
Figure 1.
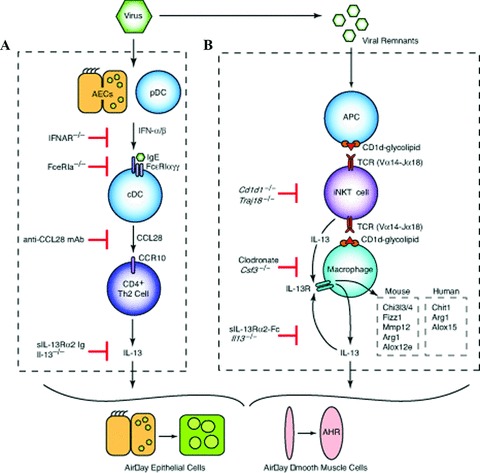
Cellular and molecular scheme for immune pathways leading to chronic lung disease after viral infection. (A) For the first phase of chronic disease, viruses cause airway epithelial cell (AEC) and plasmacytoid dendritic cell (pDC) production of type I interferon (IFN). Subsequent type I IFN receptor(IFNAR) signaling leads to upregulation of FcɛRI expression on resident lung conventional (c)DCs. In turn, FcɛRI activation by viral antigen and antiviral IgE leads to production of CCL28 and recruitment of CCR10‐expressing IL‐13‐producing T helper (Th)2 cells to the lung. The actions of IL‐13 drive differentiation of airway epithelial cell precursors toward mucous cells (mucous cell metaplasia) and airway smooth muscle cells to become more reactive to contractile agonists (airway hyper‐reactivity [AHR]). (B) For the second phase of chronic disease, viral remnants activate antigen‐presenting cells (APCs) and thereby facilitate CD1d‐dependent antigen presentation and consequent activation of invariant CD4− natural killer T (NKT) cells. NKT cells then interact directly with lung macrophages via IL‐13 production and binding to the IL‐13 receptor (IL‐13R) as well as contact between invariant Va14 T‐cell receptor (TCR) and glycolipid‐loaded CD1d. This interaction leads to increased expression of IL‐13R and production of IL‐13 that drives a positive feedback loop to amplify IL‐13 production and alternative activation of macrophages, including Chi3l3/4, Fizz1, Mmp12, Arg1, and Alox12e gene expression in mice and Chit1, Arg1, and Alox15 in humans.