

Abstract
Abnormal aggregation of islet amyloid polypeptide (IAPP) into amyloid fibrils is a hallmark of type 2 diabetes. In this study, we investigated the initial oligomerization and subsequent addition of monomers to growing aggregates of human IAPP at the residue-specific level using NMR, atomic force microscopy, mass spectroscopy, and computational simulations. We found that in solution IAPPs rapidly associate into transient low-order oligomers such as dimers and trimers via interactions between histidine 18 and tyrosine 37. This initial event is proceeded by slow aggregation into higher-order spherical oligomers and elongated fibrils. In these two morphologically distinct types of aggregates IAPPs adopt structures with markedly different residual flexibility. Here we show that the anti-amyloidogenic compound resveratrol inhibits oligomerization and amyloid formation via binding to histidine 18, supporting the finding that this residue is crucial for on-pathway oligomer formation.
Keywords: Amyloid, Atomic Force Microscopy, Mass Spectrometry (MS), Molecular Dynamics, NMR
Introduction
A number of progressive diseases, including Alzheimer and Parkinson diseases, spongiform encephalopathy, and type 2 diabetes (T2D),3 are directly associated with aggregation of specific proteins (1). Despite their diverse sequences and native structures, the aggregated proteins all become disordered and assemble into similar linear, unbranched fibrils, which are commonly referred to as amyloid (2). A characteristic x-ray diffraction pattern of amyloid fibrils suggests they all commonly share a core cross-β structure consisting of a helical array of β-sheets parallel to the fibril long axis and with strands perpendicular to the axis (3–4). Amyloid formation is generally described by a self-replicating nucleation-dependent polymerization model in which the assembly pathway includes a nucleation step and subsequent fibril growth (5–6). In the nucleated conformational conversion model, spherical, fluid-like oligomeric complexes appear to be crucial intermediates in amyloid nucleus formation (7). Increasing evidence suggests, rather than mature amyloid fibrils, the formation of these intermediate aggregates mediates high cytotoxicity (7–11). Additionally, amyloid growth may also occur in the absence of oligomeric intermediates through direct monomer addition. This suggests that an alternative amyloid growth pathway exists, which is distinct from and competes with the nucleation-dependent model for formation of potentially toxic oligomeric intermediates (12). Despite great interest in the distinct amyloid formation pathways, a comprehensive understanding of the underlying mechanisms of amyloid formation, especially at the residue specific level, is impeded by fast aggregation and co-existing oligomeric intermediates and fibrils.
Human IAPP (13), the main component of extracellular amyloid deposits found in the pancreas of most T2D patients (14), is a 37-amino acid polypeptide hormone secreted together with insulin by pancreatic β cells participating in regulating glucose metabolism. Whereas IAPP is largely unstructured and nontoxic in monomeric form, it shows high cytotoxicity when aggregated into β-sheet-rich amyloids. The relationship between IAPP amyloid formation and T2D progression is further highlighted by the observation that transgenic mice, which have the non-amyloidogenic IAPP variant will still develop T2D when fed a high-fat diet (15–16). Synthetic IAPP will form amyloid fibrils in vitro which are morphologically similar to pancreatic amyloid deposits. The kinetics of in vitro amyloid formation follows the nucleation-dependent polymerization mechanism with a distinctive lag time before fibrillogenesis (17). Two distinct phases, oligomer growth followed by fibril elongation, are reported in amyloid formation (18). During oligomer growth molten globule-like oligomers mediate a conformational transition of IAPP from nearly random coil to β-sheet for subsequent amyloid formation (17). Because early oligomers might be the most cytotoxic species that induce β-cell apoptosis (19) it is of high importance to determine the molecular mechanisms of oligomerization in the prefibrillar stage.
In this study, we combined NMR spectroscopy, atomic force microscopy (AFM), mass spectrometry (MS), and computer simulations of molecular dynamics (MD) to investigate the self-association mechanism of IAPP in the initial oligomerization stage as well as the subsequent aggregation mechanisms at residue-specific resolution. Distinct aggregation processes resulting in high-order spherical oligomers or fibrils were monitored by NMR with the corresponding morphologies analyzed by AFM. We demonstrated that both the phenolic compound resveratrol and insulin could inhibit IAPP aggregation by blocking the critical residues involved in IAPP self-association, which might represent a natural mechanism for IAPP amyloid inhibition.
EXPERIMENTAL PROCEDURES
Expression and Purification of Unlabeled and Uniformly Labeled IAPP
The amino acid sequence of human IAPP is: 1KCNTA 6TCATQ 11RLANF 16LVHSS 21NNFGA 26ILSST 31NVGSN 36TY. Recombinant IAPP was expressed and purified according to the protocol described by Lopes et al. (20–21) with the following modifications: DNA oligonucleotide containing coding sequences of an enterokinase (Ek) cleavage site and IAPP was synthesized and cloned into pBluescriptII SK(+) plasmid vector between KpnI and SalI restriction sites. The coding sequence of IAPP was optimized for expression in Escherichia coli. The sequence was then ligated into a pET32a vector, which was previously dephosphorylated and digested with KpnI and SalI (New England Biolabs, Inc). The plasmid was subsequently cloned into DH5α E. coli cells and verified by sequencing. The pET32a construct was then transformed into BL21 competent cells and grown in LB medium (for an unlabeled peptide) or M9 minimal medium (for an uniformly isotopically labeled peptide) with 100 μg/ml ampicillin at 37 °C. Expression was induced was by the addition of 1 mm isopropyl-β-d-thiogalactopyranoside (IPTG) when the culture reached an A600 of 0.8, and the cells were harvested after 6 h.
The harvested cells were centrifuged at 7,000 × g at 4 °C for 30 min, and resuspended in lysis buffer (20 mm Tris-HCl, pH 7.4, 500 mm NaCl, 1 mm imidazole,) and lysed using a French press. The suspension was centrifuged at 40,000 × g at 4 °C for 40 min and the inclusion bodies containing IAPP were collected as a pellet. The pellet was resuspended in the same buffer containing 6 m urea and sonicated to solubilize the inclusion bodies. After removing the insoluble material by centrifugation, the sample was purified using a 5 ml HisTrapTM FF column according to the steps described by Dahabada et al. (20). The IAPP was eluted from the column with lysis buffer containing 6 m urea and purified by reverse-phase HPLC on a C18 column using 0–70% acetronitrile (ACN)/water gradient with 0.045% trifluoroacetic acid (TFA) over 70 min. Matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) was used to assess the purity and verify the size of IAPP.
Thioflavin T Assay
Lyophilized IAPP was first dissolved in 100% hexafluoroisopropanol (HFIP) at a concentration of 1 mm, 30-μl aliquots were dried and lyophilized and individual aliquots were redissolved in 150 μl of buffer (10 mm sodium phosphate pH 6, 20 mm NaCl, 1 mm ThT) in the presence and absence of 100 μm resveratrol. The resulting fluorescence signal was measured immediately at excitation and emission wavelengths of 420 nm and 487 nm, respectively.
NMR Spectroscopy
All NMR experiments were performed using Bruker spectrometers operating at proton Larmor frequencies of either 600.13 or 700.25 MHz 25 °C. Uniformly 15N- or 13C, 15N-labeled IAPP were dissolved in 100% HFIP and dried for the subsequent use. [1H,15N]TROSY spectra were acquired to monitor initial oligomerization and subsequent aggregation processes. The IAPP chemical shifts as function of peptide concentration, pH, resveratrol, and insulin were monitored in a series of [1H,13C]-HSQC and [1H,15N]-HMQC experiments. Three-dimensional 15N-resolved NOESY-HSQC (τmix = 200 ms) was used to obtain {1H,1H}-NOEs. [1H,15N]-HMQC spectra of insoluble IAPP aggregates were acquired under high-resolution magic angle spinning (HR-MAS) using a Bruker 4-mm HR-MAS probe with the sample spinning at a rate of 10 kHz. The chemical shifts were referenced to internal 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) directly for 1H and indirectly for 13C and 15N channels.
Structural Modeling of IAPP in Fibrils using Molecular Dynamic Simulations
Structural models of IAPP in solution were reconstructed by MD simulations of monomeric and dimeric human IAPP with the AMBER 9 package (22) using a Generalized Born (GB) implicit solvent model (23) constrained by 13Cα, 13Cβ chemical shifts and {1H,1H}-NOEs. In 20 MD simulations (10 for monomeric and 10 for dimeric IAPP) the system was initially heated up to 600 K and cooled down to 300 K within the first 600 ps, and then maintained at 300 K for 20 ns. At 300 K, 217 NOE constraints were added to both monomeric and dimeric IAPP forms as intramolecular distance-restrained potentials. For the dimeric form additional constraints between inter-strand His-18 and Tyr-37 were applied. The trajectories from the last 10 ns of each run were concatenated and the root mean square deviation (RMSD) based on a structural clustering scheme was imposed on the ensemble to obtain the dominant structural cluster. Seven conformers of IAPP from the dominant cluster with the lowest total energy (supplemental Table S1) were selected to represent IAPP structure in solution.
Residual structural flexibility of IAPP in fibrils was modeled by MD simulations using the GROMACS suite of programs (24). A fibril subunit was extracted from the 2-fold symmetric model of the protofibrils (25). It consists of two antiparallel β-sheet layers, with each layer containing 5 peptides (see Fig. 7A). Each polypeptide chain was arranged in a β-strand-loop-β-strand fold: an N-terminal β-strand (residues 6–19), a loop (residues 20–23) and a C-terminal β-strand (residues 24–36). The fibril subunit was modeled by an all-atom OPLS force field (26) and solvated by explicit SPC water molecules in a dodecahedron box. The final simulation systems contained 10 hIAPP1–37 peptides and 17833 water molecules with 30 chloride anions.
FIGURE 7.

Resveratrol inhibits IAPP amyloid formation. A, kinetics of IAPP aggregation and inhibition effect of resveratrol on IAPP fibrillogenesis as monitored by ThT fluorescence. B, AFM topographic images of IAPP aggregates (top row) and resveratrol-treated IAPP (bottom row) visualized after 18 h (left panels) and 96 h (right panels). 200 μm IAPP was dissolved in pure H2O in the absence and presence of 100 μm resveratrol and aliquots of the aggregation reactions taken at specific time points were scanned by AFM. The image sizes are 3 μm × 3 μm.
Atomic Force Microscopy
At predefined time points during the aggregation experiments (see above), 10-μl aliquots of the hIAPP samples were collected and added to a freshly cleaned silicon wafer and dried. The samples were rinsed with deionized water, dried again and imaged at the room temperature. Images were acquired in the tapping mode using a MultiMode AFM and Nanoscope IV controller (Veeco, Santa Barbara, CA), with a PP-NCH silicon tip from NanosensorsTM, NanoWorld AG, Switzerland.
Mass Spectrometry
Lyophilizied IAPP was dissolved in 10 mm ammonium acetate (pH 6.8) to reach concentration of 300∼400 μm. The solution was directly infused into a quadrupole-Time-of-Flight (Q-TOF Premier, Waters) instrument using a syringe pump at a flow rate of 5 μl/min. ESI spectra were acquired using a spray voltage of 2.5 kV and source temperature of 80 °C.
RESULTS
Residues Involved in Initial Oligomerization
[1H,15N]-TROSY spectra of freshly prepared IAPP show well resolved narrow lines together with narrowly dispersed amide proton chemical shifts, indicative of a globally unfolded structure in solution, similar to that previously observed for rat IAPP (27–28) (Fig. 1A). In contrast to non-amyloidogenic rIAPP, which does not display concentration dependence (5, 29), chemical shift changes were observed here for hIAPP as a function of concentration (Fig. 1, A and B). This indicates that IAPP monomers were associating with each other in solution under the conditions investigated. With increasing peptide concentrations perturbations of 1H and 15N chemical shifts were mainly observed in the central region between residues His-18 and Phe-23, with maximum changes observed at His-18 and Ser-19, as well as Tyr-37 suggesting that low-order oligomerization might involve these residues.
FIGURE 1.

Initial self-association of IAPP. A, superimposed [1H,15N]-TROSY spectra of IAPP at concentrations of 25 μm (black), 100 μm (green), 200 μm (cyan), and 500 μm (red). Residues with evident concentration-dependent chemical shift changes such as His-18, Ser-19, and Tyr-37 are highlighted by rectangular boxes. All IAPP samples were prepared in 5 mm sodium phosphate, 5 mm NaCl, 3% D2O, pH 5.5. The IAPP sample at 500 μm was prepared in the presence of 30 μm resveratrol to prevent excessively fast oligomerization. B, concentration-dependent chemical shift perturbations (Δδ in ppm) were calculated as Δδ = [(ΔδH)2 + (0.1ΔδN)2]1/2, where ΔδH and ΔδN were obtained by comparing backbone 1H and 15N chemical shifts of IAPP at 100 μm (green), 200 μm (cyan), and 500 μm (red) with those at 25 μm in the [1H,15N]-TROSY spectra. C, pH-dependent chemical shifts of His-18 and Tyr-37 aromatic protons. The chemical shifts were monitored using [1H,13C]-HSQC spectra of 200 μm IAPP in 1 mm sodium formate buffer, pH was adjusted in steps using HCl. The pH titration curves were fitted according to the 1H chemical shifts (ppm) measured at pH range 1.35 to 5.62. No chemical shifts were measured at relatively high pH (shaded region) due to aggregation of IAPP. The extracted pKa values of 3 and 6.2 correspond to the titratable carboxyl group of Tyr-37 and the imidazole ring of His-18, respectively. D, mass spectra of dimers and trimers were detected in freshly prepared IAPP sample using Q-TOF ESI-MS. The spacing of 0.2 and 0.143 in the isotope patterns indicates particles with 5 and 7 charges, respectively.
To investigate the topology of IAPP association, pH-dependent chemical shift changes were monitored. The topology of the most populated dimeric oligomers can be gleaned from the observation that the spins in spatial proximity to protonatable sites often show correlated chemical shift changes as a function of pH (30–31). In the detectable pH range from 1 to 6 (limited by rapid precipitation at higher pH), 1H,13C, and 15N chemical shifts of His-18 and Tyr-37, but no other residues, are systematically perturbed as shown in Fig. 1C (supplemental Fig. S1 for overlaid two-dimensional HSQC). For aromatic 1H chemical shifts of His-18 and Tyr-37 two transitions with apparent pKa values of 6.2 and 3 corresponding to protonation of the imidazole ring of His-18 and the carboxyl group of Tyr-37, respectively, were detected. As no intra-molecular long-range NOEs were detected beyond the range of four consecutive residues, these data are suggestive of a transient close proximity of His-18 and Tyr-37 side chains from the adjacent monomers in the low-order oligomeric assembly. The detectable but significantly reduced amplitude of the chemical shift perturbation of His-18 side chain protons due to protonation of the C-terminal carboxyl group might indicate wider separation of His-18 and Tyr-37 and point to the dissociation of transient low-order oligomers at acidic pH. This is in line with an earlier observation that oligomerization and aggregation of hIAPP is suppressed at low pH (32).
Low-order Oligomeric States Resolved by Mass Spectrometry
The presence of dimers and trimers in equilibrium with monomers in freshly dissolved IAPP sample was established directly through Q-TOF ESI mass spectrometry. The ESI settings used here usually destabilize non-covalent complexes so that the monomeric IAPP species in different charge states are detected as dominant peaks in the spectra. However, even under these unfavorable conditions, peaks carrying 5+ and 7+ charges (with neighboring isotope peaks differing by 0.20 Da and 0.14 Da, respectively) were observed (Fig. 1D). The peak with a m/z ratio equal to 1561.74 Da gives a monoisotopic mass of 7803.69 Da, corresponding well to the dimeric form of IAPP (expected mass of 7803.69 Da). The monoisotopic peak of trimeric IAPP could not be distinguished from the background noise due to its low abundance. The average experimental m/z ratio of 1674.27 Da is identical to the theoretical m/z ratio of the trimer carrying 7+ charges (1674.27 Da).
Theoretical Structural Models Based on NMR Constraints
Three-dimensional models of monomeric IAPP were reconstructed using NMR constrained molecular dynamics (MD) simulations. Although the clustering analysis showed a widely dispersed distribution of the resulting conformers, ∼70% of the monomers exhibited structural similarities between residues 9 and 19 with RMSD of backbone atoms smaller than 2 Å. Fig. 2A shows 7 representative conformers superimposed using backbone atoms in this region. Averaged violations of the NOE constraints are found below 0.25 Å (supplemental Fig. S2). The distribution of the backbone φ and ψ angles (supplemental Fig. S3) indicates that residues 10–14 are the most structurally constrained and form a distorted (presumably only transiently populated) α-helix. Because of the disulfide bridge between Cys-2 and -7 a highly constrained turn at the N terminus is present. In contrast at the C terminus (residues 20–37) all conformers diverge with no residual structure similarity detected.
FIGURE 2.
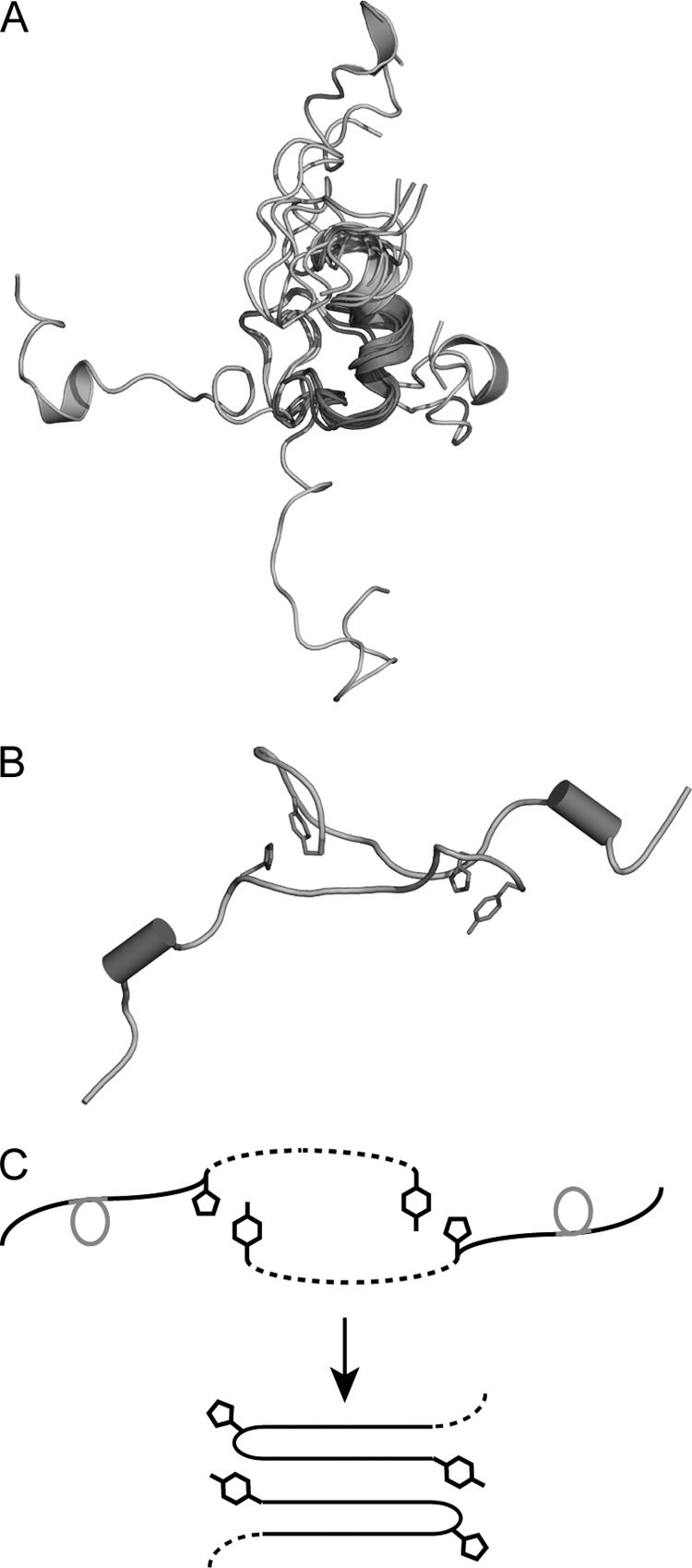
Models of monomeric and dimeric IAPP derived from NMR and constrained MD simulations. A, seven representative conformers of monomeric IAPP superimposed using the backbone atoms of residues 9–19 are shown in red. The N-terminal segment comprising residues 1–8 is highlighted in green. The conformations of the C-terminal residues shown in cyan are divergent. B, representative conformer of IAPP dimer. The short residual helical fragments are highlighted as red cylinders. The residues His-18 and Tyr-37 are shown as sticks (magenta). C, sketch diagram of potential conversion from dimers to higher level aggregates. The structured helical regions are highlighted in red, and nonstructural parts are shown as dashed lines. Side chains of His-18 and Tyr-37 mediating interactions between individual strands are shown.
We attempted to reconstruct IAPP dimers by imposing weak inter-strand constraints between side-chains of His-18 and Tyr-37. This resulted in stable dimers with antiparallel orientation of monomers stabilized by stacking of aromatic side chains of His-18 and Tyr-37. Fig. 2B shows a representative model derived from constrained MD simulations. It is interesting to note that association of monomers mediated by hydrogen bonding and ring stacking of His-18 and Tyr-37 side chains impose only a few additional contacts in the backbone region. This results in significant structural flexibility in the dimer and is consistent with NMR measurements, which indicated only small chemical shift perturbations all residues in this region aside from His-18 and Tyr-37. As illustrated in Fig. 2C, this antiparallel arrangement of the dimer can be regarded as a nascent form on the pathway to a cross-β structure (25). We hypothesize that stacking of these two pivotal side-chains in the C-terminal segment might provide a structural platform for formation of turns flanking residues 19–24. These turns may be further stabilized by intra-molecular contacts between residues in the N-terminal part. Thus, a layer of cross-β structure may develop in the subsequent conformational consolidation and rearrangement (Fig. 2C).
Residues Involved in Distinct Aggregation Pathways
To elucidate residues which interact with growing oligomers, the formation of high-order aggregates was monitored in real-time by measuring a series of [1H,15N]-TROSY spectra of aggregating IAPP. At room temperature and pH 6.5 all of our samples showed a time-dependent decrease of cross-peak intensities likely due to the slow sequestering of soluble IAPP into insoluble aggregates. However, due to pseudo-equilibrium between the association and dissociation of IAPP to and from aggregates, excessive transverse relaxation of spins at the binding interface might ensue (33–34). Thus, using the concomitant relative decrease in intensities of the corresponding cross-peaks, preferential binding of individual residues to growing surfaces of aggregates may be established (33–34).
In a variety of tested samples a mixture of two distinct aggregation processes can be discerned which result in either amorphous or filamentous aggregates respectively. These two types of aggregation are found in the most pure forms in two of our tested samples containing 200 μm and 500 μm IAPP in the same buffer. In a series of TROSY spectra of the sample of 200 μm IAPP freshly dissolved in the buffer at room temperature, the signals of all residues across the peptide decreased homogenously, indicating a lack of specific interactions with the expanding surface of aggregates, with the notable exception of His-18 (Fig. 3, A and B). The latter observation is in line with the finding that His-18 can mediate initial transient oligomerization of IAPP (Fig. 1) and might drive the subsequent oligomerization and aggregation as well. After 21 h of aggregation, almost all soluble peptide material assembled into insoluble oligomers as indicated by complete loss of signal in [1H,15N]-TROSY (Fig. 3, A and B). In the resulting precipitate variably-sized spherical oligomers were observed by AFM (Fig. 3C). The dominant species were spherical particles with a height of 6∼12 nm (supplemental Fig. S4A). A few short nascent fibrils were associated with the largest particles of the height of 20∼25 nm (Fig. 3C).
FIGURE 3.

Aggregation of IAPP into spherical oligomers as moniotored by NMR and AFM. A, loss of signal in a series of [1H,15N]-TROSY spectra of 200 μm IAPP recorded after 0, 15, and 21 h after initiation of aggregation. The aggregation process was monitored on the same 200 μm IAPP sample described in Fig. 1. B, plot of volumes of cross-peaks versus residue number as a function of time. All peak volumes in the [1H,15N]-TROSY spectra were normalized to the corresponding volumes at time 0 h. C, AFM topographic image of the resulting aggregates (left panel), and vertical elevation slices taken at the positions 1 and 2 (right panel). The image size is 5 μm × 5 μm.
In the 500 μm IAPP sample, which was pre-incubated for 2 weeks at 4 °C before the aggregation trial, a process of fibril growth by direct monomer addition was observed. The freshly prepared sample was found to be stable in solution for 2 weeks at 4 °C as shown by nearly identical 1H spectra recorded before and after incubation (supplemental Fig. S5). The aggregation of the peptide into fibrils was initiated by increasing its temperature to 25 °C. In contrast to the spherical oligomer formation involving the majority of residues in IAPP (Fig. 3), excessive transverse relaxation was extended to residues 20, 24–28, and 30–37. Morphologically, the resulting aggregates are composed primarily of fibrils as confirmed by AFM (Fig. 4). This indicates that the direct binding of the monomers to the fibril growing edge is mediated by the C-terminal residues. The apparent diameter of the fibrils shows discrete heights of 3.9 nm and 7.8 nm (supplemental Fig. S4B).
FIGURE 4.

Aggregation of IAPP into fibrils as moniotored by NMR and AFM. A, NMR signal loss in a series of [1H,15N]-TROSY spectra of 500 μm IAPP recorded at 0, 10, and 20 h after initiation of aggregation. The aggregation process was monitored on the same 500 μm IAPP sample shown in Fig. 1. B, plot of volumes of cross-peaks versus residue number as a function of time. All peak volumes in the [1H,15N]-TROSY spectra were normalized to the corresponding volumes at time 0 h. C, AFM topographic image of the resulting aggregates. The image size is 5 μm × 5 μm.
Residual Conformational Flexibility of IAPP in Spherical Aggregates and Fibrils
To better understand the structural organization of the polypeptide in oligomers and amyloid fibrils we identified structurally flexible residues of IAPP that remain in the insoluble aggregates and are detectable in HR-MAS [1H,15N]-HMQC spectra of precipitated material. The resulting NMR spectra of spherical aggregates and fibrils exhibited different sets of cross-peaks indicating different structural organization of IAPP in these two types of aggregates (Fig. 5). Although cross-peaks observed arising from insoluble aggregates were shifted relative to their positions in the corresponding solution spectrum, we unequivocally assigned the observed peaks of the fibrils by retracing their positions to the corresponding peaks in solution. The observed mobile residues are all localized at the N terminus extending to Thr-9 and including the HNϵ group of Arg-11, indicative of retained flexibility at the N-terminal region of the peptide in fully formed fibrils.
FIGURE 5.

[1H,15N]-HMQC spectra observed for spherical aggregates (A) and fibrils (B) using a high resolution MAS probe. The samples were prepared by spinning down respective spherical and fibrils aggregates and resuspending them in 50 μl of 5 mm sodium phosphate, pH 5.5, 5 mm NaCl, 3% D2O.
The significant conformational flexibility in the N terminus as observed by NMR was confirmed by MD reconstruction of atomic B-factors (root mean square fluctuations) (35) in a model of IAPP fibril subunit (25). Fig. 6 shows simulated B-factors at atomic positions in each of 10 β-strands consistently demonstrating significantly increased B-factor values in the first 9 residues of the IAPP sequence. Fluctuations of atomic positions in residues 1–9 and the side-chain of Arg-11 are significantly larger than those in the middle layers in the correspondence with the NMR data.
FIGURE 6.
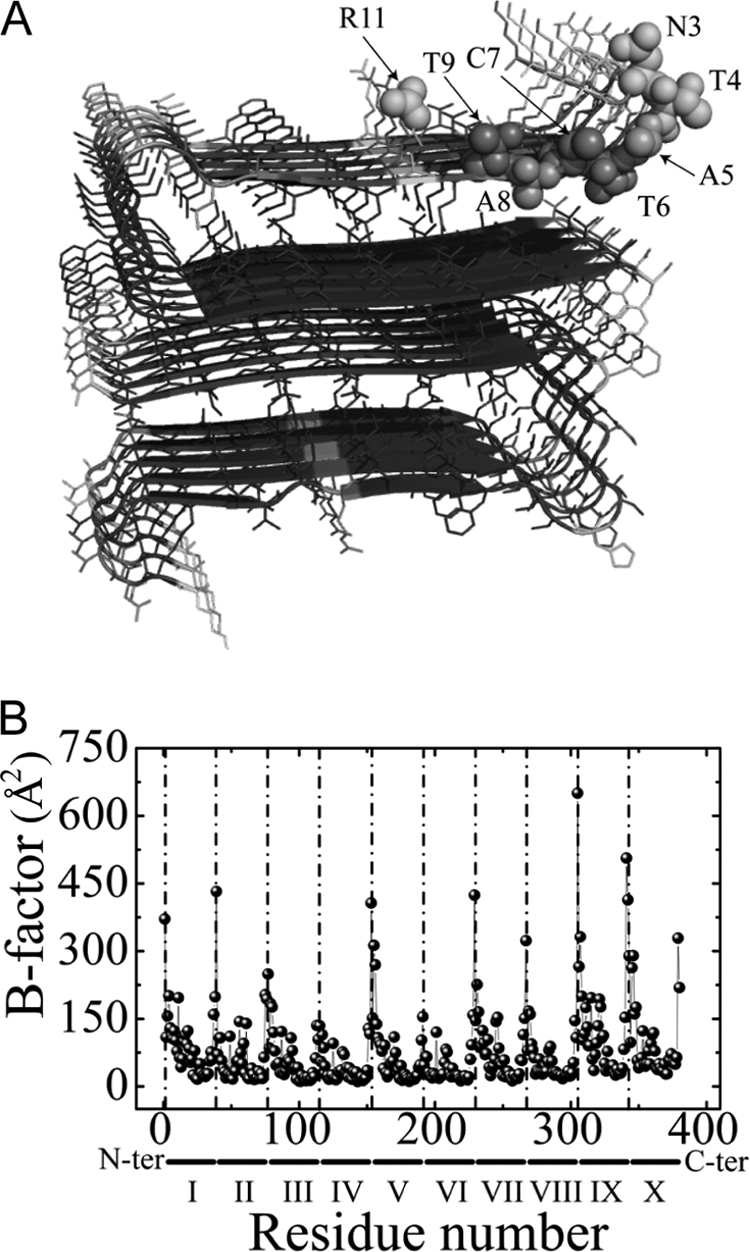
MD simulations of the IAPP fibril models. A, three-dimensional structure of the fibril model (25) continuously colored according to the simulated B-factor values (35) in the range from 50 Å2 (blue) to 400 Å2 (red). The residues with backbone amide moieties observed in the high resolution MAS spectrum of hIAPP fibrils are shown in a full heavy atoms representation. B, simulated B-factors of individual monomers as calculated based on all heavy atom positions after fitting to the initial structure. All residues of ten monomers are plotted along the horizontal axis comprising 370 residues in total. The vertical dotted lines indicate the N terminus of each monomer.
IAPP Amyloid Formation via Oligomeric Intermediates and Inhibition by Resveratrol
Because different residues of IAPP are involved in the processes of amorphous aggregation and fibril formation, it is important to elucidate the regions of IAPP targeted by known inhibitors of amyloidogenesis. IAPP amyloid formation and the effect of resveratrol were monitored by the Thioflavin T (ThT) binding assay (36–37). In the absence of resveratrol, IAPP peptides are very amyloidogenic and fully aggregate into ThT containing β-sheet-rich amyloid fibrils in about 1 h under the experimental conditions used (Fig. 7A). In contrast, formation of IAPP amyloid was suppressed in the presence of resveratrol at a 1:2 molar ratio to IAPP. ThT intensity only reached 50% and an apparent lag time of fibril formation of ∼5 min was observed (Fig. 7A, right panel).
We further investigated the effect of resveratrol on IAPP amyloid formation using AFM. To avoid excessively fast aggregation, the reaction was performed in pure water. In the absence of resveratrol, IAPP predominately aggregates into spherical species at 18h (Fig. 7B, top left). This species presumably serves as important intermediates for fibrils as they are predominantly replaced with elongated fibrils after 96 h of incubation (Fig. 7B, top right). Conversely, in the presence of resveratrol the apparent density of oligomers was observatively reduced at 18 h (Fig. 7B, low left) and never reach a similar density of fibrils formed in non-resveratrol even after 96 h (Fig. 7B, low right). These results suggest that the binding of resveratrol suppresses IAPP amyloid formation by targeting primarily amorphous spherical high-order oligomeric intermediates.
Inhibition Mechanism of Resveratrol and Insulin on IAPP Oligomerization
To understand the inhibition mechanism of resveratrol, we measured a series of [1H,13C]-HSQC spectra of IAPP with different concentrations of resveratrol (Fig. 8A). The side chain of His-18, particularly the δ2C-δ2H and ϵ1C-ϵ1H groups, showed significant chemical shift changes upon resveratrol binding, indicating that resveratrol may inhibit oligomerization via competition with other monomeric species of IAPP for His-18 (Fig. 8A). This is consistent with the finding that His-18 plays important role in initial transient oligomer formation (Fig. 1) and subsequent oligomerization (Fig. 3). In addition, the chemical shifts of Lys-1, especially αC-αH, were also perturbed indicating the presence of the second binding site for the compound (Fig. 8A).
FIGURE 8.

Analysis of resveratrol and insulin binding to IAPP with NMR. A,[1H, 13C]-HSQC spectra of 50 μm IAPP alone (red) and in the presence of 40 μm (cyan) and 80 μm (green) resveratrol were recorded in pure H2O. His-18 and Lys-1 side-chains show distinct chemical shift changes upon resveratrol titration. B, [1H, 15N]-HMQC spectra of 20 μm IAPP alone (red) and in the presence of 10 μm (cyan), 25 μm (green), and 50 μm (blue) insulin were recorded in 5 mm sodium phosphate, pH 5.5, 5 mm NaCl, 3% D2O. His-18, Ser-19, and Tyr-37, which show evident chemical shift changes upon insulin titration are highlighted by rectangular boxes.
The initial self-assembly of IAPP can also be inhibited by insulin, which is reportedly able to prevent transition of IAPP from random coil to β-sheet rich amyloid fibrils (38–40). We monitored the titration of IAPP with insulin by [1H,15N]-HMQC experiments (Fig. 8B). At pH 5.5, insulin binds to IAPP mainly via residues His-18, Ser-19 and Tyr-37, which are the identified sites of IAPP self-association (Fig. 1). The protonation of both nitrogen atoms Nδ1, Nϵ2 in the His-18 side chain plays an important role in the interaction as the corresponding peaks are significantly less perturbed at pH above the pKa of histidine. At pH 7, insulin preferentially binds to Arg-11 and its vicinal Gln-10, Leu-12, and Ala-13 instead of His-18 (supplemental Fig. S6). This suggests that insulin might function as a competitive inhibitor for the initial self-assembly of IAPP by blocking His-18 under acidic conditions. This is consistent with the finding that insulin binds to rat IAPP mainly by the positively charged side chains of Arg-11 and Arg-18 (28).
DISCUSSION
Recombinant human IAPP exhibits similar amyloidogenicity and cytotoxicity to synthetic IAPP and is suggested as a suitable model for studies of mechanisms of amyloid formation (20). In the present study we analyzed the initial low-order oligomerization of recombinant IAPP as well as the progressive growth of insoluble high-order aggregates at residue specific resolution. Low-order soluble oligomeric forms including dimers, trimers, and tetramers have been found at equilibrium with monomers in preaggregation solutions of other amyloid proteins such as Aβ, α-synuclein, and β-2-microglobulin (41–43). However, low-order oligomers of IAPP in solution were not detected in previous studies due to their instability and low population (44–45). In this study, we observed a clear concentration dependent self-association of IAPP monomers during its initial oligomerization stage with the presence of dimers and trimers as detected using mass-spectroscopy (Fig. 1). Our results suggest that the small oligomers of IAPP are at equilibrium with monomers in solution. The amount of low-order oligomers increases with the peptide concentration due to the self-association of monomers, which is consistent with and explains the previous observation that the initial rate of amyloid formation is proportional to the concentration of the peptide (46). Low-order oligomers of IAPP are mainly formed through the interactions of His-18 and Tyr-37 (Fig. 1). This serves to reduce the entropic cost of association of the intervening residues and thus facilitates subsequent aggregation in a pH-dependent manner. Because of the small interface between His-18 and Tyr-37, the transient low-order oligomers are not stable and could readily dissociate into an equilibrium with the monomers in solution.
Our results suggest that IAPP amyloid fibrillogenesis can be mediated by prefibrillar on-pathway oligomers (Fig. 7). Previous studies pointed out those oligomers that are responsible for nucleated conformational conversion in amyloid formation of prion proteins (7, 47). Micelle-like oligomers were also detected in Aβ and proposed to slowly convert into fibril nuclei (6, 48–50). For IAPP, molten globule-like self-associating oligomers are also found to be populated just prior to fibril formation (Fig. 7). These oligomers might mediate conformational transition of IAPP from nearly random coil to β-sheet followed by subsequent amyloid formation and are the primary cause of IAPP cytotoxicity (17, 51–53). AFM studies established that the growth of oligomers with estimated size from 16 to >300 monomers precedes fibril elongation, suggesting that mature fibrils are assembled directly via lateral growth of oligomers followed by the longitudinal growth of full-width oligomers (18). For two selected samples in our study, we were able to monitor two distinctly different aggregation processes in real time. These two processes involved either the growth of predominantly amorphous spherical aggregates or elongated fibrils (Fig. 3). The binding interface to spherical aggregates significantly extends to residues of the whole peptide with a special emphasis on His-18 and is not exclusively localized to the previously identified amyloidogenic regions (7–8, 40, 54–55). Further, we found that the sizes of the prefibrillar oligomers are polydispersed and only the largest oligomeric species can seed the growth of fibrils. Smaller oligomers might either gather into the large seeding nuclei or coalesce directly for fibril growth (56).
Although the amyloid could be formed by direct association of oligomers, it might also be formed by fibril elongation via direct binding of monomers at the growing surface (Fig. 4). This latter mode might be mediated by specific binding of IAPP monomers via residues 20, 24–28, 30–37 (Fig. 5B). These residues are localized at the C-terminal region with high propensity to form β-structure and closely coincide with two out of the three known amyloidogenic regions which are localized to residues 8–20, 20–29, and 30–37 (40, 54–55). The structural flexibility of IAPP in the spherical and fibril aggregates is markedly different, as shown by HR-MAS NMR spectroscopy (Fig. 5). Our results reveal that interactions driving formation of the spherical oligomers can be distinct from those involved in the direct fibril elongation via monomer binding, resulting in morphologically and structurally different aggregated forms.
It was previously shown that a phenolic compound, resveratrol, which is abundant in red wine, can inhibit IAPP aggregation on the surface of negatively charged membranes (57). Here we demonstrated that resveratrol retards aggregation of IAPP by binding to His-18 and inhibiting early formation of oligomeric intermediates. Insulin, as a known natural stabilizer of IAPP, also may share this mechanism with resveratrol by binding to His-18 and Tyr-37 under acidic conditions. These data are consistent with the important role of His-18 in oligomerization as identified in the previous studies. Recently it was shown that the polyphenolic compound EGCG could convert large, mature α-synuclein and amyloid-β fibrils into smaller, nontoxic amorphous aggregates (58). As a comparison, resveratrol blocks the self-binding sites of IAPP and as such does not show this remodeling ability. Considering the different chemical structures of resveratrol and EGCG, it would be of interest to study the binding and effect of EGCG on IAPP.
Supplementary Material
Acknowledgments
We thank Dr. Alistair Irvine and Dr. Steven Libich for careful reading of the manuscript. Fibril model structures were kindly provided by Tycko and Eisenberg. Computer time was generously provided by the HPC NTU.
This work was supported in part by AcRF Tier 2 by the MOE (T206B3210RS) (to Y. M. and K. P.) and a competitive research program grant from the Singapore National Research Foundation (NRF-CRP2-2007-02) (to M. B. C.-P.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S6.
- T2D
- type 2 diabetes
- AFM
- atomic force microscopy
- HR-MAS
- high-resolution magic angle spinning
- IAPP
- islet amyloid polypeptide
- MS
- mass spectrometry
- MD
- molecular dynamics
- NMR
- nuclear magnetic resonance
- ThT
- Thioflavin T.
REFERENCES
- 1. Dobson C. M. (2006) Protein Pept. Lett. 13, 219–227 [DOI] [PubMed] [Google Scholar]
- 2. Sawaya M. R., Sambashivan S., Nelson R., Ivanova M. I., Sievers S. A., Apostol M. I., Thompson M. J., Balbirnie M., Wiltzius J. J., McFarlane H. T., Madsen A. Ǿ., Riekel C., Eisenberg D. (2007) Nature 447, 453–457 [DOI] [PubMed] [Google Scholar]
- 3. Serpell L. C., Sunde M., Benson M. D., Tennent G. A., Pepys M. B., Fraser P. E. (2000) J. Mol. Biol. 300, 1033–1039 [DOI] [PubMed] [Google Scholar]
- 4. Sunde M., Serpell L. C., Bartlam M., Fraser P. E., Pepys M. B., Blake C. C. (1997) J. Mol. Biol. 273, 729–739 [DOI] [PubMed] [Google Scholar]
- 5. Lomakin A., Teplow D. B., Kirschner D. A., Benedek G. B. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 7942–7947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jarrett J. T., Lansbury P. T., Jr. (1993) Cell 73, 1055–1058 [DOI] [PubMed] [Google Scholar]
- 7. Serio T. R., Cashikar A. G., Kowal A. S., Sawicki G. J., Moslehi J. J., Serpell L., Arnsdorf M. F., Lindquist S. L. (2000) Science 289, 1317–1321 [DOI] [PubMed] [Google Scholar]
- 8. Hardy J., Selkoe D. J. (2002) Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 9. Caughey B., Lansbury P. T. (2003) Annu. Rev. Neurosci. 26, 267–298 [DOI] [PubMed] [Google Scholar]
- 10. Cleary J. P., Walsh D. M., Hofmeister J. J., Shankar G. M., Kuskowski M. A., Selkoe D. J., Ashe K. H. (2005) Nat. Neurosci. 8, 79–84 [DOI] [PubMed] [Google Scholar]
- 11. Haass C., Selkoe D. J. (2007) Nat. Rev. Mol. Cell Biol. 8, 101–112 [DOI] [PubMed] [Google Scholar]
- 12. Collins S. R., Douglass A., Vale R. D., Weissman J. S. (2004) PLoS Biol 2, e321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sunde M., Blake C. C. (1998) Q. Rev. Biophys. 31, 1–39 [DOI] [PubMed] [Google Scholar]
- 14. Westermark P., Grimelius L. (1973) Acta Pathol. Microbiol. Scand. A. 81, 291–300 [DOI] [PubMed] [Google Scholar]
- 15. Westermark P., Engström U., Johnson K. H., Westermark G. T., Betsholtz C. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 5036–5040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fox N., Schrementi J., Nishi M., Ohagi S., Chan S. J., Heisserman J. A., Westermark G. T., Leckström A., Westermark P., Steiner D. F. (1993) FEBS Lett. 323, 40–44 [DOI] [PubMed] [Google Scholar]
- 17. Kayed R., Bernhagen J., Greenfield N., Sweimeh K., Brunner H., Voelter W., Kapurniotu A. (1999) J. Mol. Biol. 287, 781–796 [DOI] [PubMed] [Google Scholar]
- 18. Green J. D., Goldsbury C., Kistler J., Cooper G. J., Aebi U. (2004) J. Biol. Chem. 279, 12206–12212 [DOI] [PubMed] [Google Scholar]
- 19. Haataja L., Gurlo T., Huang C. J., Butler P. C. (2008) Endocr. Rev. 29, 303–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lopes D. H., Colin C., Degaki T. L., de Sousa A. C., Vieira M. N., Sebollela A., Martinez A. M., Bloch C., Jr., Ferreira S. T., Sogayar M. C. (2004) J. Biol. Chem. 279, 42803–42810 [DOI] [PubMed] [Google Scholar]
- 21. Goldberg M. S., Lansbury P. T., Jr. (2000) Nat. Cell Biol. 2, E115–E119 [DOI] [PubMed] [Google Scholar]
- 22. Case D. A., Cheatham T. E., 3rd, Darden T., Gohlke H., Luo R., Merz K. M., Jr., Onufriev A., Simmerling C., Wang B., Woods R. J. (2005) J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Onufriev A., Bashford D., Case D. A. (2004) Proteins 55, 383–394 [DOI] [PubMed] [Google Scholar]
- 24. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., Berendsen H. J. (2005) J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
- 25. Wiltzius J. J., Sievers S. A., Sawaya M. R., Cascio D., Popov D., Riekel C., Eisenberg D. (2008) Protein Sci. 17, 1467–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peng Y., Kaminski G. A. (2005) J. Phys. Chem. B 109, 15145–15149 [DOI] [PubMed] [Google Scholar]
- 27. Lashuel H. A., Lansbury P. T., Jr. (2006) Q. Rev. Biophys. 39, 167–201 [DOI] [PubMed] [Google Scholar]
- 28. Wei L., Jiang P., Yau Y. H., Summer H., Shochat S. G., Mu Y., Pervushin K. (2009) Biochemistry 48, 2368–2376 [DOI] [PubMed] [Google Scholar]
- 29. Williamson J. A., Loria J. P., Miranker A. D. (2009) J. Mol. Biol. 393, 383–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Szyperski T., Antuch W., Schick M., Betz A., Stone S. R., Wüthrich K. (1994) Biochemistry 33, 9303–9310 [DOI] [PubMed] [Google Scholar]
- 31. Farrell D., Miranda E. S., Webb H., Georgi N., Crowley P. B., McIntosh L. P., Nielsen J. E. (2010) Proteins 78, 843–857 [DOI] [PubMed] [Google Scholar]
- 32. Chargé S. B., de Koning E. J., Clark A. (1995) Biochemistry 34, 14588–14593 [DOI] [PubMed] [Google Scholar]
- 33. Sette M., Spurio R., Van Tilborg P., Gualerzi C. O., Boelens R. (1999) Rna-a Publication Rna Society 5, 82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bodner C. R., Dobson C. M., Bax A. (2009) J. Mol. Biol. 390, 775–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tieleman D. P., Marrink S. J., Berendsen H. J. (1997) Biochim. Biophys. Acta 1331, 235–270 [DOI] [PubMed] [Google Scholar]
- 36. LeVine H., 3rd, Scholten J. D. (1999) Methods Enzymol. 309, 467–476 [DOI] [PubMed] [Google Scholar]
- 37. LeVine H., 3rd. (1999) Methods Enzymol. 309, 274–284 [DOI] [PubMed] [Google Scholar]
- 38. Jaikaran E. T., Nilsson M. R., Clark A. (2004) Biochem. J. 377, 709–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gilead S., Wolfenson H., Gazit E. (2006) Angew. Chem. Int. Ed. Engl. 45, 6476–6480 [DOI] [PubMed] [Google Scholar]
- 40. Gilead S., Gazit E. (2008) Exp. Diabetes Res. 2008, 256954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bitan G., Lomakin A., Teplow D. B. (2001) J. Biol. Chem. 276, 35176–35184 [DOI] [PubMed] [Google Scholar]
- 42. Giannakis E., Pacifico J., Smith D. P., Hung L. W., Masters C. L., Cappai R., Wade J. D., Barnham K. J. (2008) Biochim. Biophys. Acta 1778, 1112–1119 [DOI] [PubMed] [Google Scholar]
- 43. Smith A. M., Jahn T. R., Ashcroft A. E., Radford S. E. (2006) J. Mol. Biol. 364, 9–19 [DOI] [PubMed] [Google Scholar]
- 44. Vaiana S. M., Ghirlando R., Yau W. M., Eaton W. A., Hofrichter J. (2008) Biophys. J. 94, L45–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Soong R., Brender J. R., Macdonald P. M., Ramamoorthy A. (2009) J. Am. Chem. Soc. 131, 7079–7085 [DOI] [PubMed] [Google Scholar]
- 46. Kudva Y. C., Mueske C., Butler P. C., Eberhardt N. L. (1998) Biochem. J. 331, 809–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Luca S., Yau W. M., Leapman R., Tycko R. (2007) Biochemistry 46, 13505–13522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yong W., Lomakin A., Kirkitadze M. D., Teplow D. B., Chen S. H., Benedek G. B. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 150–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grant M. A., Lazo N. D., Lomakin A., Condron M. M., Arai H., Yamin G., Rigby A. C., Teplow D. B. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 16522–16527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lomakin A., Chung D. S., Benedek G. B., Kirschner D. A., Teplow D. B. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 1125–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin C. Y., Gurlo T., Kayed R., Butler A. E., Haataja L., Glabe C. G., Butler P. C. (2007) Diabetes 56, 1324–1332 [DOI] [PubMed] [Google Scholar]
- 52. Meier J. J., Kayed R., Lin C. Y., Gurlo T., Haataja L., Jayasinghe S., Langen R., Glabe C. G., Butler P. C. (2006) Am. J. Physiol. Endocrinol Metab. 291, E1317–E1324 [DOI] [PubMed] [Google Scholar]
- 53. Lomakin A., Asherie N., Benedek G. B. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 10254–10257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nilsson M. R., Raleigh D. P. (1999) J. Mol. Biol. 294, 1375–1385 [DOI] [PubMed] [Google Scholar]
- 55. Jaikaran E. T., Higham C. E., Serpell L. C., Zurdo J., Gross M., Clark A., Fraser P. E. (2001) J. Mol. Biol. 308, 515–525 [DOI] [PubMed] [Google Scholar]
- 56. Xu W., Ping J., Li W., Mu Y. (2009) J. Chem. Physics 130, 164709 [DOI] [PubMed] [Google Scholar]
- 57. Evers F., Jeworrek C., Tiemeyer S., Weise K., Sellin D., Paulus M., Struth B., Tolan M., Winter R. (2009) J. Am. Chem. Soc. 131, 9516–9521 [DOI] [PubMed] [Google Scholar]
- 58. Bieschke J., Russ J., Friedrich R. P., Ehrnhoefer D. E., Wobst H., Neugebauer K., Wanker E. E. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 7710–7715 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.