

Abstract
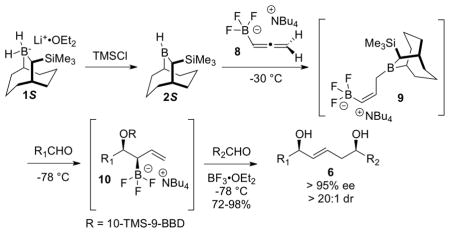
A highly stereoselective synthesis of (E)-1,5-syn-diols 6 is described. The kinetically controlled hydroboration of allenyltrifluoroborate 8 with Soderquist borane 2 provides the (Z)-allylic trifluoroborate 9, which undergoes sequential allylboration with two different aldehydes to provide (E)-1,5-syn-diols 6 in 72–98% yields with > 95% ee. and > 20:1 dr. Application of this method to the synthesis of the tetrafibricin C(23)-C(40) fragment 19 is described.
The development of bifunctionalized allylmetal reagents is of considerable interest for use in the assembly of complex structures in a step-efficient, convergent manner.1 Our laboratory has developed several 1,3-bifunctionalized chiral allylborane reagents1c,i,j for use in natural product synthesis.2 In connection with an ongoing research problem, we required a method for the stereoselective synthesis of (E)-1,5-syn-diols 6. This structural motif is present in many natural products,3 but is not accessible by using our first-generation double allylboration reagents.1c,i,j,4 Accordingly, we have developed and report herein a new double allylboration reagent for the enantio- and diastereoselective synthesis of (E)-1,5-syn-diols 6 (and its enantiomer ent-6), and apply this procedure to the highly stereoselective synthesis of the C(23)-C(40) fragment of tetrafibricin.
At the outset, we envisaged that the requisite double allylborating agent 4 could be prepared via kinetically controlled hydroboration of allene 3 with the Soderquist borane, 10-TMS-9-borabicyclo[3.3.2]decane [10-TMS-9-BBD-H, 2R], and that sequential treatment of 4 with two aldehydes would provide the targeted 1,5-diols 6 (Scheme 1).2h,i Successful implementation of this plan requires (1) that the hydroboration of 3 by the chiral borane reagent 2R be highly stereoselective; (2) that Z to E isomerization of the (Z)-allylborane 4 be slow, and (3) that the reaction of 5 with the second aldehyde, R2CHO, proceed with high fidelity through transition state TS-1 (Scheme 2). The first condition is established for allene hydroborations with the Soderquist borane, while the second is generally met with (Z)-γ-substituted allylboranes containing the 10-TMS-9-BBD auxiliary.2h,i,5
Scheme 1.

First generation strategy for synthesis of ent-6
Scheme 2.

Competitive transition states leading to ent-6 and 7
In the event, the double allylboration sequence performed with 3, 2R, and two aldehydes proceeded in good yield and enantioselectivity, but ca. 4 : 1 mixtures of ent-6 and 7 were obtained (Scheme 2).6 These results indicated that second allylboration reaction (with 5) proceeded with only a slight preference for the expected1c TS-1 compared to TS-2. While the reasons for the poor stereoselectivity of this transformation are not certain, we speculated that non-bonded interactions between the (1,3,2)-dioxaborinane unit and the R1 group might destabilize TS-1 compared to TS-2. This prompted us to develop a new reagent as an alternate to 5 with a less sterically demanding set of substituents on the secondary allylic boron atom.
Batey reported allyl- and crotylation of aldehydes using potassium allyl- and crotyltrifluoroborate reagents and suggested that an allylboron difluoride intermediate is the reactive species.7 Consequently, we imagined that allylic trifluoroborate 10 should be a viable precursor to allylboron difluoride 11 (Scheme 3). Moreover, we anticipated that 11 would undergo allylboration reactions of aldehydes with much higher stereoselectivity than with 5 owing to the smaller size of the difluoroborane unit of 11 compared to the (1,3,2)-dioxaborinane unit in 5 (e.g., compare TS-3 with TS-1). Thus, we turned to the synthesis of 10 via the kinetically controlled hydroboration of allene 8 with borane 2S (or 2R in the enantiomeric series).
Scheme 3.

Second generation strategy for synthesis of 6
Allene 8 was synthesized as described in the Supporting Information. The tetrabutylammonium counterion was used to increase the solubility of 8, 9, and 10 in the non-polar solvents used in these experiments.
The hydroboration experiments commenced by treating a 0 °C solution 8 (1.3 equiv) in CH2Cl2 with 1 equiv of 2S that was generated in situ by treatment of the borohydride 1S with TMSCl (Table 1). After a 1 h reaction time, the solution was cooled to −78 °C and then benzaldehyde (0.7 equiv) was added. The reaction was worked up oxidatively to give 1,2-diol 12 as a 5.7: 1 mixture of syn and anti diastereomers, along with the unexpected (E)-1,5-syn-diol 13 (Table 1, entry 1). The mixture of 1,2-diol diastereomers 12 provides an indirect assessment of the isomeric purity of the initial hydroboration product, 9, while the formation of 13 suggested that allylboron difluoride 11 was formed during the reaction. A significant improvement of the dr (16:1) for 12 was realized by performing the hydroboration at −10 °C for 1 h (entry 2), suggesting that the rate of boratropic isomerization of 9 can be controlled by keeping the reaction temperature below −10 °C. However, products 12 and 13 were still formed in a ca. 3:1 ratio. Addition of DIBAL to the reaction mixture to reduce any residual benzaldehyde prior to the oxidative workup did not eliminate the formation of 13 (entry 3). This experiment indicates that 13 must be formed during the reaction, and not during workup. The best dr for 12 (> 20:1) from experiments performed in CH2Cl2 was obtained when the hydroboration reaction was run at −30 °C (entries 5, 6). Evidently, under these conditions, the rate of the [1,3]-boratropic isomerization of 9 to the corresponding (E)-isomer is slow. We subsequently discovered that the competitive abstraction of fluoride ion from 10 (that generates 11 en route to 13) was strongly influenced by the reaction solvent (entries 7, 8, and 9). Under optimal conditions (entry 9), the hydroboration/allylboration sequence performed in a mixture of toluene and CH2Cl2 (15:1) led to the chemo-, enantio- and diastereoselective formation of allylic trifluoroborate 10, as evidenced by the isolation of syn-1,2-diol 12 in 87% yield, with 97% ee, and > 20:1 dr after oxidative workup.
Table 1.
Optimization of the hydroboration/allylboration
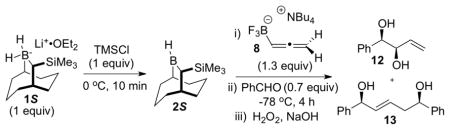 | ||||
|---|---|---|---|---|
| entry | solvent | hydroboration temp/time | % yield of 12a (syn/anti)b | ratio 12:13c |
| 1 | CH2Cl2 | 0 °C/1 h | 39 (5.7:1) | 3.0:1 |
| 2 | CH2Cl2 | −10 °C/1 h | 38 (16:1) | 2.9:1 |
| 3d | CH2Cl2 | −10 °C/1 h | 39 (12:1) | 3.3:1 |
| 4 | CH2Cl2 | −10 °C/3 h | 41 (5.1:1) | 3.2:1 |
| 5 | CH2Cl2 | −30 °C/1 h | 52 (> 20:1) | 3.7:1 |
| 6 | CH2Cl2 | −30 °C/3 h | 52 (> 20:1) | 4.3:1 |
| 7 | Et2O/CH2Cl2e | −30 °C/1 h | 50 (> 20:1) | 7.1:1 |
| 8 | toluene/THFf | −30 °C/1 h | 51 (> 20:1) | > 30:1 |
| 9 | toluene/CH2Cl2g | −30 °C/1 h | 87h (> 20:1) | > 30:1 |
Isolated yields.
Determined by 1H NMR analysis.
Determined after isolation of 12 and 13.
Dibal-H (2 equiv) added before the oxidation step.
Et2O/CH2Cl2 (2:1).
Toluene/THF (2:1).
Toluene/CH2Cl2 (15:1).
12 obtained in 97% ee, determined by Mosher ester analysis.
With confidence that intermediate 10 could be generated with high efficiency and excellent stereochemical control, we turned the reactions of this species with a second aldehyde to give (E)-1,5-syn-diols 6 (Table 2). For these purposes, the reactive allylic boron difluoride 11 was generated in situ by treatment of the solution of 10 at −78 °C with BF3·OEt2 in the presence of a slight excess of hydrocinnamaldehyde.8
Table 2.
Optimization of the double allylboration leading to 6a
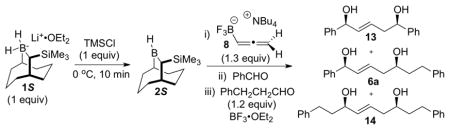 | |||||
|---|---|---|---|---|---|
| entry | PhCHO (equiv) | BF3·OEt2 (equiv) | % yield 6ab | ratio E/Zc | ratio 13/6a/14c |
| 1 | 0.75 | 1.5 | 60 | > 20:1 | 0:89:11 |
| 2 | 0.85 | 1.5 | 73 | > 20:1 | 0:100d:0 |
| 3 | 0.95 | 1.5 | 51 | > 20:1 | 8:92:0 |
| 4e | 0.85 | 1.5 | 30 | 6:1 | 0:100:0 |
| 5f | 0.85 | - | 77 | 6:1 | 0:100:0 |
Reaction conditions: solvent: toluene/CH2Cl2; hydroboration: −30 °C, 1 h; first allylboration: −78 °C, 4 h; second allylboration: −78 °C, 4 h; workup: pH 7 buffer (KH2PO4/NaOH).
Isolated yields.
Determined by 1H NMR analysis.
6a obtained with > 20:1 dr and 97% ee, determined by Mosher ester analysis.
Second allylboration: 0 °C, 2h.
Second allylboration: −78 to 20 °C, 12 h.
The amount of the aldehyde used in the first allylboration leading to 10 proved to be critical for the selective formation of (E)-1,5-syn-diol 6a (entries 1,2,3). Use of 0.75 equiv of benzaldehyde in the first allylation resulted in the formation of the 1,5-diol 14, which indicated that the allylborane 9 was not fully consumed during the first step (entry 1). On the other hand, 1,5-diol 13 was produced when a larger amount of benzaldehyde (0.95 equiv) was used in the first allylation reaction (entry 3). However, use of 0.85 equiv of benzaldehyde in the first allylation, followed by addition of 1.2 equiv of the second aldehyde and 1.5 equiv of BF3·OEt2 led to the isolation of (E)-1,5-syn-diol 6a in 73% yield, with excellent enantioselectivity (97% ee), diastereoselectivity (dr > 20:1), and E/Z ratio (> 20:1) (entry 2). When the second allylation step was performed at higher temperatures (entries 4, 5), product 6a was still obtained even in the absence of BF3·OEt2, but with a significant decrease of the E/Z ratio. The high reactivity of allylboron difluoride 11, allowing the second allylboration step to be run at −78 °C, was essential to achieve the selective E formation of 1,5-syn-diol 6a.
Additional examples of this new double allylboration sequence are provided in Scheme 4. The optimal reaction conditions defined by entry 2 of Table 2 proved to be applicable to a variety of aldehydes (aromatic, aliphatic, α, β-unsaturated) and compatible with -OBn and -OTBS protecting groups. The (E)-1,5-syn-diols 6 were obtained in 72–98% yields, > 95% ee, dr > 20:1, and E/Z > 20:1 in all cases. To the best of our knowledge, 10 is the first chiral α-substituted allyltrifluoroborate reagent to exhibit such high E/Z olefin selectivity in reactions with aldehydes.9 Both enantiomers of 1,5-diol 6 can be accessed by using either enantiomer of the borane 2S or 2R, as exemplified by the syntheses of 6d and ent-6d.
Scheme 4.

Stereoselective synthesis of (E)-1,5-syn-diol 6 a
The stimulation to develop this new procedure for the synthesis of (E)-1,5-syn-diols was provided by the structure of tetrafibricin 15, and especially the C(23)-C(40) fragment 16 (Figure 1). Tetrafibricin is a structurally unique fibrinogen receptor inhibitor isolated in 1993 from Streptomyces neyagawaensis.10 Tetrafibricin displays potent anti-aggregation properties against human platelets by blocking the glycoprotein (GP)IIb/IIIa receptor on the platelet surface, which is important for blood clotting.11 Unlike other fibrinogen receptor inhibitors (like snake venoms) 15 is non-peptidic. These features highlight 1 as a potential probe molecule for studying stroke and heart attack. The stereostructure of 15 was assigned by Kishi based on NMR database technology and NMR measurements in chiral solvents.3b Syntheses of fragments of tetrafibricin have been reported by Cossy,12 Curran,13 Friestad,14 and our group.15
Figure 1.

Tetrafibricin (15) and retrosynthetic analysis of 16
Our retrosynthetic analysis of the C(23)-C(40) fragment (16) of tetrafibricin is outlined in Figure 1. We were attracted to the possibility that 16 could be obtained in a highly convergent manner from aldehyde precursors 17 and 18, by application of the new reagent ent-9 (deriving from 2R). Indeed, by using the optimized double allylboration procedure described above, the 29S,33S-diastereomer 19, a synthetic precursor to 16 with stereochemistry identical to that proposed for tetrafibricin, was generated in 83% yield with > 20:1 diastereoselectivity and > 20:1 E/Z selectivity by the one-pot convergent coupling of 17 and 18 using ent-9 (Scheme 5). Moreover, by using the enantiomeric reagent, 9, deriving from 2S, the 29R,33R-diastereomer 20 was obtained in 78% yield, also with exceptional diastereoselectivity (dr > 20:1) and complete control of the E-olefin (> 20:1). The absolute stereochemistry of the two new hydroxyl groups in 19 and 20 was assigned by using the Mosher method, as summarized in the SI.
Scheme 5.

Syntheses of tetrafibricin fragment 19 and 20
In summary, we have developed an efficient and highly stereoselective double allylboration reaction leading to syn-1,5-diols 6 via a simple one-pot process. This method was successfully applied to the synthesis of the C(23)-C(40) tetrafibricin fragment 19 and its diastereoisomer 20, which has inverted stereochemistry at C(29) and C(33). The ability to synthesize either 19 or 20 with excellent stereochemical control simply by changing the absolute configuration of the Soderquist borane, 2, augurs well for application of this methodology for highly stereocontrolled, late stage fragment assembly reactions in the synthesis of natural products.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM038436) for support of this research, and the German Academic Exchange Service (DAAD) for a postdoctoral fellowship to A.S.
Footnotes
Supporting Information Available Experimental procedures and tabulated spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Brown HC, Narla G. J Org Chem. 1995;60:4686. [Google Scholar]; (b) Barrett AGM, Braddock CD, de Koning PD, White AJP, Williams DJ. J Org Chem. 2000;65:375. doi: 10.1021/jo991205x. [DOI] [PubMed] [Google Scholar]; (c) Flamme EM, Roush WR. J Am Chem Soc. 2002;124:13644. doi: 10.1021/ja028055j. [DOI] [PubMed] [Google Scholar]; (d) Morgan JB, Morken JP. Org Lett. 2003;5:2573. doi: 10.1021/ol034936z. [DOI] [PubMed] [Google Scholar]; (e) Pelz NF, Woodward AR, Burks HE, Sieber JD, Morken JP. J Am Chem Soc. 2004;126:16328. doi: 10.1021/ja044167u. [DOI] [PubMed] [Google Scholar]; (f) Woodward AR, Burks HE, Chan LM, Morken JP. Org Lett. 2005;7:5505. doi: 10.1021/ol052312i. [DOI] [PubMed] [Google Scholar]; (g) Sieber JD, Morken JP. J Am Chem Soc. 2006;128:74. doi: 10.1021/ja057020r. [DOI] [PubMed] [Google Scholar]; (h) Gonzalez AZ, Roman JG, Alicea E, Canales E, Soderquist JA. J Am Chem Soc. 2009;131:1269. doi: 10.1021/ja808360z. [DOI] [PubMed] [Google Scholar]; (i) Kister J, DeBaillie AC, Lira R, Roush WR. J Am Chem Soc. 2009;131:14174. doi: 10.1021/ja905494c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Chen M, Handa M, Roush W. J Am Chem Soc. 2009;131:14602. doi: 10.1021/ja904599h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Flamme EM, Roush WR. Org Lett. 2005;7:1411. doi: 10.1021/ol050250q. [DOI] [PubMed] [Google Scholar]; (b) Owen RM, Roush WR. Org Lett. 2005;7:3941. doi: 10.1021/ol0514303. [DOI] [PubMed] [Google Scholar]; (c) Hicks JD, Flamme EM, Roush WR. Org Lett. 2005;7:5509. doi: 10.1021/ol052322j. [DOI] [PubMed] [Google Scholar]; (d) Lira R, Roush WR. Org Lett. 2007;9:4315. doi: 10.1021/ol7018746. [DOI] [PubMed] [Google Scholar]
- 3.Examples of natural products containing a (E)-1,5-syn-diols motif. Amphidinol 3: Murata M, Matsuoka S, Matsumori N, Paul GK, Tachibana K. J Am Chem Soc. 1999;121:870.Tetrafibricin: Kobayashi Y, Czechtizky W, Kishi Y. Org Lett. 2003;4:93. doi: 10.1021/ol0272895.Marinomycins A-C: Kwon HC, Kauffman CA, Jensen PR, Fenical W. J Am Chem Soc. 2006;128:1622. doi: 10.1021/ja0558948.Aigialomycin F: Isaka M, Yangchum A, Intamas S, Kocharin K, Gareth Jones EB, Kongsaeree P, Prabpai S. Tetrahedron. 2009;65:4396.Marinisporolides: Kwon HC, Kauffman CA, Jensen PR, Fenical W. J Org Chem. 2009;74:675. doi: 10.1021/jo801944d.
- 4.In our previous paper (ref. 1i) we described initial efforts on the synthesis of (E)-1,5-syn diols 6 via a reagent generated by hydroboration of allenyltributylstannane with the Soderquist borane.
- 5.(a) Burgos CH, Canales E, Matos K, Soderquist JA. J Am Chem Soc. 2005;127:8044. doi: 10.1021/ja043612i. [DOI] [PubMed] [Google Scholar]; (b) Munoz-Hernandez L, Soderquist JA. Org Lett. 2009;11:2571. doi: 10.1021/ol900865y. [DOI] [PubMed] [Google Scholar]; (c) Ess DH, Kister J, Chen M, Roush WR. Org Lett. 2009;11:5538. doi: 10.1021/ol902364d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.See Supporting Information.
- 7.Batey RA, Thadani AN, Smil DV. Tetrahedron Lett. 1999;40:4289. [Google Scholar]
- 8.Batey RA, Thadani AN, Smil DV, Lough AJ. Synthesis. 2000;7:990. [Google Scholar]
- 9.Hall reported an E/Z ratio of 2.6:1 with potassium α-allylic trifluoroboronate: Carosi L, Hall DG. Angew Chem Int Ed. 2007;46:5913. doi: 10.1002/anie.200700975.
- 10.Kamiyama T, Umino T, Fujisaki N, Fujimori K, Satoh T, Yamashita Y, Ohshima S, Watanabe J, Yokose K. J Antibiot. 1993;46:1039. doi: 10.7164/antibiotics.46.1039. [DOI] [PubMed] [Google Scholar]
- 11.(a) Satoh T, Yamashita Y, Kamiyama T, Watanabe J, Steiner B, Hadvary P, Arisawa M. Thromb Res. 1993;72:389. doi: 10.1016/0049-3848(93)90239-k. [DOI] [PubMed] [Google Scholar]; (b) Satoh T, Yamashita Y, Kamiyama T, Arisawa M. Thromb Res. 1993;72:401. doi: 10.1016/0049-3848(93)90240-o. [DOI] [PubMed] [Google Scholar]; (c) Satoh T, Kouns WC, Yamashita Y, Maiyama T, Steiner B. Biochem J. 1994;301:785. doi: 10.1042/bj3010785. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Satoh T, Kouns WC, Yamashita Y, Kamiyama T, Steiner B. Biochem Biophys Res Commun. 1994;204:325. doi: 10.1006/bbrc.1994.2463. [DOI] [PubMed] [Google Scholar]
- 12.BouzBouz S, Cossy J. Org Lett. 2004;6:3469. doi: 10.1021/ol048862i. [DOI] [PubMed] [Google Scholar]
- 13.(a) Gudipati V, Bajpai R, Curran DP. Collect Czech Chem Commun. 2009;74:774. doi: 10.1135/cccc2008195. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang K, Gudipati V, Curran DP. Synlett. 2010;4:667. doi: 10.1055/s-0029-1219376. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gudipati V, Curran DP. Tetrahedron Lett. 2011;52 doi: 10.1016/j.tetlet.2011.01.086. in press, accepted manuscript. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friestad GK, Sreenilayam G. Org Lett. 2010;12:5016. doi: 10.1021/ol1021417. [DOI] [PubMed] [Google Scholar]
- 15.Lira R, Roush WR. Org Lett. 2007;9:533. doi: 10.1021/ol0629869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.