

Abstract
Approximately 81.7 million cats are in 37.5 million USA households. Shed fur can be criminal evidence due to transfer to victims, suspects, and / or their belongings. To improve cat hairs as forensic evidence, the mtDNA control region from single hairs, with and without root tags, was sequenced. A dataset of a 402 bp CR segment from 174 random-bred cats representing four USA geographic areas was generated to determine the informativeness of the mtDNA region. Thirty-two mtDNA mitotypes were observed ranging in frequencies from 0.6-27%. Four common types occurred in all populations. Low heteroplasmy, 1.7%, was determined. Unique mitotypes were found in 18 individuals, 10.3% of the population studied. The calculated discrimination power implied that 8.3 of 10 randomly selected individuals can be excluded by this region. The genetic characteristics of the region and the generated dataset support the use of this cat mtDNA region in forensic applications.
Keywords: forensic science, DNA typing, domestic cat, mitochondrial DNA, control region, Felis catus
A study by the American Pet Product Manufacturing Association (1) reported that the majority of people in the United States, 59.5%, own at least one cat or dog. The American Veterinary Medical Association (2) reports a 12.4% increase in pet ownership from 2001 where 61.1 million households owned a pet to 2006 where 68.7 million households were estimated to own a pet. A study of hair growth in the adult domestic shorthair cat determined an average growth of 32.7g of hair per kilogram of body weight per year (3). Roughly translated, the presence of one 15-pound feline inside a home can generate 223 grams of hair every year, implying many hundreds of thousands of hairs.
As any pet owner will tell you, their animals’ hair typically finds its way onto almost every household surface. This hair sheds and easily transfers during normal daily activities such as grooming, petting, or simply rubbing against clothing or furniture. A study investigating animal hair transfer during simulated criminal activity concluded that it is almost impossible to enter a house where a domestic animal lives without being contaminated by its hair (4). Another study has shown that the persistence of hair on clothing depends largely on the type of fabric (5). In general, hairs persist longer on rougher fabrics and can remain present on a given article of clothing for a period of several hours to several days. Therefore, the transfer of pet hair between a victim and or suspect of a crime has high potential, provided one or both involved parties is a pet owner or the crime occurs in an area occupied by a pet.
Although hairs are one of the most common types of biological evidence at a crime scene (6), companion animal hair evidence, particularly feline, is under-used in forensic investigations. Lack of information regarding the probative value of this evidence by law enforcement personnel is the largest reason this evidence is often disregarded (7). Traditionally, forensic examination of animal hairs has been limited to morphological studies, consisting of visual examination under a microscope. However, a positive identification of an individual can never be achieved with this type of examination, and only rarely can a certain breed of cat or dog even be categorically excluded via a morphological test (8). Even with quantitative measurements, too much variation exists in different hairs from a single individual, let alone between different individuals, for effective differentiation (6-8).
The evidentiary value of animal hairs collected in association with a crime has become more valuable by the advancement of DNA-based testing (9, 10). Today, identifying an individual from a single hair is feasible, provided a root tag or remaining follicular material is available to yield nuclear DNA for testing. Short tandem repeat (STR) markers are the standard for individual identification and studies indicate that STR genotypes can be obtained from cat hairs containing roots in approximately 50% of trials (11). However, unless hairs are plucked, they do not usually contain a viable root tag yielding nuclear DNA. Approximately 95% of hairs that are encountered in forensic casework are in the final stage of growth, the telogen phase, and usually do not contain a root tag (5, 12). Thus, a DNA source in higher abundance on trace evidence, such as mtDNA, may be the only available source for genetic analyses.
Although the statistical occurrence of root tag presence in shed cat fur has not been directly studied, cats are known to be fastidious groomers. Shed cat fur, without the root, can potentially contain sufficient epithelial cells for DNA analysis with mini-STRs, mtDNA, or other DNA markers. For instances where STR analysis fails, mtDNA sequencing of cat fur is an option as a means of inclusion or exclusion and has successfully been employed in at least one criminal investigation for homicide involving cat evidence. In this 2003 case, the State of Iowa v. Ben O’Donnell, mtDNA profiles from several cats were used as corroborative evidence to gain a conviction of the prime suspect for second-degree murder (13).
Mitochondrial gene variation has been previously investigated in cats, however, most studies have focused on DNA variation for species identification (14-17) or cat evolution (18). Within mtDNA, the control region (CR), or D-loop, is generally one of the most divergent regions and is the most appropriate place to investigate variability for individual identification (19-22). Multiple studies investigating the CR in humans confirm the region to be highly polymorphic (23-25). Studies of the CR of domestic dogs and wolves have also shown significant diversity within canids (26-31). In dogs, research has specifically evaluated the forensic utility of the region in conjunction with both breed and geographic information (32).
The entire 17,009 bp mtDNA genome for the domestic cat has been sequenced (33). The cat mtDNA CR spans approximately 1,560 base pairs and contains two distinct repetitive sequence sites flanking the highly conserved core in the CR. This repetitive region has been examined in wild felids, but not extensively in domestic cats (34). This repetitive element is common to most carnivores (35) and could complicate sequencing of the mtDNA CR region in the domestic cat due to significantly high within individual heteroplasmy (36).
Only two published studies evaluate population diversity of the CR of the domestic cat. The first study was conducted by QuestGen Forensics in Davis, CA (36), which included sequences of 167 cats from 14 pure breeds (N=86) and mixed breeds (N=81). The sequenced region encompassed an 80 bp tandem repeat that enhances polymorphism, but proved problematic for the sequence analysis of forensic samples. The study yielded 35 mitotypes with frequencies greater than 1% and 48 mitotypes with frequencies less than 1%. The second published study of the CR was conducted on domestic cats from the Tsushima islands of Japan (37). This study evaluated a 350 bp sequence, yielding 10 mitotypes from 50 Tsushima Island cats. While both of these studies prove that multiple mtDNA mitotypes do exist in the domestic cat population, neither evaluated extensive populations for further consideration in forensic applications.
Shed cat hair is an excellent source of trace evidence although its analytical value is neither appreciated nor developed to its full capacity. The primary goal of this study was to examine the mtDNA CR in the domestic cat for forensic applications. Four populations of cats (N = 174) from diverse regions of the United States were sequenced for a 402 bp region of cat mtDNA CR that excluded the problematic tandem repeats within the region. The level of heteroplasmy and the exclusionary power of the region was examined.
Materials and Methods
Hair and buccal samples were collected from 59 random-bred cats in the Davis and Sacramento, CA area by a technician at a local feline veterinary practice or by the owners of the animals. Verbal permission to collect the samples was obtained from the owners. The cat owners self-reported their pets’ breed, sex, and residence zip code. All cats were reported as domestic short, medium or long-haired cats, except three were reported as Siamese crosses. Only one cat of related individuals was included in the sampled population. Hair was collected by brushing or rubbing with clean, dry hands down the animals back and flank. Collected hairs were placed in clean coin envelopes and stored at ambient room temperature with protection from moisture and sunlight.
All hair processing and mtDNA amplifications were performed at the Forensics Unit of the UC Davis Veterinary Genetics Laboratory (VGL). The VGL has a more controlled environment to reduce concerns of contamination of low template DNA samples as they perform routine casework with low template DNA sources. Single hairs were randomly removed from the coin envelopes using forceps and visually confirmed to have a root tag. Instruments were washed and dried thoroughly between samples to prevent cross contamination. Each hair was placed in a sterile centrifuge tube. None of the hairs was washed. For samples 1-21, entire single hairs were placed in digestion buffer without modification. For samples 22-59, approximately 1 mm of both ends of the hair was removed to ensure that no root tag was included and the remainder of hair shaft was placed in digestion buffer. Each hair sample was digested to completion for 24 hrs in 200 μl of a digestion buffer containing 10mM Tris-HCl, 1mM EDTA, 0.5 mg proteinase K (Roche Applied Science, Indianapolis, IN), 0.2 M dithiothreiol (Promega Corporation, Madison, WI), 0.5 M NaCl (Teknova, Hollister, CA), and 10% SDS (Teknova). All samples were vortexed for 30 s after 1 hr of digestion to assist in mechanical breakage of hairs. Lighter pigmented and thinner hairs appeared completely digested upon visual inspection after four hours, however, all samples were digested for 24 hrs to ensure complete solubilization. After digestion, samples were heated to 95°C for 20 min, refrigerated over-night and centrifuged for 2 min at 15,000g. The supernatant was transferred to clean tubes and the pellet discarded. DNA was precipitated by standard NaCl – ethanol method (38). DNA pellets were re-suspended in 50 μl ddH2O and frozen at −80°C until analysis.
DNA was isolated from the buccal swabs (Fisherbrand™ Sterile cytology brush, Fisher Scientific, Pittsburgh, PA) using a NaOH technique (39). Buccal swab DNA from 126 additional cats from four geographically separate populations including New York (N = 27), Texas (N = 27), Hawaii (N = 59), and 13 additional cats from Davis, CA, were analyzed for the development of the larger mtDNA CR database (Table 1). The populations and the DNA isolation for these cats is previously described (40).
TABLE 1.
Mitochondrial DNA CR Mitotype statistics in cat populations.
| Mitotype | No. | Freq. | CA | Freq. | HI | Freq. | NY | Freq. | TX | Freq. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 48 | 0.276 | 16 | 0.262 | 17 | 0.288 | 6 | 0.222 | 9 | 0.333 |
| 2 | 45 | 0.259 | 10 | 0.164 | 20 | 0.339 | 10 | 0.370 | 5 | 0.185 |
| 3 | 26 | 0.149 | 7 | 0.115 | 7 | 0.119 | 7 | 0.259 | 5 | 0.185 |
| 4 | 9 | 0.052 | 6 | 0.098 | 1 | 0.017 | 1 | 0.037 | 1 | 0.037 |
| 5 | 7 | 0.040 | 3 | 0.049 | 4 | 0.148 | ||||
| 6 | 4 | 0.023 | 2 | 0.033 | 2 | 0.034 | ||||
| 7 | 3 | 0.017 | 1 | 0.016 | 2 | 0.074 | ||||
| 8 | 2 | 0.011 | 1 | 0.016 | 1 | 0.037 | ||||
| 9 | 2 | 0.011 | 2 | 0.033 | ||||||
| 10 | 2 | 0.011 | 2 | 0.033 | ||||||
| 11 | 2 | 0.011 | 2 | 0.034 | ||||||
| 12 | 2 | 0.011 | 2 | 0.034 | ||||||
| 13 | 2 | 0.011 | 2 | 0.074 | ||||||
| 14 | 2 | 0.011 | 2 | 0.033 | ||||||
| 15 | 1 | 0.006 | 1 | 0.016 | ||||||
| 16 | 1 | 0.006 | 1 | 0.016 | ||||||
| 17 | 1 | 0.006 | 1 | 0.016 | ||||||
| 18 | 1 | 0.006 | 1 | 0.017 | ||||||
| 19 | 1 | 0.006 | 1 | 0.017 | ||||||
| 20 | 1 | 0.006 | 1 | 0.017 | ||||||
| 21 | 1 | 0.006 | 1 | 0.017 | ||||||
| 22 | 1 | 0.006 | 1 | 0.017 | ||||||
| 23 | 1 | 0.006 | 1 | 0.017 | ||||||
| 24 | 1 | 0.006 | 1 | 0.017 | ||||||
| 25 | 1 | 0.006 | 1 | 0.017 | ||||||
| 26 | 1 | 0.006 | 1 | 0.016 | ||||||
| 27 | 1 | 0.006 | 1 | 0.016 | ||||||
| 28 | 1 | 0.006 | 1 | 0.016 | ||||||
| 29 | 1 | 0.006 | 1 | 0.016 | ||||||
| 30 | 1 | 0.006 | 1 | 0.016 | ||||||
| 31 | 1 | 0.006 | 1 | 0.037 | ||||||
| 32 | 1 | 0.006 | 1 | 0.016 | ||||||
|
| ||||||||||
| Totals | 174 | 1.000 | 61 | 1.000 | 59 | 1.000 | 27 | 1.000 | 27 | 1.000 |
|
| ||||||||||
| h * | 0.834 ±0.017 | 0.862 ±0.016 | 0.805 ±0.034 | 0.810 ±0.025 | 0.828±0.040 | |||||
|
| ||||||||||
| RMP% * | 16.72 | 12.82 | 21.81 | 26.20 | 20.99 | |||||
|
| ||||||||||
| Exclusion Prob.% | 83.28 | 87.18 | 78.19 | 73.80 | 79.01 | |||||
Heterozygosity (h), random match probability and exclusion probability (1 – RMP) calculated as per Stoneking et al (41).
The domestic cat mtDNA CR was amplified using PCR primers JHmtF3 - gatagtgcttaatcgtgc and JHmtR3- gtcctgtggaacaatagg (Eurofins MWG Operon, Huntsville AL) (courtesy of Dr. Joy Halverson, QuestGen, Davis, CA), which correspond with the published feline mtDNA CR sequence (Genbank no. U20753 and NC_001700) to bp 16760-16776 and bp 240 - 223, respectively, (Fig. 1). These primers amplify a 472 bp region, including the primers.
FIG. 1.
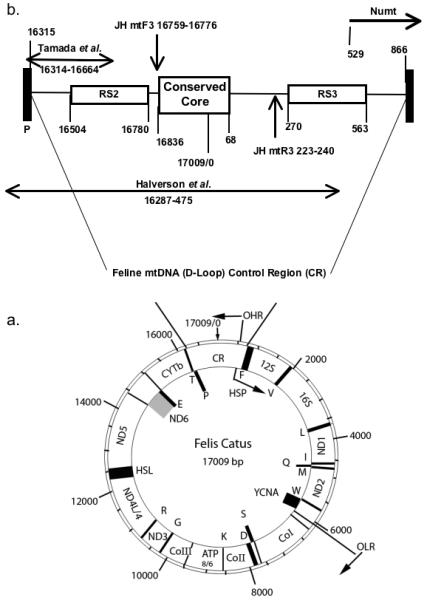
Domestic cat mtDNA control region (modified from Lopez et al., 1996). (a) Physical map of coding genes within the cat cytoplasmic mtDNA. Genes on the inner circle are transcribed from the light (L) strand. Locations of the tRNA genes (shaded boxes) conform to the canonical placental mammalian arrangement and are presented as their standard single letter abbreviations. The following additional abbreviations were used: HSP, putative heavy-strand promoter; OHR, origin of heavy-strand replication; OLR, origin of light-strand replication. (b) The domestic cat CR extends from nucleotide 16,315 to nucleotide 865, producing a 1,559 region. There is a 335-bp overlap of the CR sequence with Numt, which begins at bp 529 within the RS3 and extends to nucleotide 8,454 that includes ~80% of the COII gene. Two distinct repetitive motifs, RS2 and RS3, at opposite ends the CR contribute to the relatively large size of the cat’s mtDNA as compared to other carnivores. These sites are subject to high heteroplasmy. Placement of PCR primers for the three CR studies are noted with arrows.
PCR was performed on a PTC-100 thermal cycler (Bio-Rad, Hercules, CA). Titanium™ Taq (Clontech Laboratories, Inc., Mountain View, CA) was used as the polymerase in all protocols. The final 10 μl PCR protocol contained 0.2 mM each dNTP (ISC BioExpress, Kaysville UT), 1X Titanium Taq Buffer, 0.4 μM forward primer, 0.4 μM reverse primer, 0.75X Titanium Taq, 4 μl of sample DNA and overlaid with 15 μl Chill-out liquid wax (MJ Research Inc, Watertown, MA). The thermal cycler PCR amplification followed the following profile: a 94°C for 1 min initial denaturation followed by 35 cycles of 94°C for 1min denaturation, 56°C for 1 min annealing, 72°C for 1 min extension and a final 72°C for 10 min extension. Samples were held at 4°C until further processing. No in depth efforts, such as changing PCR volumes or template concentration were attempted to improve sequence quality. PCR products were size separated by electrophoresis using a 1% agarose gel. Results were visualized after the gels were soaked in a 1:10,000 dilution of SYBR Green I (Roche Applied Science, Indianapolis, IN) in 1X TBE overnight and documented with an Alpha Imager (Alpha Innotech Corporation, San Leandro, CA).
Remaining PCR products were transferred to a Montage PCR96 Filter Plate and processed according by manufacturer’s protocol (Millipore, Billerica, MA) for direct sequencing. Forward and reverse sequences of the purified PCR products were generated using BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) using the JHmtF3 and JHmtR3 primers in separate reactions for each sample as per manufacturer’s instructions. After cycle sequencing, the unincorporated dyes were removed using the Millipore purification system Montage Seq96 sequencing reaction clean up kit (Millipore) following manufacturer’s recommendations. Sequencing was performed on an ABI 3730 DNA Analyzer (Applied Biosystems).
Sequences were compared and evaluated using Sequencher 4.0™ software (Gene Codes Corporation, Ann Arbor, MI). Forward and reverse sequencing data were assembled into contigs, and consensus sequences were generated. Contigs with sequence ambiguities were resequenced. Manual editing was used to resolve any discrepancies between forward and reverse sequences. If minor manual editing could not resolve minor discrepancies, the sample was not used in analysis. All sequences were trimmed and aligned using both Sequencher and Bioedit (Ibis Biosciences, Carlsbad, CA) programs to a 402 bp minimal variation consensus sequence (described below) and variable nucleotide positions were identified.
Once nucleotide variants were identified in all the sequences, a minimal variation consensus sequence was constructed from the most common mitotypes and termed the “Sylvester” reference sequence (Fig. 2). All mtDNA mitotypes were then defined by their variant sites in comparison to this sequence so that the minimal number of variants would need to be recorded to describe all identified mitotypes.
FIG. 2.

Sylvester reference sequence for domestic cat mtDNA CR.
Presented is the majority rule consensus sequence generated from 1315 mtDNA CR sequences from domestic cats. The nucleotides presented in bold identify the 37 transition and transversion sites identified. Nucleotides adjacent to an insertion/deletion are underlined. Numbering is in reference to the PCR generated product/first mtDNA genomic sequence generated for the cat (33.).
The random match probability (RMP) was calculated according to Stoneking et al. (41). The exclusion probability of the feline mtDNA CR mitotype was calculated as 1 – RMP. For all four population groups, the mean number of uncorrected pair-wise differences and nucleotide diversities within and between populations was calculated in ARLEQUIN version 3.1 (42) following standard procedures (43, 44). The analysis of molecular variance (AMOVA) program in ARLEQUIN was used to estimate the degree of variation within each population and the degree of differentiation between all four populations. The program NETWORK 4.5 (Fluxus Technology Ltd., Clare, Suffolk, UK) was used to create a phylogenetic network, which incorporates all possible shortest least-complex maximum-parsimony phylogenetic trees from the dataset (45).
Results
A 402 bp sequence, extending to 409 bp sequence when including seven different identified insertions, spanning the domestic cat mtDNA CR region was successfully sequenced in 48 of 59 hair samples and 126 cats from four distinct regions in the USA (Table 1). Of 21 hairs that were not trimmed, all produced sequence; of 38 hairs that were trimmed, 27 (71%) produced sequence. The 11 hair samples that were not analyzed produced very poor quality sequence.
When compared to the “Sylvester” reference sequence, 41 variant sites were identified across the four populations. Thirty-two mitotypes were differentiated within the 174 cat sample set. The polymorphic nucleotide positions and frequencies for each of these mitotypes are described in Table 2 and Table 3, respectively. These 32 mitotypes reflect 26 transitions, 7 transversions, 7 insertions, and 1 deletion. As compared to the “Sylvester” reference sequence, the original mtDNA sequence published by Lopez et al. (33) represents a unique mitotype and has 8 variants, described as 59T, 60T, 131T, 159C, 181G, 16859T, 16973A, and 16985A, representing mitotype 33. Mitotype 33 was not considered in the analyses.
TABLE 2.
Variant nucleotide sites in feline mtDNA control region.

|
Dots (.) indicate no difference to the Sylvester sequence, a base call (according to IUPAC recommendations) shows the substitution at a distinct position, Nucleotide substitutions: R=A/G (Purine), Y=C/T (Pyrimidine), W=A/T, denoted with the symbol – Δ.
Nucleotide position counted from origin (33) at top, in reference to amplified sequence at bottom. Insertions denoted as the position number, a decimal point and the number of nucleotides inserted.
Consensus “Sylvester” reference sequence generated to produce the least number of variants to describe all other mitotypes.
TABLE 3.
Mitochondrial DNA CR Mitotype comparisons of cat populations.
| Populations | N | # Matches/ # Comparisons |
Freq. | Mean # Pairwise Differences (π) ±SD (Nucleotide Diversity) |
Range |
|---|---|---|---|---|---|
| Within groups | |||||
| Hawaii | 59 | 350/1711 | 0.20 | 4.14 ± 2.1 (0.0102 ± 0.0057) |
0-10 |
| California | 61 | 208/1830 | 0.11 | 5.05 ± 2.5 (0.0125 ± 0.0068) |
0-11 |
| Texas | 27 | 63/351 | 0.18 | 6.07 ± 3.0 (0.0151 ± 0.0082) |
0-17 |
| New York | 27 | 82/351 | 0.23 | 5.26 ± 2.6 (0.0131 ± 0.0073) |
0-10 |
|
| |||||
| Total | 174 | 2516/15051 | 0.17 | 5.04 ± 2.5 | 0-18 |
|
| |||||
| Between groups | |||||
| Hawaii/California | 120 | 531/3599 | 0.15 | 4.67 ± 2.3 (0.0114 ± 0.0062) |
0-13 |
| Hawaii/Texas | 86 | 289/1593 | 0.18 | 4.94 ± 2.4 (0.0121 ± 0.0066) |
0-18 |
| Hawaii/New York | 86 | 352/1593 | 0.22 | 4.53 ± 2.3 (0.0111 ± 0.0061) |
0-12 |
| California/Texas | 88 | 247/1647 | 0.15 | 5.38 ± 2.6 (0.0133 ± 0.0072) |
0-17 |
| California/New York | 88 | 254/1647 | 0.15 | 5.09 ± 2.5 (0.0126 ± 0.0068) |
0-13 |
| Texas/New York | 54 | 140/729 | 0.19 | 5.67 ± 2.8 (0.0141 ± 0.0076) |
0-17 |
The California population (N=61) had 20 mitotypes with 24 polymorphic sites, including 17 transitions, 4 transversions, and 3 indels. Several polymorphic sites are unique to the California population. The New York population (N=27) had 6 mitotypes and included 14 polymorphic sites consisting of 12 transitions, 1 transversion and 1 indel. No unique mitotypes were observed within this population dataset. The Hawaii population (N=59) had 16 mitotypes with 11 transitions, 2 transversions, and 5 indels, including several polymorphic sites unique to this group. The Texas population (N=27) exhibited 14 mitotypes and had 21 polymorphic sites, consisting of 18 transitions, 2 transversions, and 1 indels. No unique mitotypes were observed in the Texas population
The random match probability for the entire dataset is 16.72% for the 174 sample database examined. The overall exclusion probability of the feline mtDNA CR mitotype is 83.28%. The exclusion probability and random match probabilities of each of the individual populations is presented in Table 2. California had the lowest random match, 12.82%, and highest exclusion probabilities, 87.18%. New York had the highest random match, 26.20%, and lowest exclusion probabilities, 73.80%.
The mtDNA mitotype comparisons within and between populations is presented in Table 1. The average number of pair-wise differences between all mitotypes across all groups was 5.04 ± 2.5, reflecting an average nucleotide diversity of 0.0125 ± 0.0068. The maximum distance between mitotypes was 0.039, reflecting 16 pair-wise differences (polymorphic site differences) between mitotypes 13 and 22; the minimum value was 0.002, reflecting a single polymorphic site between several mitotypes (Fig. 3). The Texan population had the highest nucleotide diversity, 0.0151 ± 0.0082, and the Hawaiian population the lowest, 0.0102 ± 0.0057. The largest between group diversity was found between the populations from New York and Texas, 0.0141 ± 0.0076, and the smallest was between the Hawaii and New York, 0.0111 ± 0.0061.
FIG. 3.
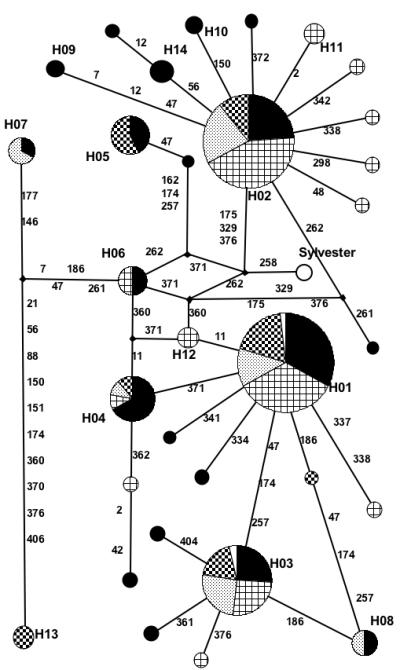
Minimal Spanning Network of mtDNA CR Haplotypes for All Cat Populations. Specific populations are represented by colors: California (solid black), Hawaii (open squares), New York (stipled), and Texas (checkered squares). Unfilled wedges in H01 and H03 represent control sample mitotypes. Bold designators (H##) indicate haplotypes. Theoretical intermediary haplotypes are identified by diamond nodes. Samples without identifiers are unique. The composite “Sylvester” reference sequence is noted. Numbers on the branches indicate the positions and amount of mutations needed to derive connecting mitotypes.
Based on calculations of minimum distances matrixes, giving substitutions, transitions, transversions, and indels equal weight, the phylogenetic relationships between mitotypes were expressed via a network created using a median joining algorithm in Figure 3. Theoretical molecular variants (MVs) or missing mitotypes that complete the mutation pattern linking all observed mitotypes are also expressed in the network. Separate network schematics were also created for each individual population as a visual representation of geographically grouped mitotype relationships (Fig. 4a-d). Common mitotypes 3 and 4 appear to be derived from mitotype 1, requiring only one and three mutations to create these mitotypes, respectively. Sixteen mitotypes are only one mutation different from the four common mitotypes, including the one mutation required to derive mitotype 4 from mitotype 1. All other mitotypes can be derived in two or three mutation events except four mitotypes: mitotypes 5, 7, 13, and 29 (one polymorphism removed from 5), which are significantly different from all other mitotypes, requiring at least six mutation events to be derived from any other mitotype in this dataset.
FIG. 4.
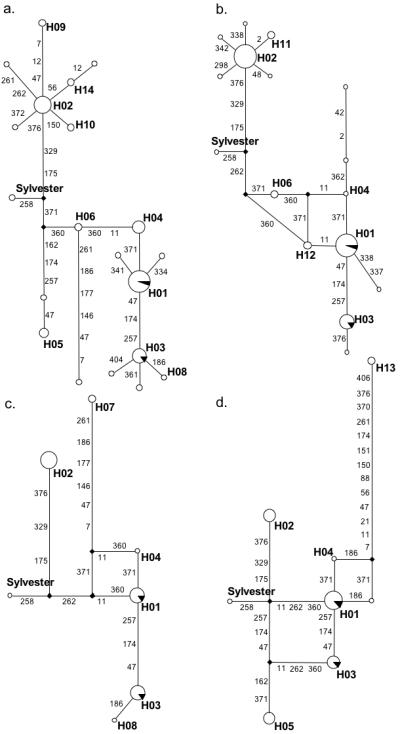
Minimal Spanning Network of mtDNA CR Haplotypes for Specific Cat Populations. a. California, b. Hawaii, c. New York, d. Texas. Black wedges represent control sample haplotypes. Bold designators (H##) indicate haplotypes. Theoretical mitotypes are designated solid diamonds. Nodes without identifiers are unique. The composite “Sylvester” reference sequence is noted. Numbers on the branches indicate the positions and amount of mutations needed to derive the connecting mitotype.
The hair samples and Davis population datasets are both from the same general geographical area in Northern California and were grouped into one population.
The four most common mitotypes were identified in all four regions of the USA. Mitotype 1, the most common mitotype, was observed in 48 individual cats (27.59%). Mitotype 2 was the second most common mitotype, found in 46 individuals (26.43%). Two other common mitotypes, 3 and 4, were identified in 14.94% and 5.17% of the population, respectively. Together, these four mitotypes represent roughly 74.13%, ranging from 64 – 89%, of the combined feline sample population and are the only mitotypes identified in all four populations.
Four additional mitotypes were found in two populations. Mitotype 5 was identified in seven cats from the California and Texas populations. Mitotype 6 was identified in four cats in the California and Hawaii populations. Mitotypes 7 and 8 were found in the New York and California populations. The remaining 24 rare mitotypes were only found in a single population, the bulk of which (N=18) were unique, 0.57% of the population each. The relative frequency of each mitotype is presented in Table 2. The rare mitotypes combined represented 17.24% of the dataset, ranging in frequency from 0 - 24.59%.
Three samples consistently produced sequences, in both forward and reverse directions, that had mixed sites, implying two or more nucleotides observed at the same position in the sequence, suggesting contamination or heteroplasmy. The samples were from the Davis area, two DNA sources were buccal swabs and one was hair. One buccal swab had one heterozygous site and was designated mitotype 18. Both inferred types from this compound mitotype would be unique. The second buccal swab sample had two heterozygous nucleotide sites. Mitotype 2 could be inferred as one mitotype, the other inferable mitotype was unique and was designated as mitotype 15. The third Davis sample, a hair sample, had three heterozygous nucleotides, as well as the mitotype 28 defining insertion. Because this sample has a unique mitotype-defining insertion, in phase with the three 5′ variant nucleotides, all 8 possible mitotypes that could be inferred would be also unique. Since the phase of the variants could not be determined, this cat was assigned only one mitotype with the undetermined nucleotides listed as purines or pyrimidines.
Discussion
Cat Hairs as Evidence
Animal hairs as trace evidence can be useful to potentially link a suspect to a victim or crime scene. A victim placed in a vehicle or held at a location where animals are found often results in the transfer of animal hair to a victim’s clothing. Cat or dog hairs have been identified on the adhesive portions of ransom and extortion notes or restraint bindings prepared by pet owners (7). The types, conditions, and numbers of hairs recovered all impact their value as evidence in a criminal investigation (7). Approximately 95% of hairs encountered in forensic casework are in the final stage of growth, the telogen phase, and usually do not contain a root tag (5, 46). DNA yields from cat hairs with excellent root tags can range from 15-30 ng, 10 times less than is typically extracted from a human hair root (47). However, because cats are fastidious groomers, their hairs may contain more epithelial cells as a source of DNA than hairs of other species such as dogs. Cats are typically only witnesses or victims of crimes, whereas dogs can also be “perpetrators” in attack or bite scenarios, but the ability to more successfully obtain nuclear DNA profiles from cat hairs due to their grooming behaviors improves the cats’ role in criminal investigations.
DNA Analyses of Cat Hair
The first DNA analysis of cat fur in a criminal investigation occurred in 1996 pertaining to a Canadian murder, Beamish vs. Her Majesty’s Court, P.E.I. A set of 10 dinucleotide feline-specific STRs (48) was used to amplify the DNA from one of 27 hairs, which had a root tag, from the lining of a jacket that was part of the evidence in the case (49). No comments were made in this publication as to any processing of the hairs. Studies regarding the development of a tetranucleotide panel of microsatellites for cat forensic genotyping produced DNA types from only 7 of 13 loci from hair of 1 of 13 cats (11). These hairs were all washed prior to amplification to reduce contamination, however, the loss of epithelial cells and some root tag material may have compromised the success of the genotyping. Both of these studies attempted to amplify nuclear DNA, which is in lower quantity, thus, STR studies may benefit from non-washed hairs. Since pet hairs are far less likely to contain foreign contaminants that affect DNA amplification, such as dye and hair care products or cross contamination from human handling, veterinary forensic protocols should perhaps consider not washing hairs. Alternatively, DNA typing both the hairs and the retained wash solutions as a comparison for contamination may be effective.
This study has shown that sufficient mtDNA sequence data can consistently be obtained from the analysis of a single cat hair, including hairs that have had the root tag intentionally removed, provided the hairs are not washed. All cat hairs with root tags provided mtDNA sequence and 71% of cat hairs with the root tag intentionally removed were successfully sequenced. No in depth efforts, such as changing PCR volumes or template concentration, were attempted to improve sequence quality or success rate, thus a higher success rate may be obtained in laboratories performing casework if PCR protocols are refined.
Cat CR mtDNA Sylvester Consensus Reference Sequence
The first complete human mtDNA genome was described nearly thirty years ago (50) and is used as the reference of comparison to describe variation in other human mtDNA mitotypes (51-53) and is now termed the revised Cambridge reference sequence, rCRS (54). The consensus cat sequence described in this study is suggested as an mtDNA CR reference sequence for the cat and is termed the “Sylvester” reference sequence. This study represents some of the initial descriptions of cat variation in the mtDNA CR and the extent of mtDNA variation is not yet known for diverse USA or worldwide populations. An established standard reference sequence should simplify mtDNA forensic nomenclature for future studies. Although convention would suggest using the first published cat sequence by Lopez et al. as the reference, this sequence is unique and was not further identified in this study. A sequence that would require the least number of variants to describe each new mitotype is more efficient. Since two mitotypes represent approximately 25% of the population each, the selection of the most common mitotype as the reference was difficult. Thus, a minimal variant consensus was generated and nomenclature is suggested to follow the standards for human forensics (55-58). The generation of a consensus sequence for the entire mtDNA of the cat from representatives of the four most common mitotypes would augment the “Sylvester” reference sequence.
Cat CR mtDNA Sub-Structuring
The cats had an average of five variant sites between mitotypes across the 402 bp region, suggesting sufficient variation for forensic applications. Humans have an average of eight variant nucleotides, but their CR covers a larger region, approximately 608 bp of the two hyper-variable sites (51), which may account for the slightly higher average. Four common mitotypes account for 63% - 85% of cats across all the populations, suggesting sub-structuring may not be a strong concern in random-bred cats of the USA. In addition, the network analysis shows that a majority of the rare mitotypes identified in the four geographical groups of cats are close descendents, suggesting they have recently derived from the more common mitotypes.
However, although many of the mitotypes overlap all populations (Fig. 3), several of the mitotype network branches are unique to a given population, such as found in types 9 - 14. Furthermore, the Maui and Davis populations both seem to have a significant radiation of unique mitotypes derived from mitotype 2. Additional mitotypes likely exist in the cat population, which would increase the discrimination power associated with this analysis. The high percentage of unique mitotypes should support the region as an exclusionary tool in forensics. The lack of overlap observed in the four examined populations, as seen by the high amount of unique mitotypes in each population, lends strong support for expanding the scope of this study in order to identify additional rare mitotypes thereby expanding the general size of the available database and to examine pedigreed cat structuring.
Cat CR mtDNA Variation
Consistent with various diverse groups of humans, transitions were the most common variation found in the cat mtDNA CR (n = 26), accounting for 79% of substitutions. Insertions and deletions accounted for 19.5% of the total variation. The indels did not necessarily occur in regions with single nucleotide homopolymeric stretches; only one insertion appeared to be the extension of a short, 5 bp, poly-T region.
Accounting for population size, the Texan population had the most variation. The Texan population is from a small town where feral cats are unmanaged - likely for many generations. The network analysis shows that several Texan cats are represented by the more rare mitotypes, suggesting these cats may have very distinct origins or have been isolated. The Hawaiian population had the least variation with only 18 polymorphic sites within 59 individuals. The Hawaiian population is also made up of feral cats, but managed by a spay - neuter - release program. The network analyses suggest the cats have closely derived mitotypes, perhaps representing descendents of a common ancestor. Thus, this population may have had the strongest founder effect or population bottleneck in this survey. Based on the Network analysis, New York’s population is more reflective of the Texan population, and the California population is most similar to that of the Hawaiian population.
The degree of mitotype diversity reported in this study is comparable to that reported by Halverson and Basten (36) in their dataset of 167 pure (N=86) and mixed breed (N=81) cats, which sequenced 945 - 1105 bp. The range in sequence size resulted from their inclusion of the long tandem repeat in the middle of this region that they identified as difficult to sequence. This area corresponds to the published cat mtDNA sequence at bp 16,287 - 16,475 and yielded 52 mitotypes counting the tandem repeat and 31 mitotypes when only the downstream region is considered. The authors do not specify the geographic region(s) or specific breeds included in their results.
The sequence studied by Halverson and Basten (36) partially overlaps the sequence in this study and the 350 bp region studied by Tamada et al. (37) from the 50 random-bred cats in the Tsushima Islands. However, the region from the current study does not overlap the Japanese sequence. The Japanese dataset corresponds to the published sequence at bp 16,314 - 16,664 and yielded 10 mitotypes, with 14 polymorphic sites. This region might exhibit increased variability if examined outside of an island population where probable founder effects may have influenced the population.
Exclusions in Cat CR mtDNA Profiling
Because mtDNA does not segregate or recombine between generations (59) and can, therefore, only be considered as a single locus, the exclusion probability of mtDNA will never approach that of nuclear DNA markers. In forensics applications, mtDNA will always be less preferable to obtaining STR data, where exclusion capacities are on the order of 2 × 10−10 (60). The calculated discrimination power of the investigated mtDNA CR implied that 8.3 of 10 randomly selected individuals can be excluded with this region, implying that 83 out of 100 individuals unrelated to a forensic or investigative sample can be correctly excluded as a possible source of that hair sample using this dataset (61). As compared to the power of STRs, the relatively low exclusion probability of this system is explained by the high frequency, ~5.2 - 27.6%, of the four common mtDNA types found across all four geographical regions and the moderate frequency, ~1.1 - 4.0%, of a few other mitotypes found in more than one population. However, greater than 50% of the mtDNA types identified in this study were rare, with 18 of the 32 identified types having only one representative sample. If the evidence hair is found to be one of the rarer mitotypes, this system is likely to provide information that is discriminating and informative, regardless of the population.
When establishing the significance of a profile match using mtDNA, the frequency of the mitotype within the source population and the population size should be considered. Since reference and evidentiary samples usually originate from the same geographic area or population, knowledge of the population sub-structure in that location is important in assessing the rarity of DNA profiles associated with evidence found at a crime scene (62). This study focused on random-bred cats, avoiding any of the 41 breeds recognized by the Cat Fanciers’ Association (63). Four geographically distinct areas of the USA were examined to get a broad view of mtDNA diversity within the United States. The exclusion capacities for the four sampled geographic regions ranged from 73.80% in the New York population to 87.18% in the California population for the 402 bp region under analysis, suggesting an average exclusion probability of 83.28%. The New York population has an exclusion probability that is lower than the other three populations, resulting from only six mitotypes belonging to this sample set and approximately 85% of the samples are represented by three mitotypes. As the New York population represents unrelated feline patients from a private veterinary practice, the sampling would be anticipated to be diverse. The second lowest exclusion probability was found in the Maui population. There is more confidence that these results are not skewed significantly by a relatively small sample size as 59 cats from this region were examined. The lower exclusion probability may be explained by the fact that as an island, Maui has a more isolated gene pool than the other three populations, reflecting a degree of founder effect influence.
The island population included in the present study, Maui, Hawaii, produced 16 mitotypes from 59 individuals. The Maui population had the second lowest exclusionary power, perhaps also suffering from founder effects. Combining the analysis of the region from the Japanese study with the analysis of the 402 bp examined in this study would potentially significantly increase the discriminatory power of the mtDNA sequencing in forensic casework.
mtDNA Laboratory Techniques
A washing procedure is generally performed on human hairs to remove contaminants that could inhibit PCR reactions or be an alternative DNA source (64). Based on sequence alignments, the feline mtDNA primers used here would be very unlikely to prime sequence amplification from non-mammalian DNA sources, however, DNA from other cats could be a potential contaminant since cats are commonly known to cross-groom and to bite during aggressive encounters and copulation. Three sequences had nucleotide sites that appeared to be heterozygous as specific sites, suggesting either contamination, which could be from various sources, or mtDNA heteroplasmy at a level of 1.72%. Two buccal swab samples had one and two heterozygous nucleotide sites, which is strong support for heteroplasmy. One case represented sequence from a hair sample. Mitotype 28 was from a household containing two cats. If contamination is the cause, cross-grooming could be a feasible source, suggesting a level of 2.08%, 1 in 48 amplified hairs. The mtDNA sequence of the companion cat could help resolve this theory. The presence of two mitotypes from cross-grooming could have more exclusionary or inclusionary power since these cats would have to be in close proximity, potentially warranting an investigation to determine the likelihood of cross-grooming as a source of contamination. The presence of heteroplasmic nucleotide positions in an investigative sample could actually increase the exclusionary power of the sample as the combination of variant nucleotides makes the sample more unique.
This study has found mtDNA profiles can be reliably generated from a single cat hair without a root tag. Routine analysis of cat fur should go beyond a morphological assessment to include mtDNA sequencing in forensic casework when warranted. The evaluation of a 402 bp mtDNA CR region in 174 samples in this study proves that sufficient variability exists within the feline mtDNA CR to make sequencing a worthwhile analysis without having to attempt to sequence the flanking repetitive regions found in the cat mtDNA genome. The consistent distribution of common and rare mitotypes suggests that strong sub-structure of cats is not suggested by mtDNA CR studies. However, the lack of homogeneity between geographically separate populations in this dataset, as supported by the multiple unique mitotypes found in each population, suggests that additional populations need to be analyzed to create a more extensive feline mtDNA database. Analysis of pedigreed samples should be included to get the most accurate picture of potential breed sub-structuring.
Acknowledgements
Cat DNA samples from Texas were provided by Dr. Margaret Slater, from New York by Dr. Betsy Arnold. We appreciate comments and suggestions to this manuscript by Ms. Leslie Bach and Ms. Elizabeth Wictum.
Footnotes
Financial support was provided in part by NIH-NCRR R24 RR016094, the UC Davis Veterinary Genetics Laboratory, and the UC Davis Forensic Sciences graduate program.
References
- 1.American Pet Product Manufacturing Association (APPMA) National pet owner’s survey. APPMA; Greenwich, CT: 2006. [Google Scholar]
- 2.American Veterinary Medical Association (AVMA) US pet ownership and demographics sourcebook. American Veterinary Medical Association; Schaumburg, IL: 2007. [Google Scholar]
- 3.Hendricks WH, Tarttelin MF, Moughan PJ. Seasonal hair growth in the adult domestic cat (Felis catus) Comp Biochem Physiol. 1996;116A(1):29–35. [Google Scholar]
- 4.D’Andrea F, Frides F, Coquoz R. Preliminary experiments on the transfer of animal hair during simulated criminal behavior. J Forensic Sci. 1998;43(6):1257–8. [Google Scholar]
- 5.Dachs J, McNaught IJ, Robertson J. The persistence of human scalp hair on clothing fabrics. Forensic Sci Int. 2003;138(1-3):27–36. doi: 10.1016/j.forsciint.2003.07.014. [DOI] [PubMed] [Google Scholar]
- 6.Bisbing RE. The forensic identification and association of human hair. Forensic Sci Handbook. 1982;1:185–221. [Google Scholar]
- 7.Deedrick DW. Hairs, fibers, crime, and evidence. FBI Forensic Sci Commun. 2000;2 [Google Scholar]
- 8.Moore JE. A key to the Identification of Animal Hairs. J Forensic Sci Soc. 1988;28:335–9. [Google Scholar]
- 9.Wilson MR, Polanskey D, Butler J, DiZinno JA, Replogle J, Budowle B. Extraction, PCR amplification and sequencing of mitochondrial DNA from human hair shafts. Biotechniques. 1995;18(4):662–9. [PubMed] [Google Scholar]
- 10.Higuchi R, von Beroldingen CH, Sensabaugh GF, Erlich HA. DNA typing from single hairs. Nature. 1988;332(6164):543–6. doi: 10.1038/332543a0. [DOI] [PubMed] [Google Scholar]
- 11.Coomber N, David VA, O’Brien SJ, Menotti-Raymond M. Validation of a short tandem repeat multiplex typing system for genetic individualization of domestic cat samples. Croatian Med J. 2007;48(4):547–55. [PMC free article] [PubMed] [Google Scholar]
- 12.Harding H, Rogers G. Physiology and growth of human hair. In: Robertson J, editor. Forensic Examination of Hair. Taylor and Francis; London, U.K.: 1999. pp. 1–62. [Google Scholar]
- 13.Halverson JL, Lyons LA. Forensic DNA identification of feline hairs: casework and a mitochondrial database. Proc Am Acad Forensic Sci. 2004;X:B150. [Google Scholar]
- 14.Bellis C, Ashton KJ, Freney L, Blair B, Griffiths LR. A molecular genetic approach for forensic animal species identification. Forensic Sci Int. 2003;134(2-3):99–108. doi: 10.1016/s0379-0738(03)00128-2. [DOI] [PubMed] [Google Scholar]
- 15.Bravi CM, Liron JP, Mirol PM, Ripoli MV, Peral-Garcia P, Giovambattista G. A simple method for domestic animal identification in Argentina using PCR-RFLP analysis of cytochrome b gene. Leg Med (Tokyo) 2004;6(4):246–51. doi: 10.1016/j.legalmed.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 16.Fridez F, Rochat S, Coquoz R. Individual identification of cats and dogs using mitochondrial DNA tandem repeats? Sci Just. 1999;39(3):167–71. doi: 10.1016/S1355-0306(99)72042-3. [DOI] [PubMed] [Google Scholar]
- 17.Nakaki S, Hino D, Miyoshi M, Nakayama H, Moriyoshi H, Morikawa T, et al. Study of animal species (human, dog and cat) identification using a multiplex single-base primer extension reaction in the cytochrome b gene. Forensic Sci Int. 2007;173(2-3):97–102. doi: 10.1016/j.forsciint.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 18.Driscoll CA, Menotti-Raymond M, Roca AL, Hupe K, Johnson WE, Geffen E, et al. The Near Eastern origin of cat domestication. Science. 2007;317(5837):519–23. doi: 10.1126/science.1139518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown WM, George M, Jr., Wilson AC. Rapid evolution of animal mitochondrial DNA. Proc Nat Acad Sci USA. 1979;76(4):1967–71. doi: 10.1073/pnas.76.4.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown WM, Prager EM, Wang A, Wilson AC. Mitochondrial DNA sequences of primates: tempo and mode of evolution. J Mol Evol. 1982;18(4):225–39. doi: 10.1007/BF01734101. [DOI] [PubMed] [Google Scholar]
- 21.Cann RL, Brown WM, Wilson AC. Polymorphic sites and the mechanism of evolution in human mitochondrial DNA. Genetics. 1984;106(3):479–99. doi: 10.1093/genetics/106.3.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cann RL, Stoneking M, Wilson AC. Mitochondrial DNA and human evolution. Nature. 1987;325(6099):31–6. doi: 10.1038/325031a0. [DOI] [PubMed] [Google Scholar]
- 23.Horai S, Hayasaka K. Intraspecific nucleotide sequence differences in the major noncoding region of human mitochondrial DNA. Am J Hum Genet. 1990;46(4):828–42. [PMC free article] [PubMed] [Google Scholar]
- 24.Vigilant L, Stoneking M, Harpending H, Hawkes K, Wilson AC. African populations and the evolution of human mitochondrial DNA. Science. 1991;253(5027):1503–7. doi: 10.1126/science.1840702. [DOI] [PubMed] [Google Scholar]
- 25.Ward RH, Frazier BL, Dew-Jager K, Paabo S. Extensive mitochondrial diversity within a single Amerindian tribe. Proc Nat Acad Sci USA. 1991;88(19):8720–4. doi: 10.1073/pnas.88.19.8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eichmann C, Parson W. Molecular characterization of the canine mitochondrial DNA control region for forensic applications. Int J Leg Med. 2007;121(5):411–6. doi: 10.1007/s00414-006-0143-5. [DOI] [PubMed] [Google Scholar]
- 27.Pfeiffer I, Volkel I, Taubert H, Brenig B. Forensic DNA-typing of dog hair: DNA-extraction and PCR amplification. Forensic Sci Int. 2004;141(2-3):149–51. doi: 10.1016/j.forsciint.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 28.Savolainen P, Lundeberg J. Forensic evidence based on mtDNA from dog and wolf hairs. J Forensic Sci. 1999;44(1):77–81. [PubMed] [Google Scholar]
- 29.Savolainen P, Rosen B, Holmberg A, Leitner T, Uhlen M, Lundeberg J. Sequence analysis of domestic dog mitochondrial DNA for forensic use. J Forensic Sci. 1997;42(4):593–600. [PubMed] [Google Scholar]
- 30.Schneider PM, Seo Y, Rittner C. Forensic mtDNA hair analysis excludes a dog from having caused a traffic accident. Int J Leg Med. 1999;112(5):315–6. doi: 10.1007/s004140050257. [DOI] [PubMed] [Google Scholar]
- 31.Wetton JH, Higgs JE, Spriggs AC, Roney CA, Tsang CS, Foster AP. Mitochondrial profiling of dog hairs. Forensic Sci Int. 2003;133(3):235–41. doi: 10.1016/s0379-0738(03)00076-8. [DOI] [PubMed] [Google Scholar]
- 32.Himmelberger AL, Spear TF, Satkoski JA, George DA, Garnica WT, Malladi VS, et al. Forensic utility of the mitochondrial hypervariable region 1 of domestic dogs, in conjunction with breed and geographic information. J Forensic Sci. 2008;53(1):81–9. doi: 10.1111/j.1556-4029.2007.00615.x. [DOI] [PubMed] [Google Scholar]
- 33.Lopez JV, Cevario S, O’Brien SJ. Complete nucleotide sequences of the domestic cat (Felis catus) mitochondrial genome and a transposed mtDNA tandem repeat (Numt) in the nuclear genome. Genomics. 1996;33(2):229–46. doi: 10.1006/geno.1996.0188. [DOI] [PubMed] [Google Scholar]
- 34.Eizirik E, Bonatto SL, Johnson WE, Crawshaw PG, Jr., Vie JC, Brousset DM, et al. Phylogeographic patterns and evolution of the mitochondrial DNA control region in two neotropical cats (Mammalia, felidae) J Mol Evol. 1998;47(5):613–24. doi: 10.1007/pl00006418. [DOI] [PubMed] [Google Scholar]
- 35.Hoelzel AR, Lopez JV, Dover GA, O’Brien SJ. Rapid evolution of a heteroplasmic repetitive sequence in the mitochondrial DNA control region of carnivores. J Mol Evol. 1994;39(2):191–9. doi: 10.1007/BF00163807. [DOI] [PubMed] [Google Scholar]
- 36.Halverson JL, Basten C. Forensic DNA identification of animal-derived trace evidence: tools for linking victims and suspects. Croatian Med J. 2005;46(4):598–605. [PubMed] [Google Scholar]
- 37.Tamada T, Kurose N, Masuda R. Genetic diversity in domestic cats Felis catus of the Tsushima Islands, based on mitochondrial DNA cytochrome b and control region nucleotide sequences. Zoolog Sci. 2005;22(6):627–33. doi: 10.2108/zsj.22.627. [DOI] [PubMed] [Google Scholar]
- 38.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acid Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oberbauer AM, Grossman DI, Eggleston ML, Irion DN, Schaffer AL, Pedersen NC, et al. Alternatives to blood as a source of DNA for large-scale scanning studies of canine genome linkages. Vet Res Commun. 2003;27(1):27–38. doi: 10.1023/a:1022006606796. [DOI] [PubMed] [Google Scholar]
- 40.Lipinski MJ, Froenicke L, Baysac KC, Billings NC, Leutenegger CM, Levy AM, et al. The ascent of cat breeds: genetic evaluations of breeds and worldwide random-bred populations. Genomics. 2008;91(1):12–21. doi: 10.1016/j.ygeno.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stoneking M, Hedgecock D, Higuchi RG, Vigilant L, Erlich HA. Population variation of human mtDNA control region sequences detected by enzymatic amplification and sequence-specific oligonucleotide probes. Am J Hum Genet. 1991;48(2):370–82. [PMC free article] [PubMed] [Google Scholar]
- 42.Excoffier LG, Laval S, Schneider S. Arlequin ver 3.0: An integrated software package for population genetics data analysis. Evol Bioinformatics Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- 43.Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105(2):437–60. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–95. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16(1):37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 46.Robertson J. Forensic examination of hair. Taylor and Francis; London, U.K.: 1999. [Google Scholar]
- 47.Menotti-Raymond M, David V, Wachter L, Yuhki N, O’Brien SJ. Quantitative polymerase chain reaction-based assay for estimating DNA yield extracted from domestic cat specimens. Croatian Med J. 2003;44(3):327–31. [PubMed] [Google Scholar]
- 48.Menotti-Raymond M, David VA, Stephens JC, Lyons LA, O’Brien SJ. Genetic individualization of domestic cats using feline STR loci for forensic applications. J Forensic Sci. 1997;42(6):1039–51. [PubMed] [Google Scholar]
- 49.Menotti-Raymond MA, David VA, O’Brien SJ. Pet cat hair implicates murder suspect. Nature. 1997;386(6627):774. doi: 10.1038/386774a0. [DOI] [PubMed] [Google Scholar]
- 50.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290(5806):457–65. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 51.Budowle B, Wilson MR, DiZinno JA, Stauffer C, Fasano MA, Holland MM, et al. Mitochondrial DNA regions HVI and HVII population data. Forensic Sci Int. 1999;103(1):23–35. doi: 10.1016/s0379-0738(99)00042-0. [DOI] [PubMed] [Google Scholar]
- 52.Miller KW, Budowle B. A compendium of human mitochondrial DNA control region: development of an international standard forensic database. Croatian Med J. 2001;42(3):315–27. [PubMed] [Google Scholar]
- 53.Parson W, Parsons TJ, Scheithauer R, Holland MM. Population data for 101 Austrian Caucasian mitochondrial DNA d-loop sequences: application of mtDNA sequence analysis to a forensic case. Int J Leg Med. 1998;111(3):124–32. doi: 10.1007/s004140050132. [DOI] [PubMed] [Google Scholar]
- 54.Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genetics. 1999;23(2):147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 55.Bar W, Brinkmann B, Budowle B, Carracedo A, Gill P, Holland M, et al. Guidelines for mitochondrial DNA typing. DNA Commission of the International Society for Forensic Genetics. Vox Sang. 2000;79(2):121–5. doi: 10.1159/000031227. [DOI] [PubMed] [Google Scholar]
- 56.Bar W, Brinkmann B, Budowle B, Carracedo A, Gill P, Holland M, et al. DNA Commission of the International Society for Forensic Genetics: guidelines for mitochondrial DNA typing. Int J Leg Med. 2000;113(4):193–6. doi: 10.1007/s004140000149. [DOI] [PubMed] [Google Scholar]
- 57.Carracedo A, Bar W, Lincoln P, Mayr W, Morling N, Olaisen B, et al. DNA commission of the international society for forensic genetics: guidelines for mitochondrial DNA typing. Forensic Sci Int. 2000;110(2):79–85. doi: 10.1016/s0379-0738(00)00161-4. [DOI] [PubMed] [Google Scholar]
- 58.Wilson MR, Holland MM, Stoneking M, DiZinno JA, Budowle B. Guidelines for the use of mitochondrial DNA sequencing in forensic science. Crime Lab Dig. 1993;20:68–77. [Google Scholar]
- 59.Giles RE, Blanc H, Cann HM, Wallace DC. Maternal inheritance of human mitochondrial DNA. Proc Nat Acad Sci USA. 1980;77(11):6715–9. doi: 10.1073/pnas.77.11.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Butler JM. STR population database analyses. In: Butler JM, editor. Forensic DNA typing: biology, technology, and genetics of STR markers. 2nd ed Academic Press; London, U.K.: 2003. pp. 473–96. [Google Scholar]
- 61.Angleby H, Savolainen P. Forensic informativity of domestic dog mtDNA control region sequences. Forensic Sci Int. 2005;154(2-3):99–110. doi: 10.1016/j.forsciint.2004.09.132. [DOI] [PubMed] [Google Scholar]
- 62.Salas A, Bandelt HJ, Macaulay V, Richards MB. Phylogeographic investigations: the role of trees in forensic genetics. Forensic Sci Int. 2007;168(1):1–13. doi: 10.1016/j.forsciint.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 63.CFA . The cat fanciers’ association cat encyclopedia. Simon & Schuster; New York, NY: 1993. [Google Scholar]
- 64.Vigilant L. An evaluation of techniques for the extraction and amplification of DNA from naturally shed hairs. Biol Chem. 1999;380(11):1329–31. doi: 10.1515/BC.1999.169. [DOI] [PubMed] [Google Scholar]