? Taken together, the data demonstrating T cell intrinsic role of NOD2 is in line with the increasing evidence that innate immune receptors can directly regulate T cell function and activation during inflammation [64].
Figure 3.
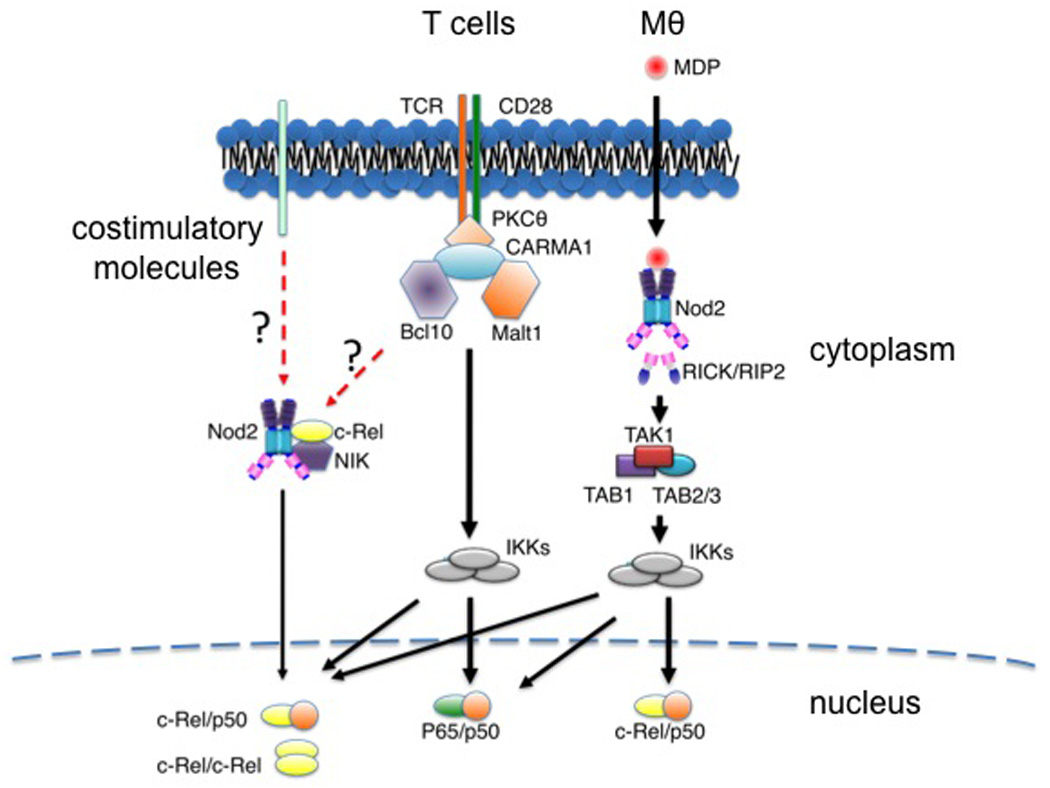
NOD2 functions in T cells and macrophages (Mθ). TCR and CD28 signaling triggers the activation of NOD2. In T cells, NOD2 interacts with NF-kB inducing kinase (NIK) and c-Rel to form a complex that promotes c-Rel nuclear accumulation. The molecular mechanisms that link TCR, CD28 and NOD2 are unknown. Similarly, it is unclear whether NOD2 function in T cells is exclusive to CD28 or is shared by other costimulatory molecules.
Concluding remarks
Mutations in NOD2 were initially identified to be associated with an increased risk for CD. Early research lead to working models of how variant NOD2 proteins lead to the enhanced inflammation: (1) NOD2 is a positive regulator of the innate immune response and defective NOD2 results in a heightened inflammatory response due to blunted bacteria clearance [54, 65] (2) NOD2 is a negative regulator of TLR signaling [66]. According to this latter model, the lack of NOD2 results in enhanced production of Th1 polarizing cytokine following TLR stimulation [66]. Despite the synthesis of these models, the mechanism by which variant NOD2 proteins lead to the enhanced inflammation associated with CD remains enigmatic. It is clear from the recent data, NOD2 has a more multifaceted role during an immune response than first imagined. According to the recent findings, the immunological output triggered by NOD2 is critically dependent on not only the stimuli but also on the responding cell types. This diverse function of NOD2, in part, could explain why CD is a multi-factorial inflammatory bowel disease. Altogether, these new and exciting discoveries raise additional questions concerning the various potential roles of NOD2 and the possibility that the functions of other NLR proteins are more diverse than previously assumed.
Acknowledgments
We apologize to investigators whose work was not explicitly cited due to space constrains. The authors' work is supported by NIH grants DK61707, AR051790, AI063331, AR059688 and DK091191. M. H. S. was supported by training grant 2T32 HL007517 from the NIH, N. K. by a Fellowship from the Uehara Memorial Foundation, Y.-G. Kim by training funds from the University of Michigan Comprehensive Cancer Center. N.W. is supported by Center for Genetics in Health and Medicine (CGHM) Postdoctoral Fellowship Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 2.Franchi L, et al. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. 2009;227:106–128. doi: 10.1111/j.1600-065X.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- 4.Ogura Y, et al. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 5.Gutierrez O, et al. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J Biol Chem. 2002;277:41701–41705. doi: 10.1074/jbc.M206473200. [DOI] [PubMed] [Google Scholar]
- 6.Rosenstiel P, et al. TNF-alpha and IFN-gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology. 2003;124:1001–1009. doi: 10.1053/gast.2003.50157. [DOI] [PubMed] [Google Scholar]
- 7.Lin ZM, et al. Expression of nucleotide-binding oligomerization domain 2 in normal human dental pulp cells and dental pulp tissues. J Endod. 2009;35:838–842. doi: 10.1016/j.joen.2009.03.047. [DOI] [PubMed] [Google Scholar]
- 8.Scurrell E, et al. Immunohistochemical detection of NOD1 and NOD2 in the healthy murine and canine eye. Vet Ophthalmol. 2009;12:269–275. doi: 10.1111/j.1463-5224.2009.00698.x. [DOI] [PubMed] [Google Scholar]
- 9.Shigeoka AA, et al. Nod1 and nod2 are expressed in human and murine renal tubular epithelial cells and participate in renal ischemia reperfusion injury. J Immunol. 2010;184:2297–2304. doi: 10.4049/jimmunol.0903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott MJ, et al. Hepatocytes express functional NOD1 and NOD2 receptors: a role for NOD1 in hepatocyte CC and CXC chemokine production. J Hepatol. 2010;53:693–701. doi: 10.1016/j.jhep.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang HJ, et al. Aspergillus fumigatus conidia upregulates NOD2 protein expression both in vitro and in vivo. Acta Pharmacol Sin. 2008;29:1202–1208. doi: 10.1111/j.1745-7254.2008.00860.x. [DOI] [PubMed] [Google Scholar]
- 12.Park JH, et al. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol. 2007;178:2380–2386. doi: 10.4049/jimmunol.178.4.2380. [DOI] [PubMed] [Google Scholar]
- 13.Inohara N, et al. Human Nod1 confers responsiveness to bacterial lipopolysaccharides. J Biol Chem. 2001;276:2551–2554. doi: 10.1074/jbc.M009728200. [DOI] [PubMed] [Google Scholar]
- 14.Girardin SE, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 15.Inohara N, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 16.Inohara N, et al. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000;275:27823–27831. doi: 10.1074/jbc.M003415200. [DOI] [PubMed] [Google Scholar]
- 17.Kim YG, et al. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to Toll-like receptor ligands. Immunity. 2008;28:246–257. doi: 10.1016/j.immuni.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 18.Opitz B, et al. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J Biol Chem. 2004;279:36426–36432. doi: 10.1074/jbc.M403861200. [DOI] [PubMed] [Google Scholar]
- 19.Hisamatsu T, et al. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124:993–1000. doi: 10.1053/gast.2003.50153. [DOI] [PubMed] [Google Scholar]
- 20.Kufer TA, et al. Role for erbin in bacterial activation of Nod2. Infect Immun. 2006;74:3115–3124. doi: 10.1128/IAI.00035-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferwerda G, et al. NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog. 2005;1:279–285. doi: 10.1371/journal.ppat.0010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Divangahi M, et al. NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. J Immunol. 2008;181:7157–7165. doi: 10.4049/jimmunol.181.10.7157. [DOI] [PubMed] [Google Scholar]
- 23.Shimada K, et al. The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniae lung infection. PLoS Pathog. 2009;5:e1000379. doi: 10.1371/journal.ppat.1000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petnicki-Ocwieja T, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A. 2009;106:15813–15818. doi: 10.1073/pnas.0907722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biswas A, et al. Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc Natl Acad Sci U S A. 2010;107:14739–14744. doi: 10.1073/pnas.1003363107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Storb R, et al. Graft-versus-host disease and survival in patients with aplastic anemia treated by marrow grafts from HLA-identical siblings. Beneficial effect of a protective environment. N Engl J Med. 1983;308:302–307. doi: 10.1056/NEJM198302103080602. [DOI] [PubMed] [Google Scholar]
- 27.Beelen DW, et al. Evidence that sustained growth suppression of intestinal anaerobic bacteria reduces the risk of acute graft-versus-host disease after sibling marrow transplantation. Blood. 1992;80:2668–2676. [PubMed] [Google Scholar]
- 28.Holler E, et al. Both donor and recipient NOD2/CARD15 mutations associate with transplant-related mortality and GvHD following allogeneic stem cell transplantation. Blood. 2004;104:889–894. doi: 10.1182/blood-2003-10-3543. [DOI] [PubMed] [Google Scholar]
- 29.Holler E, et al. Prognostic significance of NOD2/CARD15 variants in HLA-identical sibling hematopoietic stem cell transplantation: effect on long-term outcome is confirmed in 2 independent cohorts and may be modulated by the type of gastrointestinal decontamination. Blood. 2006;107:4189–4193. doi: 10.1182/blood-2005-09-3741. [DOI] [PubMed] [Google Scholar]
- 30.Penack O, et al. NOD2 regulates hematopoietic cell function during graft-versus-host disease. J Exp Med. 2009;206:2101–2110. doi: 10.1084/jem.20090623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elmaagacli AH, et al. Mutations in innate immune system NOD2/CARD 15 and TLR-4 (Thr399Ile) genes influence the risk for severe acute graft-versus-host disease in patients who underwent an allogeneic transplantation. Transplantation. 2006;81:247–254. doi: 10.1097/01.tp.0000188671.94646.16. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen Y, et al. Insufficient evidence for association of NOD2/CARD15 or other inflammatory bowel disease-associated markers on GVHD incidence or other adverse outcomes in T-replete, unrelated donor transplantation. Blood. 115:3625–3631. doi: 10.1182/blood-2009-09-243840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones JM, et al. Mortality and gross pathology of secondary disease in germfree mouse radiation chimeras. Radiat Res. 1971;45:577–588. [PubMed] [Google Scholar]
- 34.van Bekkum DW, et al. Mitigation of secondary disease of allogeneic mouse radiation chimeras by modification of the intestinal microflora. J Natl Cancer Inst. 1974;52:401–404. doi: 10.1093/jnci/52.2.401. [DOI] [PubMed] [Google Scholar]
- 35.Mizushima N, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kroemer G, et al. Autophagy and the integrated stress response. Mol Cell. 40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooney R, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 39.Homer CR, et al. ATG16L1 and NOD2 Interact in an Autophagy-Dependent Antibacterial Pathway Implicated in Crohn's Disease Pathogenesis. Gastroenterology. 2010 doi: 10.1053/j.gastro.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Travassos LH, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 41.Cadwell K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 43.Sabbah A, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10:1073–1080. doi: 10.1038/ni.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore CB, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573–577. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- 45.Adam A, et al. Muramyl peptides. Chemical structure, biological activity and mechanism of action. Mol Cell Biochem. 1981;41:27–47. doi: 10.1007/BF00225295. [DOI] [PubMed] [Google Scholar]
- 46.Ellouz F, et al. Minimal structural requirements for adjuvant activity of bacterial peptidoglycan derivatives. Biochem Biophys Res Commun. 1974;59:1317–1325. doi: 10.1016/0006-291x(74)90458-6. [DOI] [PubMed] [Google Scholar]
- 47.Strominger JL. Bacterial cell walls, innate immunity and immunoadjuvants. Nat Immunol. 2007;8:1269–1271. doi: 10.1038/ni1207-1269. [DOI] [PubMed] [Google Scholar]
- 48.van Beelen AJ, et al. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity. 2007;27:660–669. doi: 10.1016/j.immuni.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 49.Magalhaes JG, et al. Nod2-dependent Th2 polarization of antigen-specific immunity. J Immunol. 2008;181:7925–7935. doi: 10.4049/jimmunol.181.11.7925. [DOI] [PubMed] [Google Scholar]
- 50.Moreira LO, et al. Modulation of adaptive immunity by different adjuvant-antigen combinations in mice lacking Nod2. Vaccine. 2008;26:5808–5813. doi: 10.1016/j.vaccine.2008.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Asano J, et al. Nucleotide oligomerization binding domain-like receptor signaling enhances dendritic cell-mediated cross-priming in vivo. J Immunol. 2010;184:736–745. doi: 10.4049/jimmunol.0900726. [DOI] [PubMed] [Google Scholar]
- 52.Shaw MH, et al. T cell-intrinsic role of Nod2 in promoting type 1 immunity to Toxoplasma gondii. Nat Immunol. 2009;10:1267–1274. doi: 10.1038/ni.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seder RA, et al. CD28-mediated costimulation of interleukin 2 (IL-2) production plays a critical role in T cell priming for IL-4 and interferon gamma production. J Exp Med. 1994;179:299–304. doi: 10.1084/jem.179.1.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kobayashi KS, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 55.Yu XZ, et al. Role of CD28 in acute graft-versus-host disease. Blood. 1998;92:2963–2970. [PubMed] [Google Scholar]
- 56.Greenwald RJ, et al. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 57.Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9:271–285. doi: 10.1038/nri2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vang KB, et al. Cutting edge: CD28 and c-Rel-dependent pathways initiate regulatory T cell development. J Immunol. 2010;184:4074–4077. doi: 10.4049/jimmunol.0903933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deenick EK, et al. c-Rel but not NF-kappaB1 is important for T regulatory cell development. Eur J Immunol. 2010;40:677–681. doi: 10.1002/eji.201040298. [DOI] [PubMed] [Google Scholar]
- 60.Ruan Q, et al. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity. 2009;31:932–940. doi: 10.1016/j.immuni.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Long M, et al. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity. 2009;31:921–931. doi: 10.1016/j.immuni.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 62.Visekruna A, et al. c-Rel is crucial for the induction of Foxp3(+) regulatory CD4(+) T cells but not T(H)17 cells. Eur J Immunol. 2010;40:671–676. doi: 10.1002/eji.200940260. [DOI] [PubMed] [Google Scholar]
- 63.Zheng Y, et al. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.MacLeod H, Wetzler LM. T cell activation by TLRs: a role for TLRs in the adaptive immune response. Sci STKE 2007. 2007 doi: 10.1126/stke.4022007pe48. pe48. [DOI] [PubMed] [Google Scholar]
- 65.Maeda S, et al. Nod2 mutation in Crohn's disease potentiates NF-kappaB activity and IL-1beta processing. Science. 2005;307:734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 66.Watanabe T, et al. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–808. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]