

Abstract
Type 2 diabetes mellitus has been at the forefront of human diseases and phenotypes studied by new genetic analyses. Thanks to genome-wide association studies, we have made substantial progress in elucidating the genetic basis of type 2 diabetes. This review summarizes the concept, history, and recent discoveries produced by genome-wide association studies for type 2 diabetes and glycemic traits, with a focus on the key notions we have gleaned from these efforts. Genome-wide association findings have illustrated novel pathways, pointed toward fundamental biology, confirmed prior epidemiological observations, drawn attention to the role of β-cell dysfunction in type 2 diabetes, explained ~10% of disease heritability, tempered our expectations with regard to their use in clinical prediction, and provided possible targets for pharmacotherapy and pharmacogenetic clinical trials. We can apply these lessons to future investigation so as to improve our understanding of the genetic basis of type 2 diabetes.
Keywords: genome-wide association studies, type 2 diabetes, genetics, single nucleotide polymorphisms
Introduction
During the last decade there has been an outpouring of studies providing clues into the genetic architecture underlying complex diseases. Type 2 diabetes mellitus (T2D) has been at the forefront of human diseases and traits studied by new genetic analyses. Prior to genome-wide association studies (GWAS), the primary methods used to establish a link between genotype and phenotype were linkage analysis and candidate gene approaches. Linkage analysis relies on shared DNA segments inherited from common ancestors coupled with phenotypic information. This method was useful in identifying familial genetic variants with large effects, such as those giving rise to maturity-onset diabetes of the young (MODY).1 When applied to common T2D, Reynisdottir et al. identified segments in chromosomes 5 and 10 with suggestive linkage to T2D.2 The chromosome 10 region harbored TCF7L2, one of the T2D-associated genes with strongest effects, although not sufficiently high to explain the original linkage signal.3
Linkage analysis can detect rare genetic loci that strongly influence a disease, but proved limited in unveiling common genetic variants with a more modest impact on complex diseases (Figure 1). Association methods could detect genetic variants causing smaller effects. Unlike linkage analysis, in their earlier form association methods could only be applied to candidate genes: they were therefore biased in assuming that a specific locus caused disease based on biological plausibility, and limited in that they depended on prior knowledge with a consequent disregard for intergenic regions. From this work, PPARG and KCNJ11 emerged as two candidate genes both of which encode targets for anti-diabetes medications, and harbor missense variants associated with T2D.4-6
Figure 1. Frequency of genetic variation and disease susceptibility.

Figure adapted from references 112,113
A portion of “missing heritability” may be explained by low frequency variants with intermediate penetrance. The low risk-allele frequencies make the variants undetectable by current GWAS arrays. Resequencing technology and new-generation arrays may help identify these low-frequency variants; larger sample sizes may be needed to detect significant signals. The effort can be further strengthened by applying prior biological and epidemiological data to select associations.
Thanks to the completion of the Human Genome and International HapMap Projects (see below), the novel approach of searching for genetic associations in a “genome-wide” fashion came to fruition (Figure 2). Thus, scientists embarked in GWAS which allowed them to discover multiple gene variants with individually small effects. Once a specific polymorphism is associated with a disease, it is usually annotated by naming the gene in closest proximity to it. However, this does not necessarily mean that the variant in question is the molecular defect responsible for the phenotype, nor does it implicate the nearest gene; it simply flags a genomic region that harbors the causal variant, which may itself be acting at a certain distance, for instance by modulating expression of a far-away gene. Therefore while association signals are often identified by gene names, only in a few cases has a causal relationship been demonstrated, typically via fine-mapping and functional approaches.
Figure 2. Schematic of a typical GWAS design.
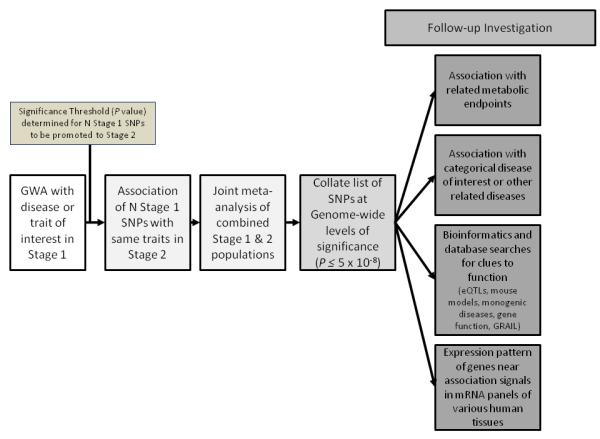
The general design of GWAS starts with a stage 1 (discovery) cohort. The top SNPs are promoted to the stage 2 (replication) cohort based on 1) a significance threshold that is usually dictated by pragmatic considerations, 2) whether there is prior knowledge of association between the disease the variant, and 3) whether an association is biologically plausible. The successfully replicated SNPs are meta-analyzed in the combined stage 1 and stage 2 cohorts. The SNPs that reach levels of genome-wide significance (P=5×10−8) are explored further using the functional analysis techniques listed.
Substantial progress in our knowledge of the genetic basis of T2D has been elucidated by T2D GWAS, but there remains a large portion of unexplained genetic heritability. This review summarizes the concept, history, and recent discoveries produced by GWAS for T2D and glycemic traits. It will emphasize what we have learned from the explosion of data in the last ten years, and how we can apply our knowledge to future investigation to gain further understanding of the genetic basis of T2D.
Launching GWAS
The ability to interrogate the entire genome was made possible by two key advances: the Human Genome Project, with a draft sequence in 2001 and near-complete sequence in 2003, and the International HapMap project, with its first phase completed in 2005, and now currently in its third phase.7-10Progress in high-throughput and affordable genotyping technology, analytical tools to assist in the data mining, cleaning and interpretation of large databases, and the assembly of international collaborations combining well-phenotyped cohorts made utilizing these advances possible.
Following the completion of the Human Genome Project, a search for genetic variation that might explain phenotypic diversity and an individual’s risk of disease ensued. Assaying single nucleotide polymorphisms (SNPs) became a mainstream way to study the association of genetic variation and disease. A SNP is a single nucleotide DNA sequence variant in the genome that differs between members of the same species or a pair of chromosomes in an individual. SNPs occur on average every 300 base pairs, have a low rate of recurrent mutation, and are most often binary in nature. Several million SNPs were discovered and deposited in public databases.11 Initially, the HapMap genotyped 3.9 million SNPs in 270 DNA samples among four different ethnic groups and defined the underlying patterns of the inheritance of genetic variation. The inheritance pattern is quantified by linkage disequilibrium (LD), which represents the likelihood that alleles of nearby SNPs will stay together and preserve their linear arrangement on a haplotype during meiosis. This likelihood is dependent on recombination rates, with recombination events more likely to separate alleles that lie further apart. In this manner, two SNPs in strong LD will be inherited together more frequently than two SNPs in weak LD. By knowing this correlation structure, investigators and chip manufacturers only have to query a smaller subset of SNPs, or“tag” SNPs, to design genotyping arrays and conduct association analyses that essentially capture the majority of remaining common genomic variation. Genetic variants that are not directly genotyped can then be imputed from the genotyped ”tag” SNPs subset. Imputation presumes the allele of a SNP at a different location inferred by its degree of LD with an allele at a directly genotyped variant.12
Initial GWAS for T2D
GWAS have led to the discovery of 38 SNPs associated with T2D, in addition to nearly two dozen SNPs associated with glycemic traits (Table 1). Notably, although these SNPs have been associated with the trait of interest beyond reasonable statistical doubt, save a few exceptions they have not yet led to the identification of the specific causal variant(−s).
Table 1.
Genetic variants associated with T2D or glycemic traits at genome-wide levels of statistical significance
| Locus | Marker | Chr | Type of mutation | Allele (Effect/ Other) |
Effect allele frequency (HapMap- CEU) |
Trait | Effect: OR/Beta (95% CI/SE) |
|---|---|---|---|---|---|---|---|
| 2000 | |||||||
| PPARG | rs18012824 | 3 | Missense: Pro12Ala |
C/G | 0.92 | T2D | 1.14 (1.08-1.20)15-17 |
| 2003 | |||||||
|
KCNJ11/
ABCC8 |
rs5219/ rs757110a,5 |
11 | Missense: Glu23Lys/Ala1369Ser |
T/C G/T |
0.50 0.40 |
T2D | 1.15 (1.09-1.21)17 |
| 2006 | |||||||
| TCF7L2 | rs7903146b,3,35 | 10 | Intronic | T/C | 0.25 | T2D FG |
1.37 (1.28-1.47)18 0.023 (0.004) |
| rs12243326b,36 | Intronic | C/T | 0.21 | 2-hr G | 0.07 (0.01) | ||
| 2007 | |||||||
| IGF2BP2 | rs440296015-17 | 3 | Intronic | T/G | 0.29 | T2D | 1.17 (1.10–1.25)18 |
| CDKAL1 | rs775484015-17 | 6 | Intronic | C/G | 0.31 | T2D | 1.12 (1.08-1.16) |
| SLC30A8 | rs1326663413,35 | 8 | Missense: Arg325Trp |
C/T | 0.75 | T2D FG |
1.12 (1.07-1.16)65 0.027 (0.004) |
| CDKN2A/B | rs1081166115-17 | 9 | 125 kb upstream | T/C | 0.79 | T2D | 1.20 (1.14-1.25) |
| HHEX | rs111187513 | 10 | 7.7 kb downstream | C/T | 0.56 | T2D | 1.13 (1.08-1.17)17 |
| FTO | rs805013615,16 | 16 | Intronic | A/C | 0.45 | T2D | 1.15 (1.09-1.22)18 |
| HNF1B | rs757210110,111 | 17 | Intronic | A/G | 0.43 | T2D | 1.12 (1.07-1.18)65 |
| 2008 | |||||||
| NOTCH2 | rs1092393118 | 1 | Intronic | T/G | 0.11 | T2D | 1.13 (1.08-1.17) |
| THADA | rs757859718 | 2 | Missense: Thr1187Ala |
T/C | 0.92 | T2D | 1.15 (1.10-1.20) |
| ADAMSTS9 | rs460710318 | 3 | 38 kb upstream | C/T | 0.81 | T2D | 1.09 (1.06-1.12) |
| JAZF1 | rs86474518 | 7 | Intronic | T/C | 0.52 | T2D | 1.10 (1.07-1.13) |
|
CDC123/
CAMK1D |
rs1277979018 | 10 | Intergenic region | G/A | 0.23 | T2D | 1.11 (1.07-1.14) |
| KCNQ1 | rs2237892c,24 | 11 | Intronic | C/T | 0.61d | T2D | 1.4 (1.34-1.47) |
| rs231362c,65 | Intronic | G/A | 0.52 | T2D | 1.08 (1.06-1.10) | ||
|
TSPAN8/
LGR5 |
rs796158118 | 12 | Intronic | C/T | 0.23 | T2D | 1.09 (1.06-1.12) |
| 2009 | |||||||
| IRS1 | rs294364127 | 2 | 502 kb upstream | C/T | 0.61 | T2D | 1.19 (1.13-1.25) |
| 2010 | |||||||
| DUSP9 | rs594532665 | X | 8 kb upstream | G/A | 0.12 | T2D | 1.27 (1.18-1.37) |
| PROX1 | rs34087435 | 1 | 2 kb upstream | C/T | 0.50 | FG T2D |
0.013 (0.003) 1.07 (1.05-1.09) |
| BCL11A | rs24302165 | 2 | 99 kb downstream | A/G | 0.46 | T2D | 1.08 (1.06-1.10) |
| G6PC2 | rs56088735 | 2 | Intronic | C/T | 0.70 | FG HOMA-B HbA1C |
0.075 (0.004) −0.042 (0.004) 0.032 (0.004) |
| GCKR | rs1260326e,17 | 2 | Missense: Leu446Pro |
T/C | 0.40 | 2-hr G | 0.10 (0.01) |
| rs780094e,35 | Intronic | C/T | 0.62 | FG FI HOMA-IR T2D |
0.029 (0.003) 0.032 (0.004) 0.035 (0.004) 1.06 (1.04-1.08) |
||
| ADCY5 | rs2877716f,36,81 | 3 | Intronic | C/T | 0.77 | 2-hr G | 0.07 (0.01) |
| rs11708067f,35 | Intronic | A/G | 0.78 | FG HOMA-B T2D |
0.027 (0.003) −0.023 (0.004) 1.12 (1.09-1.15) |
||
| SLC2A2 | rs1192009035 | 3 | Intronic | T/A | 0.85 | FG | 0.02 (0.004) |
| WFS1 | rs180121465 | 4 | Intron-exon junction |
G/A | 0.27 | T2D | 1.13 (1.07-1.18) |
| ZBED3 | rs445705365 | 5 | 41 kb upstream | G/A | 0.26 | T2D | 1.08 (1.06-1.11) |
| DGKB/TMEM195 | rs219134935 | 7 | Intergenic region | T/G | 0.47 | FG T2D |
0.03 (0.003) 1.06 (1.04-1.08) |
| GCK | rs460751735 | 7 | 36 kb upstream | A/G | 0.20 | FG HbA1C T2D |
0.062 (0.004) 0.041 (0.005) 1.07 (1.05-1.10) |
| KLF14 | rs97228365 | 7 | 47 kb upstream | G/A | 0.55 | T2D | 1.07 (1.05-1.10) |
| TP53INP1 | rs89685465 | 8 | Intronic | T/C | 0.48 | T2D | 1.06 (1.04-1.09) |
| GLIS3 | rs703420035 | 9 | Intronic | A/C | 0.53 | FG HOMA-B |
0.018 (0.003) −0.020 (0.004) |
| TLE4 g | rs1329213665 | 9 | 234 kb upstream | C/T | 0.93 | T2D | 1.11 (1.07-1.15) |
| ADRA2A | rs1088512235 | 10 | 210 kb downstream | G/T | 0.90 | FG | 0.022 (0.004) |
| CENTD2 | rs155222465 | 11 | 5′ UTR | A/C | 0.88 | T2D | 1.14 (1.11-1.17) |
| CRY2 | rs1160592435 | 11 | Intronic | A/C | 0.54 | FG | 0.015 (0.003) |
| FADS1 | rs17455035 | 11 | Intronic | T/C | 0.63 | FG HOMA-B |
0.017 (0.003) −0.020 (0.003) |
| MADD | rs794458435 | 11 | Intronic | A/T | 0.69 | FG | 0.021 (0.003) |
| MTNR1B | rs1083096335 | 11 | Intronic | G/C | 0.30 | FG HOMA-B HbA1C T2D |
0.067 (0.003) −0.034 (0.004) 0.024 (0.004) 1.09 (1.06-1.12) |
| HMGA2 | rs153134365 | 12 | 43 kb upstream | C/G | 0.10 | T2D | 1.10 (1.07-1.14) |
| HNF1A | rs795719765 | 12 | 20 kb downstream | T/A | 0.85 | T2D | 1.07 (1.05-1.10) |
| IGF1 | rs3576735 | 12 | 1.2 kb upstream | G/A | 0.90 | FI HOMA-IR |
0.01 (0.006) 0.013 (0.006) |
| C2CD4B | rs1107165735 | 15 | 21 kb downstream | A/G | 0.59 | FG | 0.008 (0.003) |
| PRC1 | rs804268065 | 15 | Intronic | A/C | 0.22 | T2D | 1.07 (1.05-1.09) |
| VPS13C | rs1727130536 | 15 | Intronic | G/A | 0.42 | 2-hr G | 0.07 (0.01) |
| ZFAND6 | rs1163439765 | 15 | 1.5 kb downstream | G/A | 0.56 | T2D | 1.06 (1.04-1.08) |
| GIPR | rs1042392836 | 19 | Intronic | A/T | 0.18 | 2-hr G | 0.11 (0.01) |
Loci are arranged by year of genome-wide significance (GWS).The SNP citation references the publications where the SNP initially associated with the trait. FG = fasting glucose (mmol/L); FI = fasting insulin (pmol/L); 2-hr G = 2-hour glucose (FG adjusted, mmol/L); HbA1C = Hemoglobin A1C (%). Effect estimates are adapted from the references for each SNP unless otherwise noted.
these two missense SNPs are in strong LD in two adjacent genes. Their protein products form the two components of the hetero-octameric beta-cell KATP channel. Recent functional analysis suggests that the missense mutation at ABCC8 is likely the causal variant for gliclazide response.
these SNPs are in strong LD in Europeans (r2=0.79);
these SNPs are in low LD in Europeans (r2=0.01) and likely represent independent association signals;
allele frequency from HapMap-JPT;
these SNPs are in strong LD in Europeans (r2=0.93) but not in Africans (r2=.43);
these SNPs are in strong LD in Europeans (r2=0.82);
formerly annotated as CHCHD9
The first GWAS for T2D was conducted in a French discovery cohort composed of 661 cases of T2D (body mass index [BMI]<30 kg/m2, first-degree family history of T2D) and 614 non-diabetic controls were genotyped on two genotyping platforms. In total, 392,935 SNPs culled from two different genotyping platforms were analyzed for association with T2D. Although two associations were not reproducible in follow-up studies (LOC387761, EXT2), this study identified novel and reproducible association signals at SLC30A8 and HHEX, and validated the well known association at TCF7L2.13 Investigators from the Icelandic company deCODEand their collaborators confirmed the association of loci SLC30A8 and HHEX with T2D and identified an additional signal in CDKAL1.14 On the same day, three collaborating groups, the Wellcome Trust Case Control Consortium (WTCCC), the Finland-United States Investigation of NIDDM Genetics (FUSION) group, and the Diabetes Genetics Initiative (DGI), published their findings replicating SLC30A8 and HHEX, and independently discovering novel associations at CDKAL1, IGF2BP2 and CDKN2A/B.15-17
More power to detect low-effect size common variants with increasing sample size
WTCCC, FUSION, and DGI ultimately combined their data to form the Diabetes Genetics Replication and Metaanalysis (DIAGRAM) consortium, which led to a substantial increase in sample size and, thus, power to detect common genetic variants with low effect size.18 Table 1 summarizes the findings of each of these cohorts and consortia.
Most T2D genetics cohorts have now coalesced to form DIAGRAM+, which achieved an effective sample size of over 22,000 subjects of European origin. In a recent report, 2,426,886 imputed and genotyped autosomal SNPs, with additional interrogation of the X-chromosome, were examined for association with T2D as a categorical phenotype. Fourteen signals, also shown in Table 1, reached genome-wide significance (P<5×10−8) in association with T2D (this threshold is determined by correction for the estimated 106 independent tests that capture common genomic variation in non-African populations19).
In addition to nine novel loci, this most recent meta-analysis allowed for the confirmation of other loci previously associated with T2D among smaller cohorts including IRS1, MTNR1B and KCNQ1.20-25 In independent work, IRS1 (encoding the insulin receptor substrate-1) had been associated with T2D, development of hyperglycemia, insulin resistance by homeostasis model assessment (HOMA-IR26), fasting glucose, and fasting insulin.27 Several GWAS for fasting glucose as a quantitative trait, described in more detail below, had already identified MTNR1B as a locus influencing fasting hyperglycemia making it a candidate locus for association with T2D.21-23 A third SNP, rs231362, is located in an intron of KCNQ1 in chromosome 11 which overlaps the KCNQ1OT1 transcript, thought to influence expression of CDKN1C which regulates β-cell development;28 an independent signal in KCNQ1 had been associated with T2D in a Japanese, Korean, Chinese and European populations.24,25 Two additional loci were near genes had been linked with T2D in previous studies, but not at genome-wide significance: HNF1A harbors rare mutations that account for MODY, and BCL11A had shown suggestive association not reaching genome-wide significance in the first DIAGRAM discovery meta-analysis.18
GWAS for continuous glycemic traits
Initial GWAS interrogated the genetic determinants of T2D as a dichotomous phenotype (disease vs. no disease), rather than examining continuous glycemic traits. Prior to the development of GWAS, Weedon et al. had examined GCK, a gene in which rare mutations cause a defect in the the rate-limiting enzyme that drives glucose metabolism in the β cell, resulting in MODY2. Using the candidate gene approach, they demonstrated that two common variants, rs1799884 and rs3757840, are associated with fasting glucose surpassing or approaching genome-wide significance (1×10−9 and 8×10−7, respectively).29
The association of GCK with fasting glucose and the success of categorically-driven GWAS primed the field to examine the genetic factors that contribute to the inter-individual variation in glycemic measures in normoglycemic subjects. Using a genome-wide approach G6PC2 and MTNR1B were associated with fasting glucose. It is postulated that G6PC2 modulates the glycolytic pathway and insulin secretion by dephosphorylating glucose-6-phosphate generated by the β-cell glucose sensor, glucokinase. In a French cohort, carriers of the A allele for rs560887 in the third intron of G6PC2 had decreased fasting plasma glucose and lower risk of developing mild hyperglycemia in 9-year follow-up.30 Shortly following the publication of these findings, a study of nondiabetic individuals from Finland and Sardinia reported an association between SNP rs563694 and fasting glucose concentrations. SNP rs563694 is in high LD (r2=0.729) with and only 11 kb away from the SNP discovered in the French cohort, rs560887.31
In early 2009, three groups concurrently described the influence of MTNR1B on quantitative glycemic traits and further characterized the functional significance of one of these variants, rs10830963.21-23 MTNR1B encodes the melatonin receptor MT2, whose endogenous ligand melatonin is a neurohormone that mediates circadian rhythmicity and appears to influence insulin secretion and glucose levels.32-34 The largest of the three studies was conducted by the Meta-Analysis of Glucose and Insulin-related traits Consortium (MAGIC). MAGIC represents an international collaborative effort to combine data from multiple GWAS to identify additional loci that affect glycemic and metabolic traits. This initial study focused on the top association signals from four individual consortia meta-analyses, with each of these consortia providing mutual replication of the top SNPs. This meta-analysis of 10 cohorts, comprised of 40,735 individuals, described the association of rs10830963 at MTNR1B with fasting glucose (P=3.2×10−50) and T2D (P=3.3×10−7) and confirmed the previously known associations of GCK and G6PC2 with fasting glucose.21,29-31 In an independent French cohort, a second variant near MTNR1B (rs1387153) was associated with T2D, fasting glucose, β-cell function by homeostasis model assessment (HOMA-B26), and glycated hemoglobin (HbA1C).22 Lyssenko et al. further characterized the physiological ramifications of carrying the risk allele in rs10830963. Carriers of the G allele showed decreased insulin secretion, increasing fasting glucose levels and a higher risk of developing T2D in an average 23.5 year follow-up period, as well as a higher proinsulin-to-insulin ratio.23
In follow-up to these initial investigations, a large-scale meta-analysis of all genome-wide data for continuous fasting and 2-hour post-glucose load traits in non-diabetic participants was performed.35,36 Twenty one GWAS of European descent including 46,186 participants analyzed for ~2.5 million genotyped or imputed SNPs coalesced around MAGIC in order to examine fasting glucose, fasting insulin, and fasting indices of β-cell function and insulin resistance (HOMA-B and HOMA-IR respectively).35 Lead SNPs were replicated among 76,558 additional individuals from 34 additional cohorts (7 of which had undergone GWAS genotyping and contributed in silico replication). The joint analysis for fasting glucose uncovered nine new loci including SNPs in or near ADCY5, MADD, CRY2, ADRA2A, FADS1, PROX1, SLC2A2, GLIS3, and C2CD4B and one SNP upstream from IGF1 in association with fasting insulin and HOMA-IR. The meta-analysis also confirmed prior associations for glycemic traits with SNPs in or near DGKB-TMEM195, GCKR, G6PC2, MTNR1B, and GCK. The 14 fasting glucose-associated loci explained a substantial proportion (10%) of the inherited variation in fasting glucose in the Framingham cohort. Additionally, an aggregate genotype score, constructed by summing the risk variants, showed a difference in fasting glucose of about 7.2 mg/dl between groups with the highest and lowest scores.35 Contrary to straightforward expectations, only five of the novel fasting glucose-associated SNPs were associated with T2D, which underscores that the mechanism by which glucose is raised, rather than elevated glucose alone, may have contributed to the progression to T2D. The three loci with the largest effect sizes on fasting glucose, GCPC2, MTNR1B, and GCK, were found to have a significant association with HbA1C at genome-wide significance thresholds 37; interestingly, G6PC2 does not increase risk of T2D. In terms of insulin resistance measures, IGF1 (rs35767) and GCKR (rs780094), were significantly associated with HOMA-IR and fasting insulin.35
In order to examine glucose and insulin levels 2 hours after an oral glucose tolerance test (OGTT), nine GWAS (n=15,234) were compiled with replication in 17 studies (n=30,620).36 This study found an association of SNP rs10423928 in GIPR (encoding the gastric inhibitory peptide receptor) with glucose levels 2 hours after an oral glucose challenge.36 Gastric inhibitory peptide is an incretin hormone released from the intestine in response to oral glucose, and it stimulates insulin secretion by the pancreatic β cell.38 GIP response is decreased in T2D.39 In further analyses supporting the known role of this pathway in glucose regulation, this intronic SNP was associated with a decreased early-phase insulin secretion, decreased insulin response, and lower insulin levels 2 hours after an oral, but not intravenous glucose load.
Genetic variants and physiological parameters
Assessing more detailed physiological parameters influenced by newly discovered genetic variants lends credibility to the original associations, besides offering a better characterization of the mechanism by which these variants might modulate glucose and insulin levels and cause T2D. Consequently, the MAGIC investigators systematically examined the influence of the genetic variants newly associated with fasting glucose, fasting insulin and/or 2-hour glucose with measures that define insulin processing, insulin secretion, and insulin sensitivity.40 With these measures, loci were categorized into five groups based on their hypothesized mechanism of action: 1) abnormal insulin processing, 2) higher proinsulin and lower insulin secretion, 3) abnormalities in early insulin secretion, 4) reduced insulin sensitivity, 5) no obvious effect on insulin processing, secretion or sensitivity. MADD (encoding a death domain-containing adaptor protein that propagates apoptotic signals) was strongly associated with proinsulin levels, but not other parameters. This indicates that the locus is implicated in isolated insulin processing only. TCF7L2, SLC30A8, GIPR and C2CD4B were associated with higher proinsulin and lower insulin secretion. The findings for TCF7L2 and SLC30A8 confirmed previous results, which appear biologically plausible given the proposed function of these genes. For instance, genetic variants in TCF7L2 are thought to adversely affect β-cell responsiveness to incretins and insulin granule exocytosis, which would impair insulin processing and decrease insulin secretion. In turn, variants at MTNR1B, FADS1, DGKB, and GCK were only associated with a lower insulinogenic index (which measures the initial phase of glucose-stimulated insulin release, by dividing the change in insulin over the first 30 minutes of an OGTT by the change in glucose over the same time frame), suggesting that variants in or near these genes impair early insulin secretion. For instance, FADS1 encodes a key enzyme in metabolism of unsaturated fatty acids, and insulin secretion differs in response to meals with varying fatty acid composition.41 The lower insulinogenic index caused by genetic variation in FADS1 indicates that variants in this gene may alter an individual’s β-cell response to meals composed of certain fatty acid content. SNPs at GCKR and IGF1 were found to influence insulin sensitivity by multiple methods whose calculation includes measurements beyond fasting glucose and insulin. A functional variant at GCKR (encoding the glucokinase regulatory protein) may act by inhibiting glucokinase in the liver, leading to increased hepatic glucose production.42 Glucose lowering by IGF1 occurs by the binding of insulin receptors, stimulation of glucose transport into fat and muscle, and thus, lowering glucose and suppression of insulin.43 Unlike GCKR, the influence of the index SNP near IGF1 on glucose homeostasis is not yet understood. This study confirmed that these variants influence insulin resistance measures that go beyond the fasting state, possibly implicating extra-hepatic tissues in the process.
What have we learned from GWAS?
In a short span of five years, GWAS investigating the links between genetics and complex traits have transformed our knowledge. As we strive to decipher how to navigate these new roads, it may be instructive to look back at what we have learned to know how to journey forward.
They illustrate novel pathways
GWAS have implicated novel pathways in the development of diabetes in humans. One of the earliest and most illustrative examples was the discovery that the variant rs13266634, which encodes a R→W change at position 325 in the β-cell zinc transporter ZnT-8 (encoded by SLC30A8) was strongly associated with T2D (OR 1.26 for the major C allele, P=5.0×10−7) in the first published GWAS for T2D.13 This association was confirmed by the UK WTCCC (P=10−3) 15, FUSION (P=10−5) 16, and DGI (P=5.3×10−8).17
SLC30A8 is expressed almost exclusively in pancreatic islets with low levels in the cortex and thyroid.44 Its protein product, ZnT-8, is a zinc transporter localized in secretion vesicle membranes that transports zinc from the cytoplasm into insulin secretory vesicles.45 Insulin is stored as a hexamer bound to two zinc ions, and ZnT-8 provides zinc to allow for insulin storage and secretion.46 ZnT-8 thus appears to be a critical component of the final biosynthetic pathway of insulin production and secretion. Interestingly, overexpression of SLC30A8 in insulinoma cells increases glucose-stimulated insulin secretion.47 Other in vitro studies demonstrate that inflammatory cytokines noted to be elevated in T2D downregulate ZnT-8 in β cells and alter β-cell function.20,48 The development of a Slc30a8 null mouse shortly after the GWAS publications further defined how these genetic variants may affect glycemic parameters in vivo, although with varying results depending on the mouse model.49,50 In the mouse models, the size and number of islets as well as insulin sensitivity were comparable between knock-out (KO) mice and wild-type littermates, though the latter showed decreased islet zinc staining. Some of the KO models demonstrated impaired glucose tolerance and fasting glucose depending on age and gender, and one of the mouse models also showed decreased plasma insulin and decreased glucose-stimulated insulin secretion. Thus, though the role of this gene in insulin packaging and storage was known from prior work, GWAS and the experiments spawned by them revealed that coding variation in SLC30A8 may increase the risk of developing T2D by causing an insulin secretory defect.
New findings point to fundamental biology
While one can easily hypothesize about the biological consequences of missense SNPs, a potential molecular mechanism is not readily apparent for most other associated SNPs, which lie in intronic or intergenic DNA segments. Therefore, GWAS have reinforced the fundamental quest to characterize the biological relevance of non-coding genomic regions.
One pertinent example concerns the intronic SNP in the gene TCF7L2, rs7903146. This SNP has the strongest effect size on T2D demonstrated for a common variant thus far (~1.40 per copy of the risk allele).51,52 Using a new method called FAIRE-seq (formaldehyde-assisted isolation of regulatory elements coupled with high-throughput sequencing), Gaulton et al. identified open chromatin sites in human pancreatic islets.53 Open chromatin sites are evolutionarily conserved and can be used to tag regions that are bound by regulatory factors.54 Of 350 SNPs in strong LD with variants associated with T2D or fasting glycemia, 38 SNPs from ten loci (TCF7L2, CDKAL1, CDKN2A/CKDN2B, IGF2BP2, CDC123/CAMK1D, THADA, FTO, SLC30A8, HNF1B, and G6PC2) were associated with an open chromatin region in islets. Upon further examination of TCF7L2, SNP rs7903146 was localized to an islet-selective open chromatin site; more importantly, the high-risk T allele is associated with a “more open” chromatin state in human islets and greater enhancer activity, as revealed by allele-specific luciferase reporter assays in two β-cell lines.53 Given that the T allele associates with an increased risk of T2D, the more open chromatin state may imply increased transcription of TCF7L2, and thus confirms prior observations that the T allele is correlated with a 5-fold increase in TCF7L2 transcripts in human islets from donors with diabetes when compared to controls.55 By pairing fundamental biology, such as open chromatin regions indicative of regulatory binding sites, with current genetic association datasets, this study demonstrated a mechanism by which an intronic polymorphism may contribute to the development of a human disease.
Genetic discoveries support prior epidemiological observations
The GWAS method has not only led us to discover new or confirm known biology, but has also underscored prior epidemiological observations by providing potential genetic links between lipid dysregulation and glycemia (FADS1, GCKR, HNF1A), circadian rhythmicity and metabolic derangements (MTNR1B, CRY2), and low birth weight with subsequent T2D risk (ADCY5). A potential connection between genetic determinants of T2D that simultaneously lower cancer risk (HNF1B, JAZF1) is discussed at length elsewhere.56
Fatty acid desaturases (FADS) convert polyunsaturated fatty acids into cell signaling metabolites, that in turn, can affect circulating lipid levels.57 Different GWAS have associated FADS1 with fasting glucose and HOMA-B (rs174550, P=2×10−15 and 5×10−13 respectively), HDL cholesterol and triglyceride levels (rs174547, P=2×10−12 and 2×10−14 respectively), and LDL cholesterol levels (rs174546, P=1×10−7).35,57,58 All of these SNPs are in complete LD (r2=1) and therefore serve as perfect proxies for each other. In a large meta-analysis of glycemic traits the T allele at rs174550 was associated with higher fasting glucose, lower HOMA-B, increased HDL and LDL cholesterol, and decreased triglyceride levels.35 Classically, T2D and impaired fasting glucose have been clustered with dyslipidemia in the metabolic syndrome; the discovery of a genetic variant that is associated with both measures provides support for this clinical correlation. More recently FADS1 was found to be associated with a statin-induced response in triglyceride and HDL cholesterol levels (P=2.6×10−6 and 6.8×10−6, respectively).59 Given that unsaturated fatty acids influence insulin secretion, this genetic variant can tightly link hyperglycemia and dyslipidemia.41 Although the same SNP appears to influence various metabolic parameters, each of these measures could actually be influenced by different SNPs in high LD with the index SNP; elucidation of molecular mechanism requires functional experiments.
HNF1A (encoding hepatocyte nuclear factor-1α) illustrates a locus that contributes to the inheritance of both a rare Mendelian disorder and common polygenic disease, while also having pleiotropic effects. Classically, MODY3 is caused by a mutation in the HNF1A gene, which impairs the dimerization of this transcription factor and thus promotes metabolic dysregulation resulting in diabetes mellitus.60 Like FADS1, SNPs in the HNF1A region have been associated with multiple related traits including T2D (P=2×10−8), C-reactive protein (P=2×10−8 to 1×10−30), coronary artery disease (P=5×10−7), and LDL cholesterol (P=2×10−8).57,61-65 In terms of lipid metabolism and inflammation, HNF1A regulates numerous genes involving lipoprotein metabolism and inflammatory markers in the liver.66 These genetic variants create another molecular link between related metabolic derangements.
MTNR1B is an equally compelling story that helps provide a genetic link between circadian rhythmicity and glucose metabolism. As discussed above, MTNR1B variants have been associated with glycemic quantitative traits including T2D, fasting glucose, HOMA-B, and HbA1C.21-23 A second circadian gene, CRY2, has also been implicated in regulating fasting glucose in non-diabetic individuals.35 Epidemiological and physiological studies in humans, as well as experimental work in animals, have established a clear relationship between alterations of the circadian system and metabolic derangements. Homozygous mutant Clock mice, which are deficient in a key circadian rhythm transcription factor, were noted to have a variation in nighttime activity, in addition to metabolic disturbances including hyperphagia, obesity, hyperlipidemia, hepatic steatosis, hyperglycemia and hypoinsulinemia.67 Furthermore, wild-type islets appear to possess a self-sustained oscillation of a circadian gene (Per2) and circadian transcription factors (CLOCK and BMAL1) that is not seen in the islets of Clock null mice. In vivo studies in Clock mutant mice and in vitro studies in islets from whole-body Clock and Bmal1 mutant mice, as well as islets from pancreas-specific Bmal1 mutant mice, demonstrated impairment in insulin secretion.68 In order to explain the higher glucose intolerance noted in the morning versus night, Boden et al. studied normal human volunteers undergoing a hyperglycemic clamp titrated to various levels of glucose control for 24 hours. By measuring insulin secretion, the investigators demonstrated that, particularly at higher glucose levels, insulin is secreted in a circadian rhythm.32 In addition, people with T2D demonstrate a decreased diurnal serum melatonin level.34 The link between genetic variants in MTNR1B and CRY2 with T2D and glycemic measures lends further credence to the relationships observed in these animal and human studies.
Finally, genetic variants in ADCY5 (encoding adenylate cyclase type 5) have been robustly associated with fasting glucose, 2-hour glucose post-OGTT and T2D.35,36 In recent work confirmed by an independent group, the same locus has been associated with low birth weight.69,70 This finding provides another molecular connection between the observed predictive power of reduced size at birth with regard to future metabolic derangements, including risk of T2D 71.
Most loci point to the β-cell
It is now clear that the risk of developing T2D is a combination of genetic risk for β-cell dysfunction superimposed on genetic and environmental factors (e.g. obesity, Western diet, sedentary lifestyle) that promote insulin resistance. Recent genetic discoveries have identified numerous variants that appear to influence insulin secretion rather than insulin resistance. For example, risk variants of CDKAL1 were associated with insulin secretion defects and impaired insulin response to both oral and IV glucose tolerance tests (IVGTT).14,72,73 Variants in SLC30A8 were associated with insulin secretion defects following OGTT.14,74 HHEX variants linked with impaired β-cell function in response to OGTT and hyperinsulinemic-euglycemic clamp.72,74,75 Finally, CDKN2A/B variants have been associated with impaired glucose-induced insulin release in healthy subjects.75 Of the ten novel variants discovered in association with fasting glycemic traits in a large meta-analysis, nine were associated with fasting glucose/HOMA-B and only one was associated with fasting insulin/HOMA-IR, measures of insulin resistance.35
There are several potential reasons for the dearth of common genetic variants related to insulin resistance found by the GWAS approach. These include study design, different heritability estimates for each of the traits, and the allelic frequency spectrum of these variants. One of the five initial high-density GWAS selected diabetic cases with BMI<30 kg/m2, thus excluding obese individuals;13 another matched cases and controls for BMI.17 These deliberate study designs were chosen to maximize the likelihood of detecting variants that increase T2D risk directly, rather than through the mediation of adiposity. Because loci that achieve genome-wide significance result from the meta-analyses of various scans, some loci that truly cause T2D via insulin resistance may show a trend toward association that fails to reach a threshold that marks them for follow-up. In this way, the studies may not identify loci associated with insulin resistance related to adiposity, but rather, detect loci related only to β-cell function. This issue has become less of a concern as more recent meta-analyses incorporate larger numbers of discovery cohorts that are not ascertained by BMI.
Investigators may bypass this limitation by performing GWAS for insulin resistance as a quantitative trait, rather than basing the discovery of insulin resistance genes in scans for T2D as a categorical trait. One measure that is readily available in large population cohorts is HOMA-IR. However, a meta-analysis of GWAS cohorts in which this phenotype was available identified many more loci influencing β-cell function than those regulating HOMA-IR, suggesting that other reasons exist for this observed discrepancy.
The degree to which each of the two traits is heritable may also contribute to the difficulties in identifying insulin resistance genes. The insulinogenic index is, on average, 10% more heritable than HOMA-IR.76 While HOMA-IR still displays substantial heritability (0.44 among the Framingham Offspring population77), there may be better insulin resistant phenotypes to examine in order to identify genetic associations. For example, in the Insulin Resistance and Atherosclerosis Study Family Study, the insulin sensitivity index (determined from IVGTT) was twice as heritable as HOMA-IR.78 This suggests that the insulin sensitivity index may be a more robust trait to examine when assessing the genetic contribution of insulin resistance; this must be balanced with the difficulty in performing the necessary physiology experiments in a sufficiently high number of individuals..
The distribution of P values in the similarly powered GWAS for HOMA-B and HOMA-IR (both derived from fasting glucose and insulin) suggests a different genetic architecture for the two traits.35 In those meta-analyses, there were many more P values that deviated from the null expectation at the top of the HOMA-B distribution than there were for HOMA-IR (Figure 3). For instance, insulin resistance variants may be fewer in number, rarer in frequency, or have a lower effect size. In addition, there may be a stronger impact of environmental interactions (e.g. diet, physical activity) with the genetic background. Exploring the lower range of the allele frequency spectrum (which requires denser genotyping arrays), integrating environmental factors in the analyses, and increasing the sample size could potentially improve the power to detect genetic variants related to insulin resistance.
Figure 3. Quantile-Quantile Plots for HOMA-B and HOMA-IR in a large meta-analysis.

Figure adapted from reference35.
The quantile-quantile (QQ) plot illustrates the observed P values in the distribution versus those that were expected under the null hypothesis of no association. The null distribution is depicted by the red diagonal line. The entire distribution of P values is in black, the exclusion of the ten newly discovered loci (DGKB-TMEM195, ADCY5, MADD, ADRA2A, FADS1, CRY2, SLC2A2, GLIS3, PROX1, C2CD4B) in green, and the exclusion of the four genome-wide significant fasting glucose-associated loci reported previously (GCK, GCKR, G6PC2 and MTNR1B) in blue. The HOMA-B P values have a larger deviation from expected compared to HOMA-IR and therefore genetic associations with HOMA-B are more likely to be detected than with HOMA-IR.
Nevertheless, several convincing associations with surrogate measures of insulin resistance (HOMA-IR and fasting insulin) have been identified, in part due to larger sample sizes and a novel study design. IRS1 is a highly attractive candidate gene for the development of insulin resistance. Rung et al. demonstrated that SNP rs2943641, 502 kb upstream of IRS1 is associated with T2D, fasting insulin and HOMA-IR. In further functional analyses, the risk allele was associated with reduced levels of IRS1 protein expression and decreased downstream effects noted by phosphatidylinositol-3-OH kinase (PI(3)K) activity (which is activated by IRS1 binding to IGF-1 receptors) in human skeletal muscle biopsies. Such functional experiments were critical in establishing that this variant, located at a non-trivial distance from IRS1, likely acts through IRS1 itself. These findings confirm what is already known biologically about the action of IRS1 in mice. Null Irs1 mice lack PI(3)K activity and demonstrate impaired glucose tolerance and insulin resistance.79
The most recent meta-analysis for fasting glycemic traits uncovered the variant rs35767 1.2 kb upstream of IGF1 as associated with fasting insulin and HOMA-IR.35 IGF1 is another excellent biological candidate for insulin resistance: it encodes the insulin-like growth factor-1, which has been shown to bind insulin receptors and enhance glucose transport in adipose and muscle tissue, while inhibiting hepatic glucose production. Both a null IGF1 mouse and a human with IGF1 gene deletion have resulted in insulin resistance that improves with IGF1 therapy.43 Therefore, it seems intuitive that genetic variation in IGF1 may perturb the expression or function of IGF1, and thereby increase insulin resistance.
In the same large meta-analysis, GCKR was associated with insulin resistance at genome-wide significance levels. Originally, the T allele of rs780094, an intronic SNP in the GCKR gene, was found to be associated with higher triglyceride levels (P=3.7×10−8), and carriers of the T allele were noted to have a trend toward lower glucose, decreased insulin resistance (HOMA-IR), and decreased T2D risk.15 Two follow-up studies confirmed these results.80,81 The directions of these trends (higher triglyceride levels but lower fasting glucose and lower insulin resistance) may be counterintuitive with what we have seen in human epidemiologic studies. This may be explained by the inherent biological mechanism of GCKR, where the functional variant may downregulate gluconeogenesis, increase VLDL-triglyceride synthesis, and also upregulate glucose utilization.42,81
It is clear that as far as the genetic architecture of insulin resistance, we have only visualized the tip of the iceberg. A potentially large proportion of the genetic foundation of T2D remains hidden owing to our difficulties in discovering insulin resistance-related genes. With well-powered samples, more refined phenotyping, assessment of gene-environment interactions, a wider range of BMI, and novel study designs, this genetic information may soon rise to the surface.
Genetic variants identified only explain ~10% of T2D heritability
T2D GWAS have been successful in identifying specific loci that contribute to the causation of the complex disease, but only roughly 10% of the heritability can be accounted for by these variants, suggesting that much remains to be discovered. In the search for the “missing heritability”, firstly the accuracy of original heritability estimates needs to be considered. The familial component caused by a shared early uterine and post-natal environment and latent epigenetic changes (inherited changes in gene expression that are not caused by genetic sequence) may produce a contribution to heritability that is not removed sufficiently by these estimates.82 In addition, GWAS use commercially available genotyping platforms whose early-generation arrays fail to adequately capture nearly 20% of common SNPs, structural variants (such as copy number variants), and variants unique to non-European populations.83 Improving upon these limitations of first-generation genotyping platforms will undoubtedly uncover other loci contributing to the disease. In addition, since a common SNP represents a large segment of genetic material, closer scrutiny of these areas by fine mapping for rare variants is required to find the potential “causal” gene that could have a larger effect than that conferred by the index variant. Through purifying natural selection, high-penetrance deleterious variants should be selectively removed from the population, so we would not expect to find common variants contributing large effect sizes to common disease, but may find large effect sizes in less common variants. Whole-genome sequencing with less costly high-thoughput methods have been developed to discover rarer variants in the human genome. The 1,000 Genomes Project (www.1000genomes.org) is an international initiative designed to catalog all variants with minor allele frequencies greater than 1% of at least 1,000 genomes, thus pushing the envelope of captured shared variation. Initial pilot analyses have successfully identified more than 9 million new polymorphisms, many insertions/deletions, and some large structural variants.84
The Metabochip is another effort to develop a single custom-made chip encompassing 200,000 SNPs culled from deeper layers of the P-value distribution in GWAS for cardiovascular disease, obesity, T2D and related traits. As the Metabochip is deployed across thousands of patients, new and rare variants whose P values are near-the-top but below threshold will be examined in association with these phenotypes. Researchers are also incorporating novel bioinformatics to examine non-additive gene × gene interactions (commonly referred as epistasis). For example, two variants may individually affect genetic risk of T2D only mildly, but together they could increase the risk significantly. In addition, examining gene × environment interactions might lead to insights into biological pathways and guide researchers to novel genes that act synergistically with environmental factors, such as physical activity or dietary composition.85 If successful, this strategy may explain why some genetic associations do not replicate across all populations, hinting to different environmental exposures. Lastly, incorporating the prior probabilities afforded by the mechanistic knowledge garnered through functional experiments in cellular or animal models, investigators may rescue variants that did not make the list of associated loci on purely statistical grounds.
Common genetic variants are not yet useful for clinical prediction
In practical terms, genetic information has been expected to enable clinicians to predict an individual’s risk of developing disease. However, thus far the clinical usefulness of genetic information has been limited. Meigs et al. examined 18 loci associated with T2D in the Framingham Offspring Study in a genotype score in association with T2D incidence, and generated C statistics to determine the individual’s future risk of diabetes.86 While the genotype score predicted diabetes better than gender alone, when adjusted for other easily obtainable clinical parameters (age, sex, family history, BMI, fasting glucose level, systolic blood pressure, HDL cholesterol, and triglyceride levels), predictive power did not improve significantly. A similar study in a larger Swedish cohort examined 16 SNPs associated with T2D and generated largely comparable results.87 In subset analyses, both studies highlighted that younger patients may benefit more from genetic testing before they manifest clinical characteristics of the disease. An updated age-stratified analysis re-examined the genotype score including 40 SNPs associated with T2D in Framingham. The researchers noted that the new genotype score improved the ability to predict the onset of T2D in participants younger than 50 years. Diabetes risk prediction using the genetic variants still needs to be confirmed by other studies, but appears potentially promising in a younger population when medical and lifestyle intervention may be the most feasible and beneficial. 88
An alternative clinical use of genetic information is determining whether genetic information may affect a person’s behavior and thus modify the risk of developing diabetes. The Chinese Da Qing Diabetes Prevention Study, the Finnish Diabetes Prevention Study, the US Diabetes Prevention Program (DPP) and the Indian Diabetes Prevention Program showed that behavioral modification can reduce one’s risk of progressing to diabetes.89-91;Ramachandran, 2006 #2944} For instance, the DPP demonstrated that lifestyle modification led to a 58% reduced incidence of the development of diabetes compared to placebo.91 In further genetic investigation in the DPP, lifestyle modification attenuated the risk of diabetes conferred by the risk variant at TCF7L2: while the placebo participants who carried the homozygous risk genotype at rs7903146 (TT) had 80% higher risk of diabetes development, those randomized to the lifestyle intervention group had no increased risk despite carrying the risk genotype, indicating that lifestyle intervention may trump the genetic risk conferred by this variant.92 This argues that if genetic information could motivate a high-risk individual to change his/her behavior, it may help prevent the eventual onset of disease. However, whether reassuring genetic information may be counterproductive in de-motivating those at lower genetic risk from engaging in healthy behaviors should be empirically tested.
The concept of disease-risk information changing one’s behavior was examined among 150 non-diabetic primary care patients.93 Those with an accurate perception of their higher risk of diabetes (as documented by the Framingham Heart Study diabetes risk score) did not intend to modify their lifestyle any more than patients with low perceived risk. Whether genetic information (rather than perceived or measured risk factors) will have greater influence is being tested. Grant et al. examined how genetic information might influence an individual’s motivation for lifestyle change.94 After questioning patients regarding how they would respond to information containing their genetic risk of T2D development, 71% of those surveyed indicated that such testing would motivate them to adopt lifestyle change. On the contrary, only 1.3% of the patients reported they would be less motivated by genetic information indicating they were at low risk. Therefore, despite the limited ability of genetic testing to predict T2D when adjusted for common risk factors, genetic information may be more powerful in persuading a patient to change his or her behavior. Further studies, including more surveyed individuals and prospective studies, are need to determine if the person’s behavior would in fact change.
Genetic findings provide an opportunity for therapeutic intervention and pharmacogenetic clinical trials
The underlying biological mechanisms which GWAS have unveiled could be targets for new pharmacotherapy. For example, prior biological knowledge regarding SLC30A8 and new genetic evidence have drawn our attention to a possible therapeutic target. A pharmacological agent that enhances the function of this transporter could hypothetically increase insulin secretion and improve β-cell function. Given the novelty of the GWAS findings, there has yet to be a therapeutic that has specifically evolved from these discoveries, but it seems that there is a wide breadth of possibilities on the horizon.
Studies to examine the pharmacogenetics of T2D have been encouraged by findings of variable pharmacotherapy response among individuals with monogenic forms of diabetes. One of the most impressive stories lies in the discovery of activating mutations in KCNJ11 (encoding the Kir6.2 subunit of the ATP-sensitive potassium channel), which causes the channels to remain open in the presence of glucose, thereby reducing insulin secretion and giving rise to permanent neonatal diabetes (PNDM).95 Infants carrying these mutations present with marked hyperglycemia or diabetic ketoacidosis at less than six months of age. Three individuals harboring mutations in KCNJ11 were successfully transitioned from insulin to a sulfonylurea with an improvement in glycemic control compared to insulin therapy.96 These findings were later confirmed by larger study populations of patients with PNDM.95,97 Similarly, the enhanced efficacy of sulfonylureas in patients with MODY3 was described with a fivefold greater response to sulfonylurea versus metformin therapy.98
Preliminary pharmacogenetic studies in polygenic diabetes have been initiated. These studies have primarily involved studying PPARG, KCNJ11, and TCF7L2, whose variants have been associated with T2D at genome-wide levels of signficance (as previously mentioned, the first two encode known targets of antidiabetic medications).
Given its therapeutic relavance in PNDM and MODY3, the influence of KCNJ11 variants has been examined in response to sulfonylurea treatment in T2D. Sesti et al. conducted a prospective trial of 524 European T2D patients who were treated with a progressive antidiabetic medication regimen which escalated with each medication failure. They started with glibenclamide, followed by metformin and then insulin. Carriers of the T2D risk K allele had a relative risk of 1.45 compared to E23E homozygotes for sulfonylurea failure.99 Feng et al. found an association of rs757110 (a SNP in the adjacent gene ABCC8, which encodes the sulfonylurea receptor SUR1) with response to sulfonurea treatment in Chinese patients with T2D, measured by a decrease in mean fasting glucose.100 The missense mutation at rs757110, S1369A, is in high LD (r2=0.87) with the E23K polymorphism, such that they are statistically indistinguishable. In contrast to Sesti et al., in the Chinese study carriers of the risk genotype showed greater responsiveness to gliclazide, which is consistent with the PNDM findings; whether such responsiveness also heralds a greater risk of long-term sulfonylurea failure must be tested in longer pharmacogenetic trials.
PPARG is an attractive genetic target because it is the known drug target of thiazolidinediones, such as troglitazone, rosiglitazone, and pioglitazone.101 Initial studies reporting that genotype at the index variant in PPARG (rs1801282, P12A) or at other nearby variants predict a better response to troglitazone therapy 102,103 have not been substantiated.104-106
Lastly, TCF7L2 has also been under investigation with regards to response to antidiabetes medication, despite its mechanism not being as well understood as that of KCNJ11/ABCC8 or PPARG. The risk variant in this gene confers a defect in insulin secretion, and possibly affects GLP-1 metabolism.107 The Go-DARTS study group has examined the variation in medication response based on TCF7L2 genotype in two settings. Firstly, they noticed an overrepresentation of the T risk allele in people with T2D treated with insulin versus diet therapy alone. In a second study, they demonstrated carriers of the risk TCF7L2 variants were more likely to fail sulfonylurea therapy than metformin.108,109
Which road do we take from here?
GWAS have led the way toward an exciting journey into understanding the genetic architecture of T2D and related traits. Critical building blocks have been established. With the development of HapMap and 1,000 Genomes, we have the ability to use imputation to combine different genotyping platforms and continue international collaborations involving multiple cohorts. Advancing technology has enabled large-scale genotyping with high quality data. Innovative statistical analyses, often publically available, have allowed the field to analyze this growing wealth of data. Strategies to identify causal variants include deploying next-generation sequencing techniques, examining the less common allelic spectrum, exploiting genetic pleiotropy, fine-mapping loci with biological relevance, and applying biological insight to the discovered associations. Ultimately, investigators strive to translate this genetic knowledge to the clinician’s bedside. Perhaps most importantly, the collaborative spirit that has permeated the field may be the most significant achievement catalyzing our enhanced understanding of the genetics underlying this complex disease.
Box 1. What have we learned from GWAS?
- They illustrate novel pathways.
- The association of a missense mutation rs13266634 in SLC30A8 (encoding Zn2+ Transporter, ZnT-8) with type 2 diabetes has highlighted the importance of Zn2+ transport in the β cell, the variant’s influence on insulin packaging and secretion, and this pathway’s potential relevance as a drug target.
- New findings point to fundamental biology.
- The intronic SNP rs7903146 in TCF7L2 is located in an open chromatin site in β cells; its risk T alllele is correlated with an increased transcription in human islets and with increased expression in cellular luciferase assays.
- Genetic discoveries support prior epidemiological observations.
- The T allele of rs17550 in FADS1 is associated with higher fasting glucose, lower HOMA-B, increased LDL and HDL cholesterol, and decreased triglycerides.
- Variants in HNF1A are associated with type 2 diabetes, C-reactive protein, coronary artery disease, and LDL cholesterol levels.
- MTNR1B and CRY2 link T2D and circadian rhythms, in support of animal and human studies.
- ADCY5 variants increase fasting and 2-hour glucose, as well as risk of type 2 diabetes; they are also associated with lower birth weight.
- Most loci point to the β cell.
- The majority of loci associated with type 2 diabetes point to primary defects in the β cell. However,
- IRS1 (encoding insulin receptor substrate-1) has been associated with T2D.
- Both IGF1 and GCKR have been associated with measures of insulin resistance (HOMA-IR and fasting insulin) at genome-wide levels of significance.
- Increasing sample size and modifying study design have helped discover variants associated with insulin resistance.
- So far genetic variants only explain 10% of T2D heritability.
- GWAS have been limited to common variants and primarily populations of European descent.
- In order to find the “missing heritability” investigators are pursuing fine-mapping around the associated regions, leveraging the 1,000 Genomes project, applying next-generation sequencing, analyzing the Metabochip, using improved informatics for gene × gene and gene × environment interactions, expanding to non-white populations, and incorporating prior biological knowledge to interpret significance of variants.
- Common genetic variants are not yet useful in clinical prediction.
- Although variants may be limited in their ability to predict type 2 diabetes, genetic information may sway a person to change lifestyle behavior that may reduce their risk of developing the disease.
- Genetic prediction may be more useful in younger age groups, before clinical risk factors develop.
- They provide an opportunity for therapeutic intervention and pharmacogenetic clinical trials.
- Pharmacogenetic studies in polygenic diabetes have studied primarily PPARG, KCNJ11, TCF7L2.
- Two correlated variants at KCNJ11 and ABCC8 are associated with sulfonylurea failure and decreased mean fasting glucose on sulfonylurea therapy.
- Carriers of risk variants at TCF7L2 are more likely to fail sulfonulyurea therapy than metformin and more likely to be on insulin therapy rather than diet alone.
Box 2. Which road do we take from here?
Using next-generation sequencing techniques to discover less common and rare variation.
Fine-mapping and applying biological insight to the discovered associations to determine the causal variant.
Exploiting genetic pleiotropy to understand how these genetic variants link common diseases.
Continuing to develop informatics methods that examine epistasis and gene × environment interaction.
Examining populations of non-European descent.
Translating genetic knowledge to the clinician’s bedside with targeted treatment algorithms and risk assessment tools that may influence patient’s behavior based on risk.
Leveraging the welcome collaborative spirit that has permeated the field to catalyze large international studies to enhance our understanding of the genetics of T2D.
Acknowledgements
L.K.B. is supported by NIH training grant 5 T32 DK007028-35. J.C.F. is supported by the Massachusetts General Hospital and a Clinical Scientist Development Award by the Doris Duke Charitable Foundation. We thank Erik Billings for help in adapting Figure 1. We thank the members of the Florez research group and the larger diabetes genetics community for fruitful and engaging discussions.
REFERENCES
- 1.Vaxillaire M, Froguel P. Genetic basis of maturity-onset diabetes of the young. Endocrinol Metab Clin North Am. 2006;35:371–384. doi: 10.1016/j.ecl.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Reynisdottir I, et al. Localization of a susceptibility gene for type 2 diabetes to chromosome 5q34-q35.2. Am J Hum Genet. 2003;73:323–335. doi: 10.1086/377139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grant SF, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–323. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 4.Altshuler D, et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;26:76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 5.Gloyn AL, et al. Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes. 2003;52:568–572. doi: 10.2337/diabetes.52.2.568. [DOI] [PubMed] [Google Scholar]
- 6.Florez JC, et al. Haplotype structure and genotype-phenotype correlations of the sulfonylurea receptor and the islet ATP-sensitive potassium channel gene region. Diabetes. 2004;53:1360–1368. doi: 10.2337/diabetes.53.5.1360. [DOI] [PubMed] [Google Scholar]
- 7.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 8.Venter JC, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 9.The International HapMap Consortium The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 10.Frazer KA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sachidanandam R, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- 12.The International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sladek R, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 14.Steinthorsdottir V, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet. 2007;39:770–775. doi: 10.1038/ng2043. [DOI] [PubMed] [Google Scholar]
- 15.Zeggini E, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott LJ, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University. Novartis Institutes of BioMedical Research Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 18.Zeggini E, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol. 2008;32:381–385. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 20.Egefjord L, et al. Zinc transporter gene expression is regulated by pro-inflammatory cytokines: a potential role for zinc transporters in beta-cell apoptosis? BMC Endocr Disord. 2009;9:7. doi: 10.1186/1472-6823-9-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prokopenko I, et al. Variants in MTNR1B influence fasting glucose levels. Nat Genet. 2009;41:77–81. doi: 10.1038/ng.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bouatia-Naji N, et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet. 2009;41:89–94. doi: 10.1038/ng.277. [DOI] [PubMed] [Google Scholar]
- 23.Lyssenko V, et al. Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat Genet. 2009;41:82–88. doi: 10.1038/ng.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yasuda K, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet. 2008;40:1092–1097. doi: 10.1038/ng.207. [DOI] [PubMed] [Google Scholar]
- 25.Unoki H, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet. 2008;40:1098–1102. doi: 10.1038/ng.208. [DOI] [PubMed] [Google Scholar]
- 26.Matthews DR, et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 27.Rung J, et al. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat Genet. 2009;41:1110–1115. doi: 10.1038/ng.443. [DOI] [PubMed] [Google Scholar]
- 28.Kassem SA, et al. p57(KIP2) expression in normal islet cells and in hyperinsulinism of infancy. Diabetes. 2001;50:2763–2769. doi: 10.2337/diabetes.50.12.2763. [DOI] [PubMed] [Google Scholar]
- 29.Weedon MN, et al. A common haplotype of the glucokinase gene alters fasting glucose and birth weight: association in six studies and population-genetics analyses. Am J Hum Genet. 2006;79:991–1001. doi: 10.1086/509517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bouatia-Naji N, et al. A polymorphism within the G6PC2 gene is associated with fasting plasma glucose levels. Science. 2008;320:1085–1088. doi: 10.1126/science.1156849. [DOI] [PubMed] [Google Scholar]
- 31.Chen WM, et al. Variations in the G6PC2/ABCB11 genomic region are associated with fasting glucose levels. J Clin Invest. 2008;118:2620–2628. doi: 10.1172/JCI34566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boden G, Ruiz J, Urbain JL, Chen X. Evidence for a circadian rhythm of insulin secretion. Am J Physiol. 1996;271:E246–252. doi: 10.1152/ajpendo.1996.271.2.E246. [DOI] [PubMed] [Google Scholar]
- 33.Van Cauter E. Putative roles of melatonin in glucose regulation. Therapie. 1998;53:467–472. [PubMed] [Google Scholar]
- 34.Peschke E, et al. Diabetic Goto Kakizaki rats as well as type 2 diabetic patients show a decreased diurnal serum melatonin level and an increased pancreatic melatonin-receptor status. J Pineal Res. 2006;40:135–143. doi: 10.1111/j.1600-079X.2005.00287.x. [DOI] [PubMed] [Google Scholar]
- 35.Dupuis J, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saxena R, et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet. 42:142–148. doi: 10.1038/ng.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soranzo N, et al. Common variants at ten genomic loci influence hemoglobin A1C levels via glycemic and non-glycemic pathways. Diabetes. doi: 10.2337/db10-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gautier JF, Choukem SP, Girard J. Physiology of incretins (GIP and GLP-1) and abnormalities in type 2 diabetes. Diabetes Metab. 2008;34(Suppl 2):S65–72. doi: 10.1016/S1262-3636(08)73397-4. [DOI] [PubMed] [Google Scholar]
- 39.Nauck MA, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab. 1986;63:492–498. doi: 10.1210/jcem-63-2-492. [DOI] [PubMed] [Google Scholar]
- 40.Ingelsson E, et al. Detailed physiologic characterization reveals diverse mechanisms for novel genetic Loci regulating glucose and insulin metabolism in humans. Diabetes. 59:1266–1275. doi: 10.2337/db09-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao C, Giacca A, Carpentier A, Lewis GF. Differential effects of monounsaturated, polyunsaturated and saturated fat ingestion on glucose-stimulated insulin secretion, sensitivity and clearance in overweight and obese, non-diabetic humans. Diabetologia. 2006;49:1371–1379. doi: 10.1007/s00125-006-0211-x. [DOI] [PubMed] [Google Scholar]
- 42.Beer NL, et al. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet. 2009;18:4081–4088. doi: 10.1093/hmg/ddp357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clemmons DR. Role of insulin-like growth factor iin maintaining normal glucose homeostasis. Horm Res. 2004;62(Suppl 1):77–82. doi: 10.1159/000080763. [DOI] [PubMed] [Google Scholar]
- 44.Murgia C, et al. Diabetes-linked zinc transporter ZnT8 is a homodimeric protein expressed by distinct rodent endocrine cell types in the pancreas and other glands. Nutr Metab Cardiovasc Dis. 2009;19:431–439. doi: 10.1016/j.numecd.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 45.Chimienti F, Devergnas S, Favier A, Seve M. Identification and cloning of a beta-cell-specific zinc transporter, ZnT-8, localized into insulin secretory granules. Diabetes. 2004;53:2330–2337. doi: 10.2337/diabetes.53.9.2330. [DOI] [PubMed] [Google Scholar]
- 46.Chimienti F, Favier A, Seve M. ZnT-8, a pancreatic beta-cell-specific zinc transporter. Biometals. 2005;18:313–317. doi: 10.1007/s10534-005-3687-9. [DOI] [PubMed] [Google Scholar]
- 47.Chimienti F, et al. In vivo expression and functional characterization of the zinc transporter ZnT8 in glucose-induced insulin secretion. J Cell Sci. 2006;119:4199–4206. doi: 10.1242/jcs.03164. [DOI] [PubMed] [Google Scholar]
- 48.El Muayed M, et al. Acute cytokine-mediated downregulation of the zinc transporter ZnT8 alters pancreatic beta-cell function. J Endocrinol. 206:159–169. doi: 10.1677/JOE-09-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pound LD, et al. Deletion of the mouse Slc30a8 gene encoding zinc transporter-8 results in impaired insulin secretion. Biochem J. 2009;421:371–376. doi: 10.1042/BJ20090530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nicolson TJ, et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes. 2009;58:2070–2083. doi: 10.2337/db09-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cauchi S, et al. TCF7L2 is reproducibly associated with type 2 diabetes in various ethnic groups: a global meta-analysis. J Mol Med. 2007;85:777–782. doi: 10.1007/s00109-007-0203-4. [DOI] [PubMed] [Google Scholar]
- 52.Florez JC. The new type 2 diabetes gene TCF7L2. Curr Opin Clin Nutr Metab Care. 2007;10:391–396. doi: 10.1097/MCO.0b013e3281e2c9be. [DOI] [PubMed] [Google Scholar]
- 53.Gaulton KJ, et al. A map of open chromatin in human pancreatic islets. Nat Genet. 42:255–259. doi: 10.1038/ng.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wallrath LL, Lu Q, Granok H, Elgin SC. Architectural variations of inducible eukaryotic promoters: preset and remodeling chromatin structures. Bioessays. 1994;16:165–170. doi: 10.1002/bies.950160306. [DOI] [PubMed] [Google Scholar]
- 55.Weedon MN, et al. A common variant of HMGA2 is associated with adult and childhood height in the general population. Nat Genet. 2007;39:1245–1250. doi: 10.1038/ng2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frayling TM, Colhoun H, Florez JC. A genetic link between type 2 diabetes and prostate cancer. Diabetologia. 2008;51:1757–1760. doi: 10.1007/s00125-008-1114-9. [DOI] [PubMed] [Google Scholar]
- 57.Kathiresan S, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41:56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sabatti C, et al. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat Genet. 2009;41:35–46. doi: 10.1038/ng.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barber MJ, et al. Genome-wide association of lipid-lowering response to statins in combined study populations. PLoS One. 5:e9763. doi: 10.1371/journal.pone.0009763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hua QX, et al. Diabetes-associated mutations in a beta-cell transcription factor destabilize an antiparallel “mini-zipper” in a dimerization interface. Proc Natl Acad Sci U S A. 2000;97:1999–2004. doi: 10.1073/pnas.97.5.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Erdmann J, et al. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. 2009;41:280–282. doi: 10.1038/ng.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elliott P, et al. Genetic Loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA. 2009;302:37–48. doi: 10.1001/jama.2009.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reiner AP, et al. Polymorphisms of the HNF1A gene encoding hepatocyte nuclear factor-1 alpha are associated with C-reactive protein. Am J Hum Genet. 2008;82:1193–1201. doi: 10.1016/j.ajhg.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ridker PM, et al. Loci related to metabolic-syndrome pathways including LEPR,HNF1A, IL6R, and GCKR associate with plasma C-reactive protein: the Women’s Genome Health Study. Am J Hum Genet. 2008;82:1185–1192. doi: 10.1016/j.ajhg.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Voight BF, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Odom DT, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378–1381. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Turek FW, et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308:1043–1045. doi: 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marcheva B, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 466:627–631. doi: 10.1038/nature09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Freathy RM, et al. Variants in ADCY5 and near CCNL1 are associated with fetal growth and birth weight. Nat Genet. 42:430–435. doi: 10.1038/ng.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Andersson EA, et al. Type 2 diabetes risk alleles near ADCY5, CDKAL1 and HHEX-IDE are associated with reduced birthweight. Diabetologia. 53:1908–1916. doi: 10.1007/s00125-010-1790-0. [DOI] [PubMed] [Google Scholar]
- 71.Barker DJ, et al. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993;36:62–67. doi: 10.1007/BF00399095. [DOI] [PubMed] [Google Scholar]
- 72.Pascoe L, et al. Common variants of the novel type 2 diabetes genes CDKAL1 and HHEX/IDE are associated with decreased pancreatic beta-cell function. Diabetes. 2007;56:3101–3104. doi: 10.2337/db07-0634. [DOI] [PubMed] [Google Scholar]
- 73.Palmer ND, et al. Quantitative trait analysis of type 2 diabetes susceptibility loci identified from whole genome association studies in the Insulin Resistance Atherosclerosis Family Study. Diabetes. 2008;57:1093–1100. doi: 10.2337/db07-1169. [DOI] [PubMed] [Google Scholar]
- 74.Staiger H, et al. Polymorphisms within novel risk loci for type 2 diabetes determine beta-cell function. PLoS One. 2007;2:e832. doi: 10.1371/journal.pone.0000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grarup N, et al. Studies of association of variants near the HHEX, CDKN2A/B, and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10,705 Danish subjects: validation and extension of genome-wide association studies. Diabetes. 2007;56:3105–3111. doi: 10.2337/db07-0856. [DOI] [PubMed] [Google Scholar]
- 76.Florez JC. Newly identified loci highlight beta cell dysfunction as a key cause of type 2 diabetes: where are the insulin resistance genes? Diabetologia. 2008;51:1100–1110. doi: 10.1007/s00125-008-1025-9. [DOI] [PubMed] [Google Scholar]
- 77.Panhuysen CI, et al. A genome scan for loci linked to quantitative insulin traits in persons without diabetes: the Framingham Offspring Study. Diabetologia. 2003;46:579–587. doi: 10.1007/s00125-003-1066-z. [DOI] [PubMed] [Google Scholar]
- 78.Bergman RN, et al. Minimal model-based insulin sensitivity has greater heritability and a different genetic basis than homeostasis model assessment or fasting insulin. Diabetes. 2003;52:2168–2174. doi: 10.2337/diabetes.52.8.2168. [DOI] [PubMed] [Google Scholar]
- 79.Araki E, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372:186–190. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- 80.Sparso T, et al. The GCKR rs780094 polymorphism is associated with elevated fasting serum triacylglycerol, reduced fasting and OGTT-related insulinaemia, and reduced risk of type 2 diabetes. Diabetologia. 2008;51:70–75. doi: 10.1007/s00125-007-0865-z. [DOI] [PubMed] [Google Scholar]
- 81.Orho-Melander M, et al. Common missense variant in the glucokinase regulatory protein gene is associated with increased plasma triglyceride and C-reactive protein but lower fasting glucose concentrations. Diabetes. 2008;57:3112–3121. doi: 10.2337/db08-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maher B. Personal genomes: The case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 83.Florez JC. Clinical review: the genetics of type 2 diabetes: a realistic appraisal in 2008. J Clin Endocrinol Metab. 2008;93:4633–4642. doi: 10.1210/jc.2008-1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Via M, Gignoux C, Burchard EG. The 1000 Genomes Project: new opportunities for research and social challenges. Genome Med. 2:3. doi: 10.1186/gm124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thomas D. Gene-environment-wide association studies: emerging approaches. Nat Rev Genet. 11:259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Meigs JB, et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med. 2008;359:2208–2219. doi: 10.1056/NEJMoa0804742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lyssenko V, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359:2220–2232. doi: 10.1056/NEJMoa0801869. [DOI] [PubMed] [Google Scholar]
- 88.de Miguel-Yanes JM. Genetic risk reclassification for type 2 diabetes by age below or above 50 years using 40 type 2 diabetes risk single nucleotide polymorphisms. Diabetes Care. 2010 doi: 10.2337/dc10-1265. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pan XR, et al. Effects of diet and exercise in preventing NIDDM in people with impaired glucose tolerance. The Da Qing IGT and Diabetes Study. Diabetes Care. 1997;20:537–544. doi: 10.2337/diacare.20.4.537. [DOI] [PubMed] [Google Scholar]
- 90.Tuomilehto J, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344:1343–1350. doi: 10.1056/NEJM200105033441801. [DOI] [PubMed] [Google Scholar]
- 91.Knowler WC, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Florez JC, et al. TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. N Engl J Med. 2006;355:241–250. doi: 10.1056/NEJMoa062418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hivert MF, Warner AS, Shrader P, Grant RW, Meigs JB. Diabetes risk perception and intention to adopt healthy lifestyles among primary care patients. Diabetes Care. 2009;32:1820–1822. doi: 10.2337/dc09-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grant RW, et al. The clinical application of genetic testing in type 2 diabetes: a patient and physician survey. Diabetologia. 2009;52:2299–2305. doi: 10.1007/s00125-009-1512-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pearson ER, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med. 2006;355:467–477. doi: 10.1056/NEJMoa061759. [DOI] [PubMed] [Google Scholar]
- 96.Sagen JV, et al. Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes. 2004;53:2713–2718. doi: 10.2337/diabetes.53.10.2713. [DOI] [PubMed] [Google Scholar]
- 97.Babenko AP, et al. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med. 2006;355:456–466. doi: 10.1056/NEJMoa055068. [DOI] [PubMed] [Google Scholar]
- 98.Pearson ER, et al. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet. 2003;362:1275–1281. doi: 10.1016/S0140-6736(03)14571-0. [DOI] [PubMed] [Google Scholar]
- 99.Sesti G, et al. The E23K variant of KCNJ11 encoding the pancreatic beta-cell adenosine 5′-triphosphate-sensitive potassium channel subunit Kir6.2 is associated with an increased risk of secondary failure to sulfonylurea in patients with type 2 diabetes. J Clin Endocrinol Metab. 2006;91:2334–2339. doi: 10.1210/jc.2005-2323. [DOI] [PubMed] [Google Scholar]
- 100.Feng Y, et al. Ser1369Ala variant in sulfonylurea receptor gene ABCC8 is associated with antidiabetic efficacy of gliclazide in Chinese type 2 diabetic patients. Diabetes Care. 2008;31:1939–1944. doi: 10.2337/dc07-2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lehmann JM, et al. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 102.Kang ES, et al. Effects of Pro12Ala polymorphism of peroxisome proliferator-activated receptor γ2 gene on rosiglitazone response in type 2 diabetes. Clin Pharmacol Ther. 2005;78:202–208. doi: 10.1016/j.clpt.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 103.Wolford JK, et al. Sequence variation in PPARG may underlie differential response to troglitazone. Diabetes. 2005;54:3319–3325. doi: 10.2337/diabetes.54.11.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bluher M, Lubben G, Paschke R. Analysis of the relationship between the Pro12Ala variant in the PPAR-γ2 gene and the response rate to therapy with pioglitazone in patients with type 2 diabetes. Diabetes Care. 2003;26:825–831. doi: 10.2337/diacare.26.3.825. [DOI] [PubMed] [Google Scholar]
- 105.Snitker S, et al. Changes in insulin sensitivity in response to troglitazone do not differ between subjects with and without the common, functional Pro12Ala peroxisome proliferator-activated receptor-γ2 gene variant: results from the Troglitazone in Prevention of Diabetes (TRIPOD) study. Diabetes Care. 2004;27:1365–1368. doi: 10.2337/diacare.27.6.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Florez JC, et al. Effects of the type 2 diabetes-associated PPARG P12A polymorphism on progression to diabetes and response to troglitazone. J Clin Endocrinol Metab. 2007;92:1502–1509. doi: 10.1210/jc.2006-2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pearson ER. Translating TCF7L2: from gene to function. Diabetologia. 2009;52:1227–1230. doi: 10.1007/s00125-009-1356-1. [DOI] [PubMed] [Google Scholar]
- 108.Kimber CH, et al. TCF7L2 in the Go-DARTS study: evidence for a gene dose effect on both diabetes susceptibility and control of glucose levels. Diabetologia. 2007;50:1186–1191. doi: 10.1007/s00125-007-0661-9. [DOI] [PubMed] [Google Scholar]
- 109.Pearson ER, et al. Variation in TCF7L2 influences therapeutic response to sulfonylureas: a GoDARTs study. Diabetes. 2007;56:2178–2182. doi: 10.2337/db07-0440. [DOI] [PubMed] [Google Scholar]
- 110.Winckler W, et al. Evaluation of common variants in the six known maturity-onset diabetes of the young (MODY) genes for association with type 2 diabetes. Diabetes. 2007;56:685–693. doi: 10.2337/db06-0202. [DOI] [PubMed] [Google Scholar]
- 111.Gudmundsson J, et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet. 2007;39:977–983. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- 112.McCarthy MI, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 113.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]