

Abstract
Aim
Atrial natriuretic peptide (ANP) is released from the heart in response to hypoxia and helps mitigate the development of pulmonary hypertension. However, the mechanism of hypoxia-induced ANP release is not clear. The cardiac atria are the primary source of ANP secretion under normal conditions, but right ventricular ANP expression is markedly up-regulated during adaptation to hypoxia. We sought to better understand mechanisms of cardiac ANP release during adaptation to hypoxia
Main methods
We measured hypoxia-induced ANP release from isolated perfused rat hearts obtained from normoxia and hypoxia-adapted rats before and after removal of the atria.
Key findings
In both normoxia- and hypoxia-adapted hearts, ANP levels in the perfusate increased within 15 minutes of hypoxia. Hypoxia-induced ANP release was greater from hypoxia-adapted than normoxia-adapted hearts. Baseline and hypoxia-induced ANP release were considerably greater with the atria intact (213 ± 29 to 454 ± 62 and 281 ± 26 to 618± 87 pg/ml for normoxia- and hypoxia-adapted hearts respectively, P < 0.001 for both) than with atria removed (94 ± 17 to 131 ± 32 and 103 ± 26 to 201 ± 55 pg/ml, respectively, P < 0.002 for both). Hypoxia-induced ANP release was reduced over 80% by removing the atria in both normoxia- and in hypoxia-adapted hearts. Acute hypoxia caused a transient increase in lactate release and reductions in pH and left ventricular generated force, but similar changes in lactate and pH did not affect ANP release under normoxic conditions.
Significance
We conclude that the right ventricle is not a major source of cardiac ANP release in normoxia or hypoxia-adapted rats.
Keywords: atrial natriuretic peptide (ANP), hypoxic pulmonary hypertension, isolated perfused heart, cardiac atria, hypoxia
Introduction
Atrial natriuretic peptide (ANP) is a cardiac hormone that is synthesized and stored primarily in the cardiac atria where it is released in response to atrial distension [1], tachycardia [2] or acute hypoxia [3]. Because of its potent diuretic and vasorelaxant properties, and its antagonistic effect on the renin-angiotensin system, ANP is thought to play an important role in intravascular volume homeostasis [4]. ANP is also a potent inhibitor of pulmonary vasoconstriction and several lines of evidence suggest that it plays an important role in protecting against the development of hypoxic pulmonary hypertension [5]. Elevation of circulating ANP levels, either by infusion of exogenous ANP [6] or as the result of transgenic overexpression of endogenous ANP [7] blunt the development of pulmonary hypertension, right ventricular hypertrophy and muscularization of pulmonary arteries during chronic hypoxia. Conversely, monoclonal antibodies against ANP can exaggerate pulmonary hypertensive responses to subacute and chronic hypoxia [8] and mice with gene-targeted disruption of ANP or its primary receptor, natriuretic peptide receptor-A (NPR-A), have increased right ventricular pressure and mass and develop greater pulmonary hypertensive and right ventricular hypertrophy in response to chronic hypoxia [9]. Finally, inhibition of phosphodiesterase-5 activity ameliorates hypoxic pulmonary hypertension in normal mice but not in mice with genetic disruption of NPR-A, suggesting that ANP-NPR-A signaling is the primary mechanism by which cGMP inhibits the development of hypoxic pulmonary hypertension [10].
Circulating ANP levels are elevated in response to acute and chronic hypoxia. This, likely occurs as the result of both reduced pulmonary clearance [11] and increased cardiac secretion [12]. Decreased pulmonary clearance occurs as the result of a decrease in expression of pulmonary natriuretic peptide clearance receptor (NPR-C), but the mechanisms responsible for increase cardiac ANP secretion are unclear. Numerous factors may contribute to the release of ANP during exposure to acute hypoxia. Hypoxic has a direct stimulatory effect on ANP release from atrial cardiomyocytes in vitro and hypoxic pulmonary vasoconstriction increases right ventricular and right atrial stretch by increasing intra-chamber pressure [3]. Chronic hypoxia is also associated with the release of a variety of substances that have been shown to stimulate cardiac ANP secretion such as vasopressin and endothelin [13].
The relative contribution of the cardiac atria and ventricles to ANP secretion during chronic hypoxia is uncertain. Under baseline conditions, ANP and ANP mRNA levels in the cardiac atria are 1-2 orders of magnitude greater than in the ventricles. The cardiac atria synthesize large quantities of ANP that are stored in secretory granules and secreted via a regulatory pathway, whereas the ventricles secrete ANP via a constitutive pathway and have little pre-synthesized ANP stores. Thus, under normal conditions the atria are considered the primary source of ANP release [14]. However, ventricular ANP expression is markedly up-regulated in cardiac hypertrophy [15], and in some models of congestive heart failure, ANP secretion from the left ventricle can approximate or exceed that of the atria [16]. In one study of rats exposed to chronic hypoxia, steady state ANP mRNA levels in the right ventricle increased 160-fold and right ventricular ANP tissue levels increased 9-fold [17]. In comparison, right atrial ANP mRNA levels did not change significantly and right atrial ANP concentration decreased [17]. In the present study, we hypothesized that if the right ventricle contributes to increased ANP secretion during adaptation to hypoxia, then ventricular ANP release should be greater in hearts obtained from hypoxia-adapted rats than from those obtained from normoxia rats. To test this hypothesis, we measured baseline and hypoxia-induced ANP release from the hearts of normoxia and hypoxia-adapted rats with the atria intact and with atria removed.
Methods
Environmental Exposures
Male Sprague-Dawley rats (Harlan Sprague Dawley, Inc., Indianapolis, IN) weighing between 270 and 380 g were placed in hypobaric chambers for 3 weeks. Hypobaric conditions were achieved by applying a continuous vacuum and adjusting an air intake valve to maintain intra-chamber pressure at 380 mmHg (0.5 atm). Airflow through the chamber was kept at 10-15 l/minute to prevent accumulation of CO2 and NH3. Intra-chamber pressure was monitored via a pressure gauge in the wall of the chamber. Chambers were opened three times weekly to clean animal cages and to replace food and water. Rats were fed standard rat chow and were allowed to take food and water ad libitum. Normoxic controls were kept in identical cages adjacent to the hypoxic chambers. For ease of identification these rats are referred to as “normoxia-adapted” in the tables, figures and figure legends.
Isolated Perfused Heart
Rats were heparinized with 2500 IU of sodium heparin, intraperitoneally. After 15 min. they were anesthetized with 50 mg pentobarbital. A subcostal incision was made, the diaphragm transected and the ribs incised and spread to expose the heart. The heart was rapidly removed and placed in ice cold Krebs-Henseleit-Bicarbonate buffer to arrest beating. The aorta was mounted on an isolated perfusion Langendorff apparatus and the coronary vessels perfused via retrograde perfusion with Krebs-Henseleit-Bicarbonate buffer at a rate of 10 ml/minute. A balloon-tipped catheter constructed from plastic wrap and polyethylene tubing PE60 containing Krebs- Henseleit-Bicarbonate buffer was inserted into the left ventricle via the mitral valve. Left ventricular pressure was monitored with this catheter connected to a pressure transducer (Gould Instrument Systems, Valley View, OR). Another transducer, connected to the side arm of the aortic perfusion catheter monitored coronary perfusion pressure. Volume in the balloon was adjusted to set left ventricular end diastolic pressure at 5 mmHg. The heart was allowed to stabilize for 30 min. From this point onward the volume in the catheter balloon remained constant. The heart was electronically paced at a rate at 300 bpm using a Grass stimulator (Model SD9J, W. Warwick, RI) at twice the minimum capture voltage. Heart rate, left ventricular peak systolic pressure (LVPSP), left ventricular end diastolic pressure (LVEDP) and coronary perfusion pressure (CPP) were monitored continuously and recorded using a Biopac (Model # MP100A, Santa Barbara, CA) analog to digital converter interfacing with an IBM compatible computer, using Acknowledge software (Biopac Systems, Santa Barbara, CA). Left ventricular generated pressure (LVGP defined as LVPSP-LVEDP) and maximum LV dP/dt (± LV dP/dtmax) were used as indices of myocardial function.
Experimental Protocol
Hearts were initially perfused with Krebs-Henseleit-Bicarbonate buffer (NaCl 118 mM; KCl 4.7 mM; CaCl2 1.75 mM; NaHC03 21 mM, MgS04 1.2 mM, KH2P04 1.2 mM, EDTA 0.5 mM, and glucose 11 mM). Buffer was prepared fresh daily using deionized water, and was aerated with 95% O2 and 5% CO2. Normally 95% O2 is used to oxygenate the perfusate in the Langendorf isolated heart because of the lack of hemoglobin. The pH of the aerated buffer solution was adjusted to 7.4. Following a 30 minute stabilization period in which hearts were perfused with Krebs-Henseleit-Bicarbonate aerated with 95% O2/5% CO2 (normoxia), hearts were exposed to two 15 minute periods of acute hypoxia, interrupted by 30 minutes of normoxia (See Figure 1). For acute hypoxic exposure, hearts were perfused with Krebs-Henseleit-Bicarbonate buffer aerated with either 5% or 21 % O2, 5% C02, balance N2 at 37° C for 15 min. The order of hypoxia was randomized such that half of the hearts were perfused with the 5%O2 perfusate first followed by 30 minutes of normoxia and then 15 minutes of the 21% O2 perfusate and half of the hearts were exposed to the 21% O2 pefusate for 15 minutes followed by 30 minutes of normoxia and then 15 minutes of the 5% O2 perfusate (See Figure 1). The results of both protocols were combined in analyzing ANP concentrations in the effluent in response to acute hypoxia since the magnitude of hypoxia had no effect on ANP release (See Figure 4). Normoxia and hypoxia-adapted controls hearts were subjected to the same protocol described above but without exposures to 5% or 21% O2 (See Figure 1). After the last exposure to hypoxia, the hearts were returned to normoxic perfusate for 30 min and then the atria were carefully removed and the perfusion cycle was repeated (See Figure 1). At predetermined intervals (1,3,8,15,30 minutes during normoxia and 1,3,8,15 minutes during hypoxia) 10 ml of the heart perfusate was collected in tubes containing 500 KIU aprotinin (Sigma Chemical, St Loius MO) and frozen for later ANP analysis ( See Figure 1). At the same time, 1.5 ml aliqout of perfusate was collected and analysed for O2, pH, glucose and electrolytes using a Nova Stat Profile Plus 9 analyzer (Waltham, MA).
Figure 1.
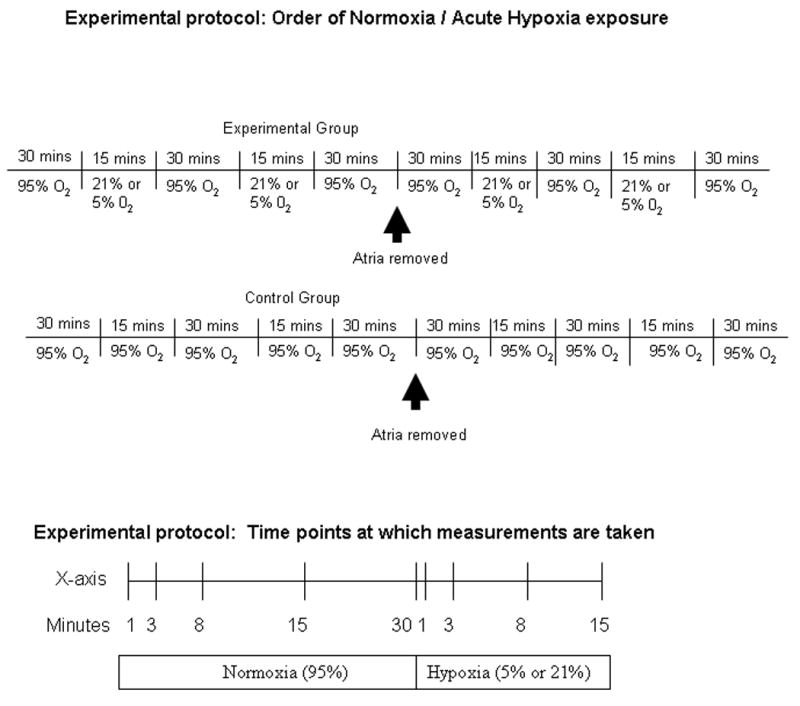
Schematic diagram outlining experimental protocol.
Figure 4.
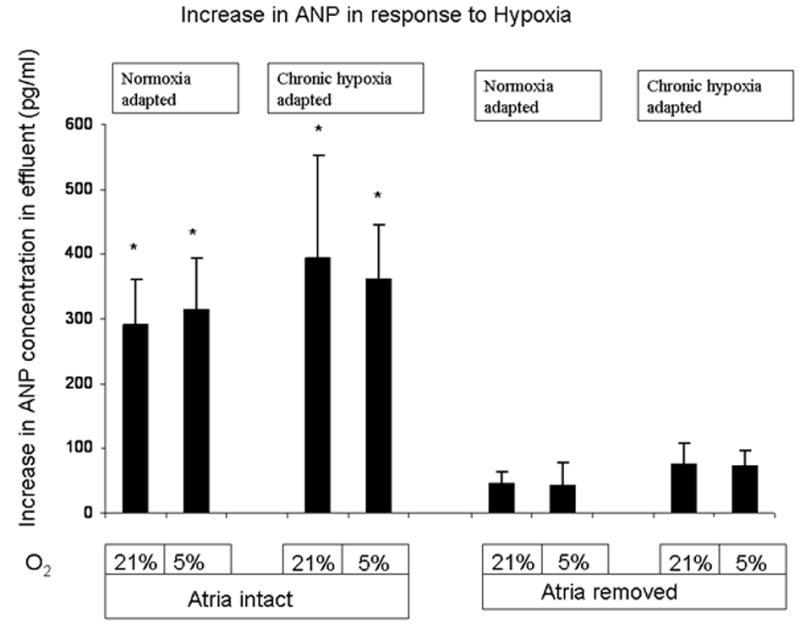
Acute hypoxia caused a significant increase in effluent ANP levels in hearts with the atria attached but not in hearts with the atria removed. * P < 0.05 (n= 3 to 12). This was not affected by the degree of acute hypoxia as there was no difference in response between 5% and 21% O2. The increase in ANP levels were calculated by comparing the mean ANP level after 15 minutes of 5 % and 21% O2 with the mean ANP levels measured at the end of the two 30 minute exposures of 95% O2
ANP Measurements
Samples were prepared for ANP analysis by concentrating samples with a Sep-Pak C18 column (Waters Corp, Milford MA). Columns were prepared with methanol, washed with water. Samples were acidified with 4% acetic acid, placed on columns, washed with 4% acetic acid and eluted with 90% ethanol/ 9.6% water/ 0.4% acetic acid. The eluate was dried in a Savant Speed-Vac Concentrator (Farmingdale NY). ANP levels were determined by ELISA (Caymen Chemical, Ann Arbor MI).
Results
Effect of Chronic Hypoxia on Ventricular Mass and Pressures
Three weeks of hypobaric hypoxia increased right ventricular mass normalized to left ventricle plus septum (RV/LV+S) or body weight (RV/BW) approximately 2-fold, consistent with the development of marked right ventricular hypertrophy (Table 1). Acute hypoxia decreased left ventricular systolic pressure (LVSP) and increased left ventricular diastolic pressure (LVDP) resulting in a marked drop in left ventricular generated pressure (LVGP) (Figure 2 and Table 2). Chronic hypoxia had no effect on left ventricular mass (Table 1) and did not affect left ventricular contractility or contractile response to acute hypoxia. Left ventricular pressures were not statistically different after removal of the cardiac atria and no differences in LVSP, LVDP, or LVGP were seen between hearts isolated from normoxic or hypoxia-adapted rats at anytime point measured (Table 2).
Table 1.
Effect of Chronic Hypoxia Exposure in rats on Right Ventricular Mass Normalized to Left Ventricle Plus Septum Mass and Body Weight compared to Normoxia exposure.
| n | Body Weight (gm) | RV Weight (mg) | LV+S Weight (mg) | RV/(LV+S) | RV/BW | (LV+S)/BW | |
|---|---|---|---|---|---|---|---|
| Normoxia-Adapted Rats | 11 | 336 ± 8.4 | 211.3 ± 15.9 | 862.7 ± 39.3 | 0.24 ± 0.01 | 0.62 ± 0.04 | 2.5 ± 0.07 |
| Hypoxia-Adapted Rats | 14 | 312 ± 5.9 | 316 ± 12.8 | 776.5 ± 12.5 | 0.41 ± 0.01* | 1.21 ± 0.08* | 2.5 ± 0.05 |
Values are mean ± SE. RV- right ventricle, LV+S – left ventricle plus septum, BW – body weight.
P < 0.05 vs normoxia.
Figure 2.
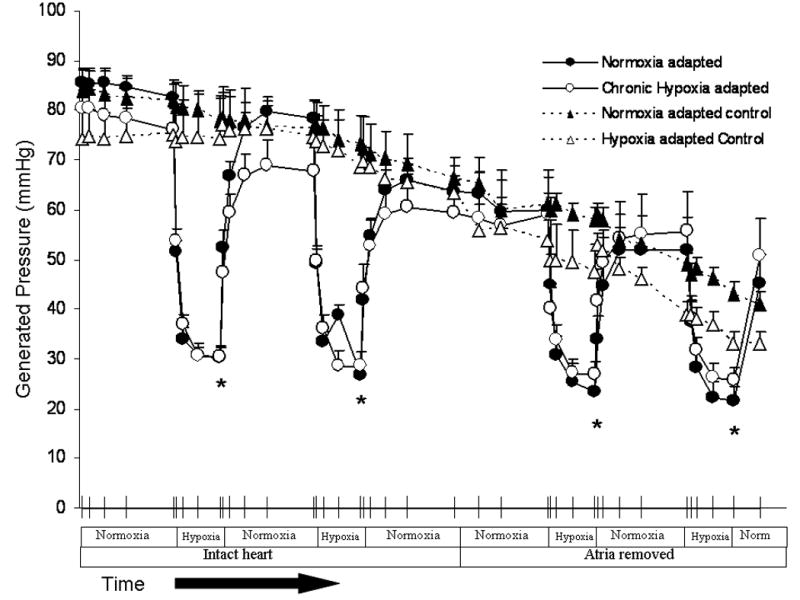
Acute hypoxia causes a significant drop in left ventricular generated pressure compared to hearts that were exposed to normoxia alone. * P < 0.05 (n= 3 to 12). No significant difference in hearts from normoxia-adapted rats versus hearts from hypoxia-adapted rats. Left ventricular generated pressure was not significantly altered by removal of the atria.
Table II.
Effect of Acute Hypoxia on Left Ventricular Systolic, Diastolic, and Generated Pressures.
| Atria Intact | Atria Removed | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| mmHg | 95% O2 | Acute Hypoxia | 95% O2 | Acute Hypoxia | 95% O2 | Acute Hypoxia | 95% O2 | Acute Hypoxia | 95% O2 | |
| Normoxia-adapted rats n=12 | LVSP | 85.2±2.2 | 46.1±2.1* | 81.6±3.9 | 43.6±2.9* | 68.3±6.8 | 47.5±5.9* | 64.7±6.2 | 46.4±6.3* | 70.2±9.0 |
| LVDP | 2.5±1.3 | 15.7±2.0* | 3.5±1.2 | 16.8±3.3* | 8.4±3.5 | 22.3±5.1* | 12.8±4.5 | 24.9±6.4* | 18.6±6.9 | |
| LVGP | 82.6±2.6 | 30.3±2.4* | 78.3±3.6 | 26.8±2.0* | 59.9±6.8 | 23.3±3.9* | 51.9±6.6 | 21.5±3.1* | 45.2±5.9 | |
| Hypoxia-adapted rats n=11 | LVSP | 80.6±5.0 | 49.7±3.1* | 78.8±6.3 | 51.0±5.5* | 69.0±12.2 | 50.4±6.7* | 70.6±11.8 | 55.8±9.7* | 70.3±10.9 |
| LVDP | 5.3±1.9 | 18.7±3.5* | 11.0±3.3 | 22.6±4.5* | 16.1±5.7 | 26.7±7.1* | 20.4±7.0 | 30.7±7.4* | 24.0±7.7 | |
| LVGP | 76.2±4.1 | 30.3±2.0* | 67.8±5.5 | 28.5±2.9* | 59.1±9.0 | 26.7±2.8* | 55.6±8.1 | 25.6±2.7* | 50.7±7.6 | |
Values are mean ± SE. LVSP - left ventricular systolic pressure, LVDP left ventricular diastolic pressure, LVGP left ventricular generated pressure.
P < 0.05. Acute Hypoxia significantly reduced Left Ventricular Systolic, Diastolic, and Generated Pressures compared to normoxia in both normoxia -adapted rats* P < 0.05 (n= 12) and hypoxia-adapted rats* P < 0.05 (n= 11). Of note, removing the atria from the ventricle did not significantly reduce Left Ventricular Systolic, Diastolic, and Generated Pressures
Effect of Chronic Hypoxia on Baseline and Hypoxia-Induced ANP Release
Secretion of ANP was assessed by measuring the concentration of ANP in the effluent of isolated perfused hearts. Baseline ANP secretion was nearly identical in normoxia-adapted and hypoxia-adapted hearts. There was a trend toward a decrease in ANP perfusate levels during the first 8 minutes after hearts were hung, followed by a steady state ANP secretion between 8 and 30 minutes (Figure 3). Acute hypoxia increased ANP release at least 2-fold. There was no difference in ANP release between 5% and 21% O2. ANP release in response to acute hypoxia was greater in hypoxia-adapted hearts than in normoxia-adapted hearts, although absolute differences in effluent ANP concentration between groups reached statistical significance only at the 8 minute time point (Figure 3).
Figure 3.
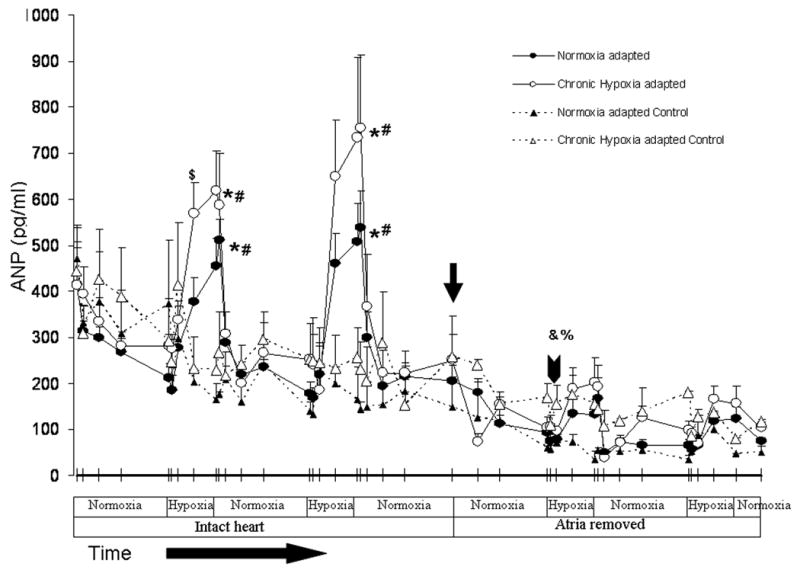
Acute hypoxia (5/21%) causes a significant increase in ANP levels compared to 95% O2 controls *P < 0.05 (n= 3 to 12). There was a significant reduction in ANP levels in intact hearts compared to hearts with the atria removed # P < 0.05 (n= 3 to 12). Of note there was a significant increase in ANP levels in the perfusate effluent at 8 minutes in the first hypoxic exposure in hypoxiaadapted rats compared to normoxia-adapted rats, but not any other time points $P < 0.05 (n= 11 to 12) . There is a significant drop in baseline ANP effluent levels following a 30 minute exposure to 95% O2 after the atria had been removed (see arrowhead) in both normoxia- and hypoxia-adapted rats compared to baseline ANP effluent levels following a 30 minute exposure to 95% with atria intact (see arrow) O2 %P < 0.05, &P < 0.05 (n= 3 to 12).
Thirty minutes after removal of both cardiac atria, baseline ANP secretion fell from 205 pg/ml to 94 pg/ml in control hearts and from 249pg/ml to 103pg/ml in the hypoxia-adapted hearts. This resulted in a 52 and 59% decrease in basal ANP release in normoxic and hypoxia-adapted hearts, respectively (Figure 3). With atria attached, the mean increase in ANP concentration after switching to perfusate equilibrated with 21 or 5% oxygen was 291.4 and 313.8 pg/ml, respectively for normoxic hearts and 394.8 and 360.6 pg/ml, respectively for hypoxia-adapted hearts (Figure 4). With atria removed, the mean increase in ANP effluent concentration for 21 and 5% oxygen was 44.6 and 42.2 pg/ml in normoxic hearts and 75.4 and 73.8 pg/ml in hypoxia-adapted hearts (Figure 4). Thus, removal of the cardiac atria decreased hypoxia-induced ANP release 85-87% in normoxia hearts and 80-81% in hypoxia-adapted hearts. With atria removed, no significant differences were seen in hypoxia-induced ANP release between normoxia and hypoxia-adapted hearts (Figure 3 and 4).
Effect of Hypoxia on Acid-base Disturbance and Transplasmalemmal Electrolyte Flux
In order to determine if acute hypoxia increased ANP release by causing an acute intracellular acidosis or changes in intracellular electrolyte concentrations, we measured changes in lactate, pH, Na+, K+, and Ca++ concentration in the effluent from isolated perfused hearts. Acute hypoxia caused an immediate and marked increase in lactate production. Lactate levels in the effluent of normoxic hearts increased nearly 9-fold within minutes of switching to perfusate equilibrated with 21 or 5% O2 (Figure 5). Hypoxia-induced lactate release was lower in hypoxia-adapted hearts. Lactate levels in the effluent of hypoxia-adapted hearts were approximately half those of normoxic hearts during each hypoxic challenge (Figure 5). Despite the difference in lactate levels, no differences in effluent pH were seen between normoxia and hypoxia-adapted hearts following exposure to acute hypoxia (Figure 6). Also, there was no difference in glucose consumption between normoxia and hypoxia-adapted hearts in response to acute hypoxia (Figure 7). In order to determine if acute hypoxia resulted in disturbances in electrolyte concentration, we examined the effect of acute hypoxia on Na+, K+, and Ca++ secretion. Effluent levels of these cations were the same in normoxia and hypoxia-adapted hearts under normoxic and acutely hypoxic conditions (data not shown).
Figure 5.
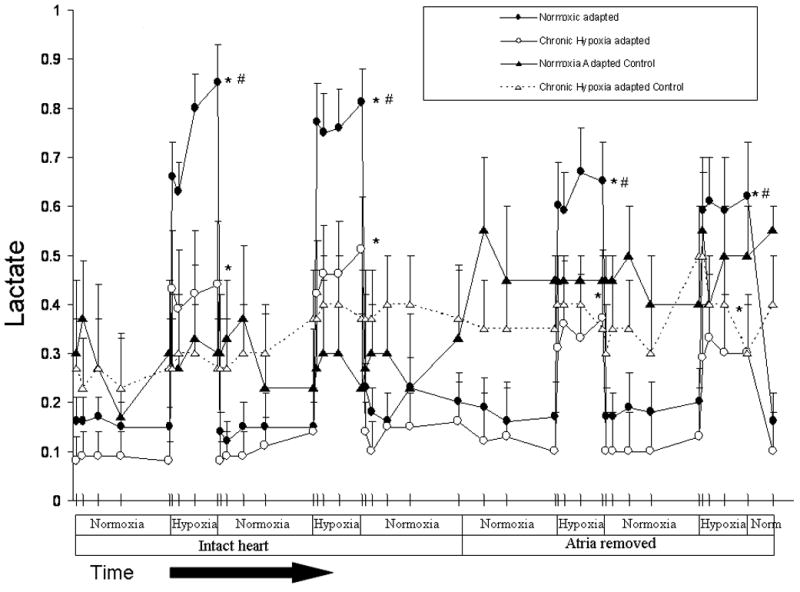
Acute hypoxia (5 or 21% O2) increased lactate production in normoxia- and hypoxia-adapted rats compared to 95% O2 * P < 0.05 (n= 3 to 12). Hypoxia induced increases in lactate release tended to be less after removal of the atria, but no significant differences were found. Hypoxia-induced lactate release was significantly greater in normoxia-adapted rats compared to hypoxia-adapted rats. # P < 0.05 (n= 11 to 12)
Figure 6.
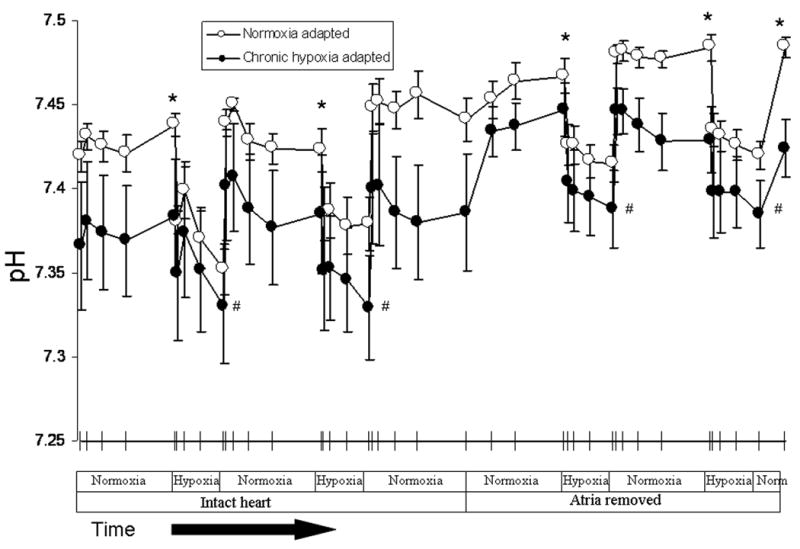
Effluent pH was lower in hearts obtained from hypoxia-adapted rats than normoxia-adapted rats * P < 0.05 (n= 11 to 12). Acute hypoxia decreased effluent pH in both groups # P < 0.05 (n= 11 to 12), but there was no difference in the two groups when comparing the drop in pH that occurred in response to acute hypoxia.
Figure 7.
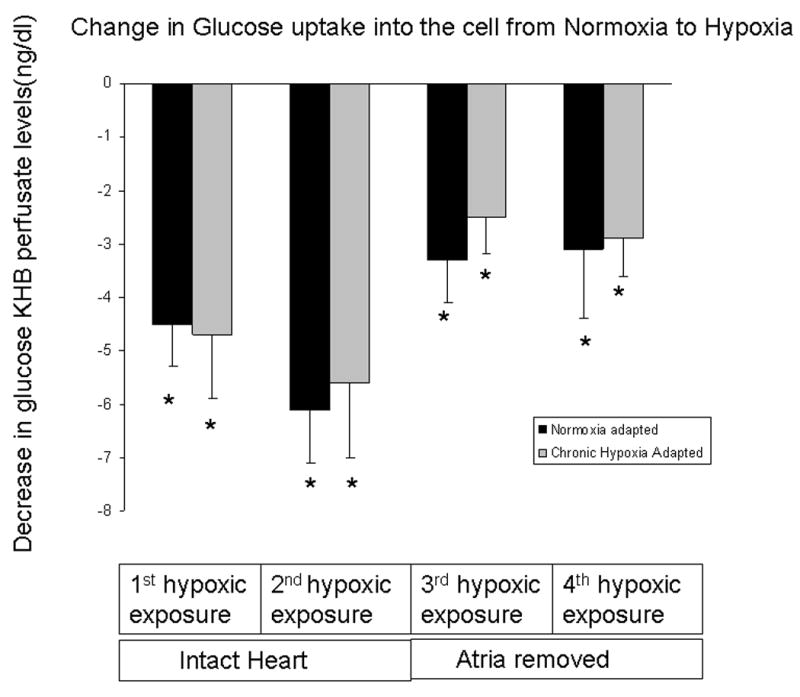
There is a significant decrease of glucose concentration in perfusate effluent in response to acute hypoxia suggesting an increase in glucose uptake compared to 95%O2 (i.e hearts that were exposed to normoxia alone) * P < 0.05 (n= 3 to 12). There was no difference in hypoxia-induced fall in glucose between normoxia-adapted and hypoxia-adapted rats.
Discussion
In the present study we examined hypoxia-induced ANP release in isolated perfused hearts obtained from normoxic rats and rats adapted to chronic hypoxia. In intact rats, chronic hypoxia decreases right atrial ANP levels with little affect on atrial ANP mRNA levels whereas right ventricular ANP content and steady state ANP mRNA levels increase dramatically [17]. The physiological significance of these changes is unclear. The fall in right atrial ANP concentration has been attributed to increased ANP secretion without sufficient increase in synthesis [20]. These changes suggest that atrial ANP release during hypoxia may be diminished, but this was not the case in our study. In fact, hypoxia-induced ANP release was greater in hearts obtained from hypoxia adapted rats and the great majority of the increase in ANP release derived from the atria. These findings suggest that atrial ANP release is increased during adaptation to hypoxia and that the RV is not an important source of ANP release under normoxic or hypoxic conditions. In the present study, we measured ANP in the effluent of isolated perfused hearts and did not examine tissue protein or mRNA levels. In previous studies [12], we found marked increases in RV ANP mRNA and tissue levels using the same hypoxic exposures as described here. Our experiments do not preclude the possibility that increased RV production of ANP occurs in the RV and is not released into the pulmonary circulation. They do, however, strongly suggest that the RV is not a major contributor to the increase in plasma ANP levels that occurs during adaptation to chronic hypoxia.
The relative contribution of atrial and ventricular ANP release during hypoxia has not been defined. In a previous study, Arad et al. found that 90% of the increase in ANP release stimulated by ischemia-reperfusion in isolated perfused rat hearts was of atrial origin [21], similar to the 80-85% increase in ANP release observed in our study when atria were intact. The ability of the atria to rapidly release large amounts of ANP is likely due to their ability to store pre-synthesized ANP in secretory granules [22]. In contrast, studies of cultured ventricular cardiomyocytes have shown that ANP is secreted by a constitutive pathway and only about 10% of newly synthesized ANP is stored [23]. Studies in intact animals however, suggests the presence of ANP granules in hypertrophied ventricular tissue [24] and that an appropriate stimulus can enhance ANP release from ventricular cells by a regulatory pathway analogous to that in atria. Previous studies have demonstrated that the amount of ventricular ANP released into the perfusion fluid increases with ventricular hypertrophy [15]. Moreover, physical exercise causes release of ANP from the hypertrophic ventricle with depletion of endocardial left ventricular stores [15]. In the present study, we observed a small increase in ANP release in response to acute hypoxia after the atria were removed. This corresponds to previous experiments that showed a non-sustained increase from 30 fmol/ml at baseline to 50 fmol/ml in response to acute hypoxia [25]. Thus, the ventricles are capable of contributing to ANP release in response to acute hypoxia, but the contribution is minor compared to that of the atria and does not increase appreciably with the development of right ventricular hypertrophy during adaptation to chronic hypoxia, despite previous reports of as much as a 9-fold increase in right ventricular tissue levels [17].
A reduction in cardiac ANP release following removal of the atria may not have occurred solely as the result of losing the atrial source of ANP. For example LVGP was slightly lower toward the end of the experiment when the atria were removed and it is possible that the decline in LVGP decreased ventricular ANP release. However, the decline in LVGP over the course of the experiment was gradual and the difference in LVGP before and after removal did not reach statistical significance, whereas the change in basal and hypoxia-induced ANP release was markedly lower immediately after atrial removal and represented a statistically significant change. It is also possible that depletion of ventricular ANP stores could have occurred during the first 2 hypoxic challenges before the atria were removed. Our experimental design did not allow us to randomize the order in which ANP release was measured, i.e. once the atria were removed they could not be reattached. Although this approach resulted in hypoxia-induced ANP release from the ventricles with atria removed always being measured after ANP release from ventricles with atria intact, it allowed us to compare serial measurements in the same heart. While we cannot exclude the possibility that ANP release from the ventricles may have been greater if it was measured at the beginning of the study, we feel that the abrupt fall in cardiac ANP release immediately following atrial removal was the result of losing the atrial ANP source and not the result of the gradual loss of LVGP or ventricular ANP stores.
In intact animals, hypoxia-induced ANP release may occur as the result of right atrial stretch secondary to hypoxic pulmonary vasoconstriction [20]. However, reduction in oxygen tension has been shown to increase cardiac ANP release independent of changes in pulmonary artery pressure and cardiac tissue remodeling [17,26]. For example, acute hypoxia causes a greater release of ANP than volume loading in rabbits and lambs, in vivo, and stimulates the release of ANP from isolated rat and rabbit hearts, in vitro, in the absence of atrial distension or changes in heart rate. In an earlier study, we found that hypoxia also causes ANP release from primary cultures of cardiac myocytes [3]. Interestingly, this effect was seen in atrial, but not in ventricular cardiocytes. Taken together, these findings suggest that hypoxia may be able to stimulate ANP release directly from the atria, independent of its effect on right atrial stretch [26].
There are several potential mechanisms by which reduced oxygen tension could stimulate ANP release from atrial cardiocytes. In the present study, we sought to ensure that ANP release was not the result of cardiac injury from ischemia or acidosis. ANP release increased substantially during acute hypoxia, but was similar in (21% O2) and severe (5% O2) hypoxia and rapidly returned to normal upon re-exposure to normoxia. Hearts demonstrated a slow decline in LVGP in all experiments including controls. This decline appeared to be a general weakening of myocardial contractility over time and was not accelerated by exposure to intermittent acute hypoxia and did not result in a decline in ANP response to acute hypoxia. Thus, hypoxia induced ANP release could not be readily attributed to myocardial injury. Acute hypoxia resulted in a significant drop in effluent pH that was associated with a significant increase in glucose uptake and increased lactate production. The increase in glucose uptake seen in the cardiac myocytes during hypoxia is well described and is likely mediated by enhanced function of the facilitative glucose transporters [27]. Interestingly, the drop in pH and glucose consumption in response to acute hypoxia was similar in both groups of rats despite the lower lactate levels in the hypoxia-adapted rats. Cardiac myocytes protect against increases in intracellular hydrogen ion concentration [H+] by exchanging H+ for Na+, and then exchanging Na+ for Ca++ [21]. Influx of extracellular Na+ and Na+-Ca++ exchange modulate ANP secretion induced by hyperosmolar conditions [28] and could play a similar role in hypoxia-induced ANP secretion [29]. However, the Na+, K+ and Ca++ effluent levels were the same in normoxic and hypoxia-adapted hearts.
Our findings do not reveal a mechanism by which acute hypoxia causes ANP release in the isolated perfused heart. However, numerous mechanisms by which hypoxia could stimulate ANP release from atrial myocytes have been proposed, including activation of PKC [32], a pulmonary neural reflex [33], increases in adrenergic activity [34], and increased circulating levels of adenosine [35], arginine vasopressin [36], and endothelin-1 [13]. The isolated perfused heart model eliminates autonomic reflexes and permits control of hemodynamic factors, but some of the paracrine mechanisms that have been proposed to contribute to hypoxia-induced ANP release may remain intact in this preparation. Further studies are needed to elucidate the role of these mechanisms in hypoxia-induced cardiac ANP secretion.
In summary, the cardiac atria appear to be the primary source of ANP released in response to acute hypoxia both in normoxic rats and in those adapted to chronic hypoxia. Increased expression of ANP that has been described in the hypertrophic RV of hypoxia-adapted rats does not appear to contribute substantially to cardiac ANP release. Hypoxia-induced ANP release appears to result from a direct effect of reduced oxygen tension on cardiac myocytes rather than cardiac injury or changes in extracellular pH or electrolyte concentration.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Edwards BS, Zimmerman RS, Schwab TR, Heublein DM, Burnett JC., Jr Atrial stretch, not pressure, is the principal determinant controlling the acute release of atrial natriuretic factor. Circ Res. 1988;62(2):191–5. doi: 10.1161/01.res.62.2.191. [DOI] [PubMed] [Google Scholar]
- 2.Nishimura K, Ban T, Saito Y, Nakao K, Imura H. Atrial pacing stimulates secretion of atrial natriuretic polypeptide without elevation of atrial pressure in awake dogs with experimental complete atrioventricular block. Circ Res. 1990;66(1):115–22. doi: 10.1161/01.res.66.1.115. [DOI] [PubMed] [Google Scholar]
- 3.Klinger JR, Pietras L, Warburton R, Hill NS. Reduced oxygen tension increases atrial natriuretic peptide release from atrial cardiocytes. Exp Biol Med (Maywood) 2001;226(9):847–53. doi: 10.1177/153537020122600907. [DOI] [PubMed] [Google Scholar]
- 4.de Bold AJ. Atrial natriuretic factor: an overview. Fed Proc. 1986;45(7):2081–5. [PubMed] [Google Scholar]
- 5.Jin HK, Yang RH, Thornton RM, Chen YF, Jackson R, Oparil S. Atrial natriuretic peptide lowers pulmonary arterial pressure in hypoxia-adapted rats. J Appl Physiol. 1988;65(4):1729–35. doi: 10.1152/jappl.1988.65.4.1729. [DOI] [PubMed] [Google Scholar]
- 6.Jin H, Yang RH, Chen YF, Jackson RM, Oparil S. Atrial natriuretic peptide attenuates the development of pulmonary hypertension in rats adapted to chronic hypoxia. J Clin Invest. 1990;85(1):115–20. doi: 10.1172/JCI114400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klinger JR, Warburton RR, Pietras LA, Smithies O, Swift R, Hill NS. Genetic disruption of atrial natriuretic peptide causes pulmonary hypertension in normoxic and hypoxic mice. Am J Physiol. 1999;276(5 Pt 1):L868–74. doi: 10.1152/ajplung.1999.276.5.L868. [DOI] [PubMed] [Google Scholar]
- 8.Raffestin B, Levame M, Eddahibi S, Viossat I, Braquet P, Chabrier PE, Cantin M, Adnot S. Pulmonary vasodilatory action of endogenous atrial natriuretic factor in rats with hypoxic pulmonary hypertension. Effects of monoclonal atrial natriuretic factor antibody. Circ Res. 1992;70(1):184–92. doi: 10.1161/01.res.70.1.184. [DOI] [PubMed] [Google Scholar]
- 9.Klinger JR, Warburton RR, Pietras L, Oliver P, Fox J, Smithies O, Hill NS. Targeted disruption of the gene for natriuretic peptide receptor-A worsens hypoxia-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2002;282(1):H58–65. doi: 10.1152/ajpheart.2002.282.1.H58. [DOI] [PubMed] [Google Scholar]
- 10.Zhao L, Mason NA, Strange JW, Walker H, Wilkins MR. Beneficial effects of phosphodiesterase 5 inhibition in pulmonary hypertension are influenced by natriuretic Peptide activity. Circulation. 2003;107(2):234–7. doi: 10.1161/01.cir.0000050653.10758.6b. [DOI] [PubMed] [Google Scholar]
- 11.Klinger JR, Arnal F, Warburton RR, Ou LC, Hill NS. Downregulation of pulmonary atrial natriuretic peptide receptors in rats exposed to chronic hypoxia. J Appl Physiol. 1994;77(3):1309–16. doi: 10.1152/jappl.1994.77.3.1309. [DOI] [PubMed] [Google Scholar]
- 12.Hill NS, Klinger JR, Warburton RR, Pietras L, Wrenn DS. Brain natriuretic peptide: possible role in the modulation of hypoxic pulmonary hypertension. Am J Physiol. 1994;266(3 Pt 1):L308–15. doi: 10.1152/ajplung.1994.266.3.L308. [DOI] [PubMed] [Google Scholar]
- 13.Skvorak JP, Sutton ET, Rao PS, Dietz JR. Mechanism of anoxia-induced atrial natriuretic peptide release in the isolated rat atria. Am J Physiol. 1996;271(1 Pt 2):R237–43. doi: 10.1152/ajpregu.1996.271.1.R237. [DOI] [PubMed] [Google Scholar]
- 14.Ruskoaho H, Lang RE, Toth M, Ganten D, Unger T. Release and regulation of atrial natriuretic peptide (ANP) Eur Heart J. 1987;8(Suppl B):99–109. doi: 10.1093/eurheartj/8.suppl_b.99. [DOI] [PubMed] [Google Scholar]
- 15.Ruskoaho H, Kinnunen P, Taskinen T, Vuolteenaho O, Leppaluoto J, Takala TE. Regulation of ventricular atrial natriuretic peptide release in hypertrophied rat myocardium. Effects of exercise. Circulation. 1989;80(2):390–400. doi: 10.1161/01.cir.80.2.390. [DOI] [PubMed] [Google Scholar]
- 16.Thibault G, Nemer M, Drouin J, Lavigne JP, Ding J, Charbonneau C, Garcia R, Genest J, Jasmin G, Sole M, et al. Ventricles as a major site of atrial natriuretic factor synthesis and release in cardiomyopathic hamsters with heart failure. Circ Res. 1989;65(1):71–82. doi: 10.1161/01.res.65.1.71. [DOI] [PubMed] [Google Scholar]
- 17.Stockmann PT, Will DH, Sides SD, Brunnert SR, Wilner GD, Leahy KM, Wiegand RC, Needleman P. Reversible induction of right ventricular atriopeptin synthesis in hypertrophy due to hypoxia. Circ Res. 1988;63(1):207–13. doi: 10.1161/01.res.63.1.207. [DOI] [PubMed] [Google Scholar]
- 20.McKenzie JC, Tanaka I, Inagami T, Misono KS, Klein RM. Alterations in atrial and plasma atrial natriuretic factor (ANF) content during development of hypoxia-induced pulmonary hypertension in the rat. Proc Soc Exp Biol Med. 1986;181(3):459–63. doi: 10.3181/00379727-181-rc3. [DOI] [PubMed] [Google Scholar]
- 21.Arad M, Zamir N, Horowitz L, Oxman T, Rabinowitz B. Release of atrial natriuretic peptide in brief ischemia-reperfusion in isolated rat hearts. Am J Physiol. 1994;266(5 Pt 2):H1971–8. doi: 10.1152/ajpheart.1994.266.5.H1971. [DOI] [PubMed] [Google Scholar]
- 22.Ruskoaho H. Atrial natriuretic peptide: synthesis, release, and metabolism. Pharmacol Rev. 1992;44(4):479–602. [PubMed] [Google Scholar]
- 23.Bloch KD, Seidman JG, Naftilan JD, Fallon JT, Seidman CE. Neonatal atria and ventricles secrete atrial natriuretic factor via tissue-specific secretory pathways. Cell. 1986;47(5):695–702. doi: 10.1016/0092-8674(86)90512-x. [DOI] [PubMed] [Google Scholar]
- 24.Ding J, Thibault G, Gutkowska J, Garcia R, Karabatsos T, Jasmin G, Genest J, Cantin M. Cardiac and plasma atrial natriuretic factor in experimental congestive heart failure. Endocrinology. 1987;121(1):248–57. doi: 10.1210/endo-121-1-248. [DOI] [PubMed] [Google Scholar]
- 25.Toth M, Vuorinen KH, Vuolteenaho O, Hassinen IE, Uusimaa PA, Leppaluoto J, Ruskoaho H. Hypoxia stimulates release of ANP and BNP from perfused rat ventricular myocardium. Am J Physiol. 1994;266(4 Pt 2):H1572–80. doi: 10.1152/ajpheart.1994.266.4.H1572. [DOI] [PubMed] [Google Scholar]
- 26.Baertschi AJ, Adams JM, Sullivan MP. Acute hypoxemia stimulates atrial natriuretic factor secretion in vivo. Am J Physiol. 1988;255(2 Pt 2):H295–300. doi: 10.1152/ajpheart.1988.255.2.H295. [DOI] [PubMed] [Google Scholar]
- 27.Zhang JZ, Behrooz A, Ismail-Beigi F. Regulation of glucose transport by hypoxia. Am J Kidney Dis. 1999;34(1):189–202. doi: 10.1016/s0272-6386(99)70131-9. [DOI] [PubMed] [Google Scholar]
- 28.Schiebinger RJ, Joseph CM, Li Y, Cragoe EJ., Jr Mechanism of hyperosmolality stimulation of ANP secretion: its dependency on calcium and sodium. Am J Physiol. 1995;268(3 Pt 1):E476–83. doi: 10.1152/ajpendo.1995.268.3.E476. [DOI] [PubMed] [Google Scholar]
- 29.Pei JM, Kravtsov GM, Wu S, Das R, Fung ML, Wong TM. Calcium homeostasis in rat cardiomyocytes during chronic hypoxia: a time course study. Am J Physiol Cell Physiol. 2003;285(6):C1420–8. doi: 10.1152/ajpcell.00534.2002. [DOI] [PubMed] [Google Scholar]
- 30.Maddock RJ. The lactic acid response to alkalosis in panic disorder : an integrative review. J Neuropsychiatry Clin Neurosci. 2001;13(1):22–34. doi: 10.1176/jnp.13.1.22. [DOI] [PubMed] [Google Scholar]
- 31.Gladden LB. Lactate metabolism: a new paradigm for the third millennium. J Physiol. 2004;558(Pt 1):5–30. doi: 10.1113/jphysiol.2003.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baertschi AJ, Teague WG. Alveolar hypoxia is a powerful stimulus for ANF release in conscious lambs. Am J Physiol. 1989;256(4 Pt 2):H990–8. doi: 10.1152/ajpheart.1989.256.4.H990. [DOI] [PubMed] [Google Scholar]
- 33.Baertschi AJ, Jiao JH, Carlson DE, Campbell RW, Teague WG, Willson D, Gann DS. Neural control of ANF release in hypoxia and pulmonary hypertension. Am J Physiol. 1990;259(3 Pt 2):H735–44. doi: 10.1152/ajpheart.1990.259.3.H735. [DOI] [PubMed] [Google Scholar]
- 34.Lew RA, Baertschi AJ. Mechanisms of hypoxia-induced atrial natriuretic factor release from rat hearts. Am J Physiol. 1989;257(1 Pt 2):H147–56. doi: 10.1152/ajpheart.1989.257.1.H147. [DOI] [PubMed] [Google Scholar]
- 35.Ogunyemi DA, Koos BJ, Arora CP, Castro LC, Mason BA. Adenosine modulates hypoxia-induced atrial natriuretic peptide release in fetal sheep. Am J Physiol. 1995;269(1 Pt 2):H282–7. doi: 10.1152/ajpheart.1995.269.1.H282. [DOI] [PubMed] [Google Scholar]
- 36.Cheung CY. Autonomic and arginine vasopressin modulation of the hypoxia-induced atrial natriuretic factor release in immature and mature ovine fetuses. Am J Obstet Gynecol. 1992;167(5):1443–53. doi: 10.1016/s0002-9378(11)91731-1. [DOI] [PubMed] [Google Scholar]