

Abstract
Tyrosine hydroxylase is the rate-limiting enzyme of catecholamine biosynthesis; it uses tetrahydrobiopterin and molecular oxygen to convert tyrosine to DOPA. Its amino terminal 150 amino acids comprise a domain whose structure is involved in regulating the enzyme’s activity. Modes of regulation include phosphorylation by multiple kinases at 4 different serine residues, and dephosphorylation by 2 phosphatases. The enzyme is inhibited in feedback fashion by the catecholamine neurotransmitters. Dopamine binds to TyrH competitively with tetrahydrobiopterin, and interacts with the R domain. TyrH activity is modulated by protein-protein interactions with enzymes in the same pathway or the tetrahydrobiopterin pathway, structural proteins considered to be chaperones that mediate the neuron’s oxidative state, and the protein that transfers dopamine into secretory vesicles. TyrH is modified in the presence of NO, resulting in nitration of tyrosine residues and the glutathionylation of cysteine residues.
Keywords: Tyrosine hydroxylase, dopamine biosynthesis, protein kinases, protein nitration, protein glutathionylation, protein-protein interactions, 14-3-3 protein, α-synuclein
Tyrosine hydroxylase (TyrH) is the rate-limiting enzyme of catecholamine synthesis. It catalyzes the hydroxylation of tyrosine to L-DOPA (1). The catecholamines dopamine, epinephrine and norepinephrine are the products of the pathway, important as hormones and neurotransmitters in both the central and peripheral nervous systems. In the latter, they are synthesized in the adrenal medulla (1, 2). The biosynthetic pathway is illustrated in Figure 1. These catechol monoamines play roles in many brain functions, such as attention (3), memory (4), cognition (5), and emotion (6, 7). As the hormone of the fight-or-flight response, epinephrine produced in the adrenal gland affects many tissues throughout the body (8). Therefore deficits and surfeits in the levels of the catecholamines have many repercussions, perhaps including high blood pressure, bipolar disorder, addiction, and dystonias (9–11). Because of this, the activity of TyrH as the slowest enzyme in the pathway is of great interest in many fields of biomedical research.
Figure 1.

The biosynthetic pathway for the catecholamine neurotransmitters. Phenylalanine hydroxylase converts phenylalanine to tyrosine, tyrosine hydroxylase hydroxylates tyrosine to L-DOPA. DOPA is converted to dopamine by aromatic amino acid decarboxylase. Dopamine-β-hydroxylase hydroxylates dopamine to norepinephrine, which is methylated to epinephrine by phenylethanolamine N-methyltransferase. Tyrosine hydroxylase is the rate-limiting enzyme of the pathway.
Given the importance of the activity of TyrH, the complexity of its regulation is not surprising. Control of its expression by transcriptional mechanisms is a very active field of research, as is the relatively new field of its degradation in the proteosome after ubiquitination (12). This review, however, will focus on mechanisms of regulation that occur after the synthesis of TyrH and before its destruction. It will discuss the short-term reversible biochemical processes and conformational changes that TyrH undergoes while the enzyme is active and being modulated in response to the metabolic state of the neuron or the chromaffin cell.
When need for neurotransmitter increases at a catecholaminergic synapse, TyrH is activated to make more DOPA, which after decarboxylation to dopamine is transferred into the synaptic vesicle by the vesicular monoamine transporter (VMAT). Catecholamine synthesis then continues in vesicles via the actions of dopamine-β-hydroxylase and phenylethanolamine-N-methyltransferase. Influx of calcium causes the emptying of the vesicles into the synaptic cleft, and the nervous signal is passed on. Some catecholamine is taken up by the presynaptic neuron but more must be synthesized for the next transmission. TyrH activity must be sustained until the need is lessened, and its activity must be turned off when the need for neurotransmitters has passed. The post-translational mechanisms that accomplish all this include phosphorylation by kinases and dephosphorylation by phosphatases, feedback inhibition, oxidation by nitrites, and inclusion in protein complexes. Some of these complexes alter the activity of TyrH and some of them simply place the enzyme near other related proteins. The review will attempt to cover these steps using the following organization:
Background: Description of the overall structure of the aromatic amino acid hydroxylases
-
Effects that result from changes in the regulatory domain
-
2.1
Phosphorylation and feedback inhibition by catecholamines
-
2.2
Binding by catecholamines
-
2.3
R domain differences between rat and human TyrH
-
2.1
-
Protein complexes
-
3.1
14-3-3 proteins
-
3.2
α-synuclein
-
3.3
PP2A, AADC, GTPCH, VMAT, DJ-1
-
3.1
Nitration/thiolation of TyrH
Note: this review will focus heavily on experiments done in vitro with purified proteins whenever possible. For an excellent and comprehensive review of TyrH regulation by phosphorylation that encompasses in vivo and in situ studies the reader is referred to the 2004 article by Dunkley et al. (13).
1. Background
TyrH is a member of a family of enzymes that also contains the aromatic amino acid hydroxylases (AAAHs) phenylalanine hydroxylase (PheH) and tryptophan hydroxylase (TrpH). PheH and TrpH will not be discussed in depth in this review. Nonetheless it is worth describing the entire family, because some of their structural similarities will be included in the discussion of TyrH regulation. All three enzymes perform hydroxylation of the aromatic ring of an amino acid. They all use diatomic oxygen and reduced biopterin in a reaction with a bound iron atom. The iron atom is held in place in the active site cleft by two histidine residues and a glutamate residue, and it must be in the ferrous state to carry out catalysis. Each of the three enzymes has a very similar active site. Similarities include the iron and its ligands, plus homologous residues for binding tetrahydrobiopterin and the aromatic amino acid (14, 15). The active sites of TyrH and PheH from crystallographic data, overlaid, are shown in Figure 2.
Figure 2.
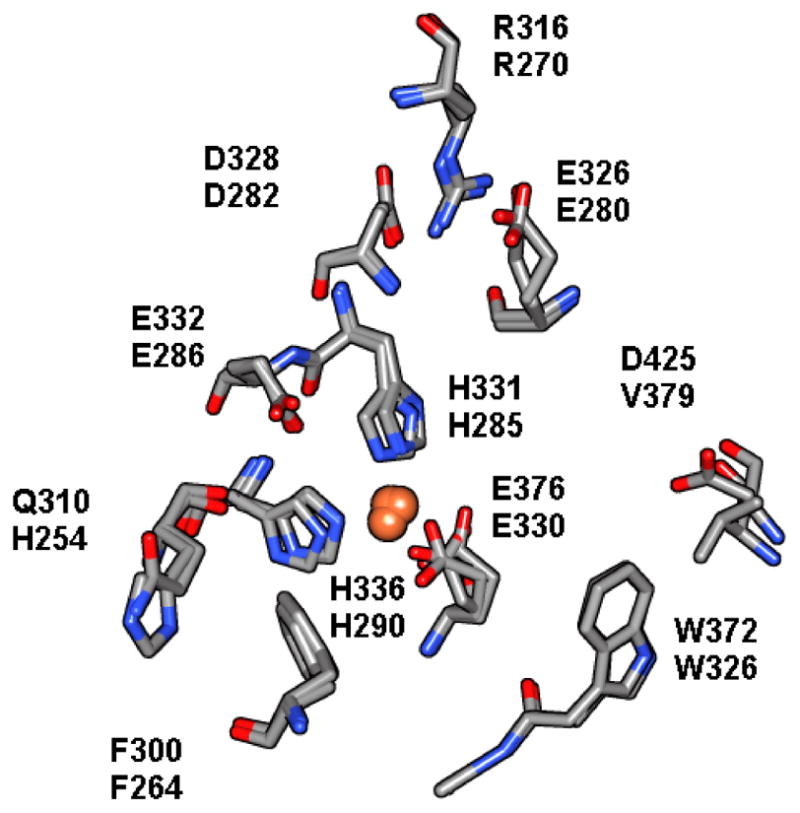
The active sites of PheH (1DMW) and TyrH (1TOH) are overlaid on each other and the residues that play identifiable roles in catalysis are highlighted. The top # in each pair of residues is the residue in TyrH and the bottom # is the residue in PheH. In particular, histidines 331 and 336 (285 and 290 in PheH) and glutamate 376 (330) are the ligands to the iron atom. Residues on the left in this view, gln310/his254, phe300/264, and glu332/286 form the binding site for tetrahydrobiopterin, and on the right, arg316/270, asp328/282/ and asp425/val379 form the binding site for the aromatic amino acid.
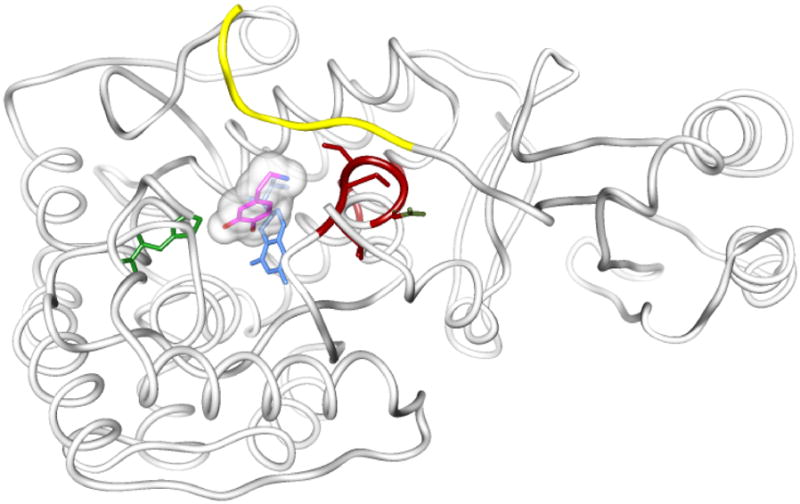
In addition to these similarities in the active site, the family shares other features of three-dimensional structure. Each enzyme has a multi-domain structure, with an amino-terminal regulatory domain (R) of varying size from 100–150 amino acid residues, followed by a catalytic domain (C) of about 330 residues, and a coiled-coil domain at the carboxyl terminus of about 20 amino acids. All three enzymes form tetramers. The C domains display 50% identity in primary structure, while the R domains are as little as 15% similar (16); rat PheH and TyrH are ~75% similar in their C domains. A scheme illustrating the domains of the AAAHs is shown in Figure 3. Also shown are the domains of TyrH and PheH via coloration of their crystal structures (17, 18). An alignment of the amino acid sequences of TyrH and PheH by CLUSTALW is shown in Figure 4.
Figure 3.

The domain structure of the aromatic amino acid hydroxylases. At top: the AAAHs consist of three domains: regulatory, catalytic, and tetramerization. The amino terminal regulatory domains differ in length (~150, TyrH; ~115, PheH; and ~105, TrpH) and contain regulatory serine residues at different positions. The portions of the R domains that are less similar among the three enzymes and that contain the regulatory serine residues are in yellow. The portions of the R domains that display some homology are in lilac. The catalytic cores are similar in length and sequence; they appear in light blue. The leucine-zipper-style tetramerization domains are at the very carboxyl termini, and are not emphasized in this figure.
Left: The crystal structure of PheH (1PHZ), color coded to highlight the domain structure. The catalytic core is shown in blue, and the iron with its ligands are displayed as a sphere and stick models. The regulatory domain is above and offset to the left of the catalytic domain, and is bicolored. The R domain region that is somewhat homologous to the R domains of TyrH and TrpH is shown in lilac. Amino terminal to it, one long strand shown in yellow traverses the space to the active site and covers its opening. This region contains residues 20 to 34 of PheH. This crystal structure does not show the position of the amino terminal 19 amino acids, presumably due to flexibility.
Right: The crystal structure of TyrH (1TOH). Because the R domain of TyrH has not been crystallized, presumably due to its flexibility, only the catalytic domain and the tetramerization domain are seen. The active site iron and its ligands are displayed as a sphere and stick models. This protein is missing its 155 amino terminal amino acids, therefore, its R domain is absent. The two proteins are viewed from approximately the same angle to facilitate comparison.
Figure 4.

Clustal W alignment of the amino acid sequences of the rat aromatic amino acid hydroxylases. An asterisk below the three sequences denotes identity, and a period indicates similarity. The C domain begins near the end of the third set of rows, that is, near position 105 for TrpH, 164 for TyrH, and 117 for PheH, where the sequence VPWFP appears in bold type for all three enzymes.
Despite little similarity in R domains, all three hydroxylases are phosphorylated at serine residues that lie in the R domains. They are all phosphorylated by cAMP-dependent protein kinase (PKA) (19). When TyrH is phosphorylated by PKA, it is less susceptible to feedback inhibition by catecholamines (20), and when PheH is phosphorylated by PKA, lower levels of phenylalanine are required for substrate activation (21). While there is a fine crystal structure of the C domain of TyrH (17), the R domain of TyrH has never been crystallized, presumably due to its flexibility. Attempts to crystallize it with ligands have been unsuccessful. Therefore some study of the R domain of TyrH relies on the crystals of PheH (18), because the structure of PheH has been solved, though it is missing its amino terminal 18 residues. These are the structures shown in Figure 3. PheH is activated by phosphorylation at ser16 (missing from the crystal structure). Although the serine of interest is missing the structure is still very informative. The R domain of PheH, a lobe consisting of 2 α-helices and 4 strands of β-sheet, lies mainly above and to the side of the opening to the active site. However, a short portion of the R domain (from residues 19 to 33) spans the distance from that somewhat removed position to the opening of the active site, lying across it and possibly restricting access to the active site. Although no crystal structures prove it, it is logical to hypothesize that phosphorylation moves the R domain out of the opening of the active site, and dephosphorylation by a phosphatase returns it to its obstructive position. A simple graphical representation of this model, applied to TyrH, appears in Figure 5.
Figure 5.
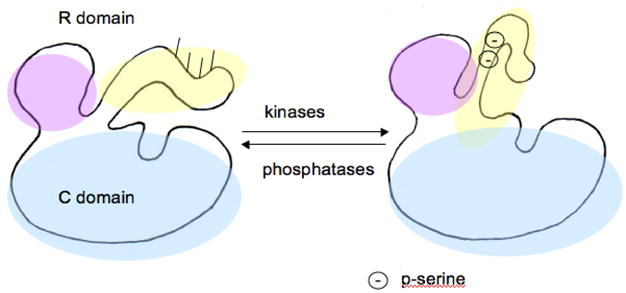
A model for the mechanism of activation by phosphorylation of serine residues in the R domain of TyrH. The catalytic core is indicated in light blue. The R domain is in lavender with the mobile portion in yellow. It contains serines that become phosphorylated by kinases, at positions 8, 19, 31, and 40. Upon phosphorylation of the R domain it moves out of its position obstructing access to the active site. Phosphatases then return the R domain to its inactivating position. Only 2 serine phosphates are shown for clarity.
A further difference in R domains among the family of enzymes is that humans have four isozymes of TyrH, which differ in their R domains. Human TyrH isozyme 1 (hTH1) is very similar (93% similar and 89% identical, and identical in length) to rTyrH, the enzyme which has received the most study. The human isozymes 2, 3, and 4 differ in their R domains, as pre-mRNA splicing results in additional amino acids following met30. Since the additional amino acids are included in the region of the R domain where phosphorylation occurs, there is an added level of complexity in structure for human TyrH.
The first section of this review will concern the structural changes in the R domain of TyrH. TyrH is phosphorylated at serine residues in the R domain. Because TyrH regulation by feedback inhibition by the catecholamines is dependent on phosphorylation of ser40, binding of dopamine et al. will be covered in the same section. The first section will also describe R domain differences between rat TyrH and human TyrH. The review will next recount reports of TyrH and protein binding partners and complexes. A family of scaffolding proteins, the 14-3-3 proteins, found in myriad organisms and cells, binds to many proteins that have been phosphorylated and activate them or stabilize them (22). TyrH and TrpH were among the first enzymes discovered to be activated by the 14-3-3 proteins (23). Experiments using immunochemical techniques have found that TyrH may be in complex with other proteins as well. Finally, a new field of TyrH regulation has emerged in the last decade which has not yet been compiled into a review, that of nitration and glutathionylation (24). Therefore the last section of this review will summarize those findings.
2. Effects that result from changes in the regulatory domain
2.1 Phosphorylation and feedback inhibition by catecholamines
TyrH is different from the other family members in that it has four serine residues in its R domain that become phosphorylated by protein kinases in vivo and in vitro rather than only one. A short summary follows: rat TyrH (rTyrH) is phosphorylated in vivo at ser8, -19, -31, and -40, in response to various nervous stimuli or signaling molecules (position 8 contains a threonine in some species, including human TyrH). The kinases include PKA, PKC, CaMKII, MAPKAP-K2, ERK1, ERK2, MSK1, and PRAK, among others (Figure 6). The in vivo results of those phosphorylation reactions have been well documented (13, 14) and include activation of TyrH. In vivo and in crude lysate samples, rTyrH activity is activated perhaps 20-fold by phosphorylation by PKA and 1.5 to 3-fold by phosphorylation via CaMKII or ERK, respectively (13, 14). In contrast, in vitro studies to determine the mechanism of activation for each serine phosphorylation have recorded less activation: 1) No mechanism of activation has been identified for phosphorylation at position 8 (13, 19). 2) PRAK phosphorylates ser19 alone, and CaMKII phosphorylates ser19 and ser40 with a strong preference for ser19 (13, 14). No effect on Vmax values, KM values, or Kd values for the neurotransmitters has been recorded for ser19 phosphorylation. However, rTyrHpser19pser40 (TyrH phosphorylated at both ser19 and ser40) is activated 1.5 to 2-fold if the enzyme is assayed in the presence of the 14-3-3 proteins (discussed in section 3.1) (13, 14). 3) Ser31 is phosphorylated by ERK1 and 2 and Cdk5, causing a two-fold lowered KM value for tetrahydrobiopterin (13, 14). 4) Ser40 is phosphorylated mainly by PKA with the result of a 2-fold decrease in KM value for tetrahydrobiopterin, a slight increase in Vmax value, and a 300-fold decrease in binding affinity for the catecholamine neurotransmitters, as judged by Kd values obtained from on rates vs off rates of the catecholamines (13, 14). Ser40 is also phosphorylated by MAPKAPK-2, which also labels some ser19 but to a lesser extent (13, 14). Reversal of activation of TyrH occurs when the phosphatase PP2A (and PP2C to a lesser extent) removes the phosphates from ser19, -31, and -40. (25, 26). The major questions concerning TyrH and its multiple phosphorylatable serine residues are considered in turn in the following paragraphs.
Figure 6.
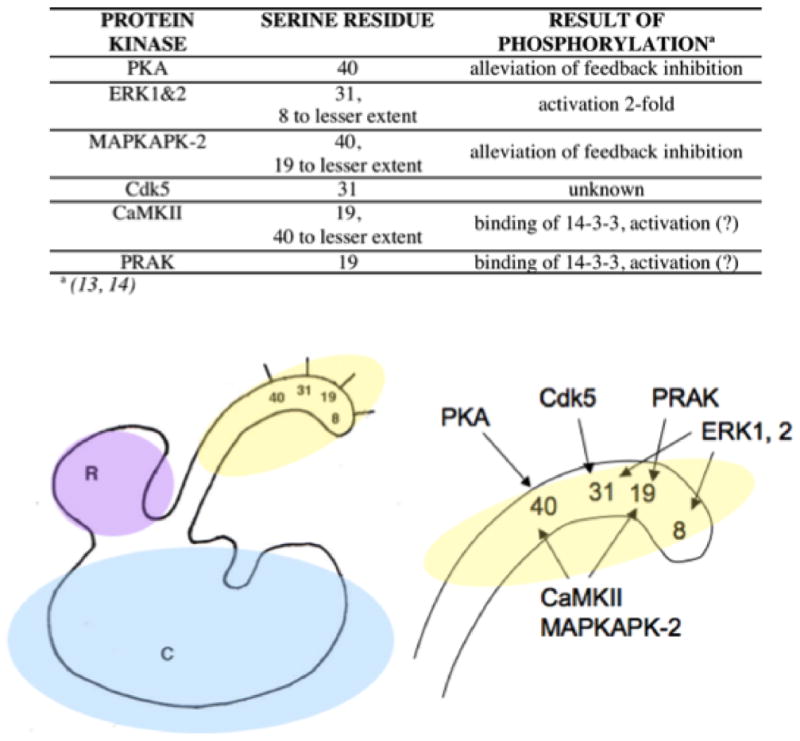
Simplified map of the reactivity of some protein kinases with the serine residues of the R domain of TyrH. Ser40 is modified by PKA, CaMKII, and MAPKAPK-2. Ser31 is modified by ERK1 & 2 and Cdk5. PRAK labels ser19, as do CaMKII, and MAPKAPK-2. Ser8 is modified by ERK1 but it is not certain that the reaction has an effect on TyrH activity. Extensive coverage of these phosphorylation events have been covered in previous reviews (14, 25)
Does multiple phosphorylation result in additional activation or release from feedback inhibition?
Why TyrH is phosphorylated in multiple positions is a question that has engaged a number of laboratories. One assumes that a protein would not acquire 3 to 4 different serine residues as modification sites for different kinases, responding to different stimuli, using ATP in every phosphorylation event, without some advantageous molecular effect. Furthermore, since phosphorylation of ser40 by PKA results in at least 20-fold activation, it seems doubtful that 1.5-fold or 2-fold activation after phosphorylation at the other serines tells the whole story of their mechanism, unless it is to fine-tune the effects that are coarsely produced by phosphorylation of ser40 (27, 28).
A common approach when seeking the molecular mechanism for regulation of a protein by phosphorylation is to employ phosphomimetic variants, that is, variants with serine mutated to glutamate or aspartate. Since phosphorylated TyrH is difficult to obtain in fully and discretely (at one site only) labeled forms, glutamate and aspartate substitution variants have been used, including rTyrHser8glu, -ser19glu, -ser31glu, and -ser19glu/ser40glu (29). While some glutamate or aspartate-substituted enzyme variants do not mimic phosphorylation, rTyrHser40glu is a good mimic of rTyrHpser40 (30, 31), displaying resistance to inhibition by DA to the same extent as rTyrHpser40 (20), and rTyrHser31glu is a good mimic of rTyrHpser31 (29). The double variant rTyrHser19glu/ser40glu was studied because CaMKII and MAPKAPK-2 label both ser19 and -40, so perhaps both must be phosphorylated for full effect from those kinases (32, 33). However, rTyrHser19glu/ser40glu showed the same slight decrease in KM value for tetrahydrobiopterin and slight increase in Vmax compared to the values for wild-type rTyrH as expected for rTyrHpser40 (31) and did not display any additional alterations that would explain why phosphorylation of both serine residues is different than phosphorylating just one. None of the other ser-to-glu variants had altered Michaelis constants compared to wild-type TyrH, nor did any of them show decreased affinity for catecholamines (29), suggesting that phosphorylation at ser19 and ser31 have a different activation mechanism than phosphorylation at ser40.
Does multiple phosphorylation result in stabilization of the most activated phosphorylated form of the enzyme?
To determine whether phosphorylation stabilizes TyrH, the stability of the phosphomimetic variants has been tested. Rat TyrHser19glu and –ser31glu were more stable to denaturation at elevated temperature but the effect was not dramatic, a 1.6 to 1.7-fold slower inactivation rate. Rat TyrHser40glu was less stable, denaturing 1.6 times faster than wild-type (29). This suggests that phosphorylation at ser19 and ser31 may serve to stabilize TyrHpser40. Rat TyrHpser40 was less stable than unphosphorylated rTyrH when studied by circular dichroism (34), but IR conformational analysis found rTyrHpser40 to be more stable than unphosphorylated rTyrH (35), so there is some controversy about whether TyrHpser40 is more or less stable than unphosphorylated TyrH.
Does multiple phosphorylation result in alteration of substrate specificity for other kinases?
Since phosphorylation attaches a negative charge to a position that had been merely polar (i.e., phosphoserine vs. serine), another possibility for multiple/prior phosphorylation is that once-phosphorylated TyrH might be a better or worse substrate for other kinases. For example, phosphorylation at ser31 by ERK or at ser19 by CaMKII might alter the KM value for TyrH as a substrate for PKA. Relative Vmax/KM (V/K) values are indicators of relative substrate specificity (36), so if a phosphomimetic variant at position 19 or 31 has an altered V/K value as a substrate for PKA, that would indicate a change in substrate specificity for TyrHpser19 or –pser31. However, when the V/K values for the glutamate substitution variants rTyrHser8glu, -ser19glu, and -ser31glu, were measured, none was a better or worse substrate for PKA (37). For PKA the V/K values for wild type rTyrH, rTyrHser19glu and rTyrHser31glu were all around 120 min−1 μM−1. Therefore phosphorylation at ser19 or ser31 does not affect PKA’s activity at ser40. The results were similar for ERK2; in this experiment the possible impact of prior phosphorylation at ser19 and ser40 on substrate specificity of TyrH for ERK2 was considered, since ERK2 phosphorylates ser31. The V/K values for rTyrH, rTyrHser19glu, rTyrHser40glu, and the double variant rTyrHser19glu/ser40glu, respectively, were all between 0.5 and 0.6 min−1 μM−1. Apparently prior phosphorylation by PKA does not alter ERK2 reactivity with TyrH.
Does a first phosphorylation step by a single kinase result in faster reaction at a second serine?
The previous experiments showed that the kinetic parameters of PKA, CaMKII, MAPKAPK-2, and ERK were not altered by phosphorylation at a different serine by a different kinase. In contrast, using mass spectrometry to determine the rates of phosphorylation of wild-type rTyrH, rTyrHser40ala, and rTyrHpser40 by CaMKII, Bevilaqua et al. calculated that ser40 is more available for phosphorylation if ser19 is already phosphorylated; that is, the rate of ser40 phosphorylation by CaMKII is 2-to-3-fold higher if ser19 is already phosphorylated (28). Presumably the resultant rTyrHpser19pser40 is then released from feedback inhibition by catecholamines; rTyrHser19glu/ser40glu has the same lowered affinity for DOPA as does rTyrHser40glu (29). At the time of this writing, this result is the most convincing example for hierarchical phosphorylation of TyrH. However, using an ATP to phosphorylate ser19 in order to speed up phosphorylation at ser40 when another kinase exists that only phosphorylates ser40 (PKA) seems inefficient. One possibility is that ser19 phosphorylation occurs to proactively stabilize TyrHpser40. The answer to this question may lie in an added feature of ser19 phosphorylation, that is, the subsequent binding of the 14-3-3 proteins. This feature will be discussed in the section on protein complexes.
Does phosphorylation result in demonstrable physical changes?
Bevilaqua et al. (28) have demonstrated rTyrH R domain structural changes upon phosphorylation using gel filtration chromatography; they also demonstrated that phosphorylation of ser19 causes an alteration of structure similar to that caused by phosphorylation of ser40. The authors suggested a hinged-movement structure for the R domain of TyrH, similar to the one in Figure 5, in which the movement is greater when caused by pser40 than by pser19 because ser40 is closer to the pivot point (28).
Proteolysis has also been used to probe the structure of the R domain of TyrH and changes upon phosphorylation. It has long been known that limited proteolysis of the AAAHs separates the R and C domains (38, 39). Rat TyrH missing 157 and rat PheH missing 141 amino terminal residues retain full activity. More limited proteolysis of TyrH has provided information concerning changes upon phosphorylation. McCulloch (40) showed that rTyrH is cut at 4 positions, namely, arg33, arg37, arg38, and arg49, by trypsin. Rat TyrHpser40 is cut by trypsin faster than non-modified TyrH, meaning that the R domain undergoes a conformational change upon ser40 phosphorylation to a more open position more accessible by the protease. Proteolysis by the Staphylococcus V8 protease, which cuts at positions 27 and 50, was used to probe for alterations in structure due to introduction of negative charges at positions 19, 31, and 40 (29). The glutamate variants rTyrHser19glu, -ser31glu and –ser40glu were used as mimics of phosphorylated TyrH. Only rTyrHser31glu was digested at a different rate than wild-type enzyme (digest was slower), demonstrating that there is a conformational change upon the introduction of a negative charge at position 31. That series of experiments also showed that rTyrH and rTyrHser31glu were digested more quickly by the V8 protease if tyrosine was present. More information about changes associated with tyrosine binding is found below in the section on conformational changes to the catalytic domain.
Summary Section 2.1: Phosphorylation and feedback inhibition by catecholamines
TyrH is activated after phosphorylation of any of three serine residues in its regulatory domain. Ser40 is phosphorylated mainly by PKA, resulting in a decrease in affinity for catecholamines. Ser31 is phosphorylated by several kinases, resulting in a decrease in KM value for tetrahydrobiopterin. Ser19 is phosphorylated by enzymes that modify only ser19 or both ser19 and -40, and does not result in activation in the absence of other factors, which will be discussed later under protein interactions. Phosphorylation of ser19 by CaMKII accelerates phosphorylation of ser40 by the same kinase. Any other result of multisite phosphorylation has not yet been established, although stabilization and tighter binding to chaperone proteins are possibilities.
2.2 Binding by catecholamines
Dopamine, norepinephrine, and epinephrine are all feedback inhibitors of TyrH, and the biggest alteration of TyrH activity upon ser40 phosphorylation is the change in Kd value for catecholamines. DA affinity for TyrH is 300-fold decreased when the enzyme is phosphorylated (41). Questions concerning feedback inhibition by dopamine, epinephrine, and norepinephrine are considered in turn in the following paragraphs.
Does catecholamine binding result in demonstrable physical changes?
The limited proteolysis experiments described above also provided insights into alterations of structure that occur upon dopamine binding (40). McCulloch showed that when dopamine was bound to TyrH, cleavage was much slower than if the enzyme was catecholamine-free. This demonstrates that there is another conformational change in the R domain upon dopamine binding making the peptide less available to trypsin (40). As previously mentioned, proteolysis of TyrHser31glu by the S. aureus V8 protease was slower than proteolysis of wild-type enzyme, but in the presence of dopamine the digest was even slower, demonstrating that the conformational alteration caused by dopamine is separate from the conformational change caused by a negative charge at ser31 (29).
What are the molecular interactions between TyrH and the catecholamines?
All three catecholamines bind to the active site via the iron atom, but they only bind if the iron is oxidized; therefore binding of catecholamine inhibitors is not a simple equilibrium (30). One might have assumed that the catecholamines bind at the amino acid site, since they are similar in structure to phenylalanine or tyrosine. The proteolysis experiments clearly show that dopamine binds differently than tyrosine. When catecholamines are included in steady-state kinetic assays of TyrH they display competitive binding with respect to tetrahydrobiopterin (42). Several reports propose two binding sites for dopamine with the lower affinity site allowing competitive inhibition with tetrahydrobiopterin (43, 44). Crystal structures show the ligands dopamine, norepinephrine, and epinephrine bound to PheH (there is no crystal structure of TyrH with catecholamines bound) (45). In those structures the catechol moiety is bound in bidentate fashion to the active site iron. Figure 7 shows the overlaid structures of PheH with dopamine (45), PheH with thienylalanine and tetrahydrobiopterin (46), and PheH with its R domain intact (18). (There is no crystal structure of PheH with phenylalanine bound to use for comparison, but thienylalanine is a good analog.) It is clear that in the crystal-packed enzyme, the catecholamine site slightly overlaps the pterin site, not the amino acid site, and the amino end of the catecholamine is close to the R domain, easily close enough for a salt bridge. The amino acid substrate is bound to a separate site from these two sites; the analog is bound presumably where phe would bind in the active site pocket, and where tyr would bind in the active site of TyrH. This arrangement would explain how tyrosine and dopamine could both bind in the active site but have different effects on the proteolytic pattern of the R domain. It appears that the R domain has at least three conformations; it may be held tightly closed against the C domain when catecholamines are bound, have another form when tyrosine is bound, and a third when it is phosphorylated. A model representing the variability of structure in the R domain is drawn in Figure 8. The difficulty of crystallizing the R domain suggests that there are others, perhaps many others, when neither substrate nor inhibitor is bound.
Figure 7.

On left, PheH with tetrahydrobiopterin, dopamine, and thienylalanine bound. Three structures were overlaid to create these images: 1PHZ, which contains the R domain; 4PAH, which contains catecholamine; and 1KW0, which contains tetrahydrobiopterin, and thienylalanine, a phenylalanine analog. The pterin is in blue, the dopamine is in orange, and the thienylalanine is in green. Clearly the dopamine and biopterin sites overlap, and the aromatic amino acid site is separate.
At right are the same structures in a higher magnification, turned slightly to illustrate the proximity of the catecholamine to the R domain.
Figure 8.

Drawing of three different possible regulatory domain configurations. The regulatory domain is lavender with a mobile yellow loop, and the catalytic domain is light blue. Dopamine is represented by a blue circle. Left, unphosphorylated TyrH; the R domain is very flexible. Lower right, TyrH with dopamine bound, chelated to active site iron. The R domain is in a more rigid conformation, less accessible to proteases, and obstructing entrance of substrates. Upper right, TyrHpser40 with salt bridge between some acidic residue and phosphoserine40, exposing the entrance to the active site. TyrHpser40 has released the catecholamine which was bound prior to phosphorylation, the flexible loop is in a different configuration that makes arg37 and arg38 less accessible to trypsin but exposing arg33 and arg49.
Is catechol binding to the iron in the TyrH active site the only interaction between the two molecules?
Catecholamines are bound to the iron in the active site of PheH crystals, but there is reason to believe that the other end of the molecule also interacts with the R domain of TyrH. Deletion mutants and site-directed mutants have been studied in the attempt to pinpoint the R domain residue(s) that interact with dopamine. Nakashima et al. (47) have focused on the arginine residues at position 37 and 38. They studied site-directed mutants containing single or double mutations of these arginine residues to glycine or glutamate. When both were mutated TyrH activity was not decreased as much by dopamine, judging by activities of the TyrH variants preincubated with 1, 10, and 100 μM dopamine, but Ki values were not measured. This is consistent with the catechol end of dopamine binding to the iron, but the amino end not being able to form an association with the R domain when arg37 and arg38 are missing. Piper and Daubner (48) made deletion variants of rTyrH lacking the first 32 (TyrHΔ32), the first 68 (TyrHΔ68), the first 76, or the first 120 amino acids. The deletion variants were tested for inhibition by preincubation with stoichiometric amounts of dopamine; TyrHΔ32 was 90% inhibited by dopamine, but TyrHΔ68 and the other truncates were not inhibited (unpublished observations). Furthermore, when dopamine binding and release rates were investigated using the method of Ramsey and Fitzpatrick (41), dopamine was not released from TyrHΔ32 but was rapidly released from TyrHΔ68. That TyrHΔ32 interacts with dopamine but TyrHΔ68 does not suggests that some amino acid residue or some three-dimensional structure between positions 32 and 68 is important for dopamine binding. This is consistent with the Nakashima result that arg37 and arg38 are involved (47). However, it is not clear how arginine residues would link the dopamine amino group to the R domain, since all would presumably be positively charged at pH 7. A thorough study of the Kd values of an array of catechols to TyrH suggested that the amino group of dopamine is imperative for tight binding and the carboxyl group is not needed. Dopamine binds 1000-fold more tightly than DOPA, and dihydroxyphenylacetate binds 100-fold times less tightly than DOPA (30). Perhaps arg37 and arg38 are important for the tertiary structure of the R domain. Supporting this idea, the molecular mechanics and dynamics work of Alieva et al. (49) proposed that arg37 is necessary for an important β-turn in the R domain. A carboxylate residue could bind the catecholamine amino group, but alanine and glutamine substitution for the negatively-charged residues between position 32 and 68 (TyrHglu43, -asp44, -glu48, and glu-50) did not identify any carboxylate in the region important for dopamine binding. These TyrH variants were all inhibited by preincubation with stoichiometric amounts of dopamine but Kd or Ki values were not measured (Daubner, unpublished observations).
Recently TyrH has been studied using hydrogen-deuterium exchange followed by proteolysis and mass spectrometry (50). The technique identifies portions of proteins that readily exchange hydrogens from the peptide backbone with deuterium in solvent, and in this way, identifies regions of proteins that are more flexible (51). This technique established that 2 peptides encompassing residues 35–71 were less likely to incorporate deuterium if TyrH was preincubated with dopamine, that is, these peptides are more shielded from solvent when dopamine is bound. The finding corroborates the proteolysis experiments that showed that dopamine-bound R domain was less available to trypsin or V8 protease. It also corroborates the Nakashima conclusion, since arg37 and arg38 lie within the peptide whose structure becomes less exposed in the presence of dopamine. The peptides containing residues 35–71 were not detected when the experiment was done with TyrHpser40, so a comparison of this peptide to its phosphorylated form is not possible. A peptide in the catalytic domain showed changes after phosphorylation. The peptide containing residues 295–299 (leu29-ser296-ala297-arg298-asp299) became more exposed to deuterated solvent upon treatment with PKA, presumably because the movement of the R domain away from the opening to the active site exposed that peptide. This peptide’s position with respect to the R domain in PheH is shown in Figure 9.
Figure 9.
Peptides of PheH corresponding to the peptides of TyrH identified by hydrogen-deuterium exchange. The ribbon is derived from crystals of PheH missing 19 amino terminal amino acids (1PHZ). Overlaid on that structure are the structures of PheH with dopamine (5PAH), BH4 and thienylalanine (1KW0), with only the small molecules shown. Thienylalanine is in green, dopamine in magenta with its amino nitrogen in blue, and tetrahydrobiopterin is blue. The region of the R domain that is homologous to TyrH’s gly36-glu50 is colored yellow. The region of PheH that is homologous to TyrH positions 295–299 is colored red.
Summary Section 2.2: Binding of catecholamines
TyrH phosphorylation at ser40 results in a lowered affinity for catecholamines. Dopamine binds to TyrH in the active site in a location that overlaps tetrahydrobiopterin; dopamine binding to TyrH results in an altered conformation for the R domain that prohibits entry of substrates into the active site. Arg37 and arg38 are necessary for inhibition by dopamine, perhaps by determining the overall three-dimensional structure of the R domain. Inhibition by the catechols is dependent on the presence of an amino group, not a carboxylate group.
2.3 R domain differences between rat TyrH and human TyrH
Human TyrH is more complex than the rat enzyme. Humans have 4 isoforms of TyrH that differ in primary sequence just prior to ser31, the result of different splicing of the pre-mRNA. Referred to as hTH1, hTH2, etc, they differ in the length of the R domain. hTH1 is the same size as rat TyrH and is very similar in amino acid sequence. hTH2 contains 4 additional amino acids after met30 (VRGQ), hTH3 has 27 additional amino acids (GAPGPSLTGSPWPGTAAPAASYTPTPR), and hTH4 has all 31 additional amino acids (VRGQGAPGPSLTGSPWPGTAAPAASYTPTPR) (52, 53). A drawing of the differences in the R domain of the human enzymes is shown in Figure 10. The insertion of new amino acids between met30 and ser31 alters the protein just prior to the serine that is phosphorylated by ERK1 and 2, and also increases the spacing between ser19 and ser40. It also alters the numbering of ser31 and ser40, but the homologs of ser31 and ser40 in isoforms 2–4 will be refered to as ser31 and ser40 for the sake of clarity. No complete model has yet emerged to explain the physiological advantage for the existence of the four human isoforms. They have comparable Vmax and KM values. They are all inhibited by dopamine, binding 2 to 3-fold tighter than the rat enzymes, and all bind DOPA approximately 6-fold more tightly than the rat enzyme (54). Because of the loss of the ERK consensus sequence in hTH2, its homolog to ser31 is not phosphorylated by ERK (55), but the repercussions of that loss are not known.
Figure 10.
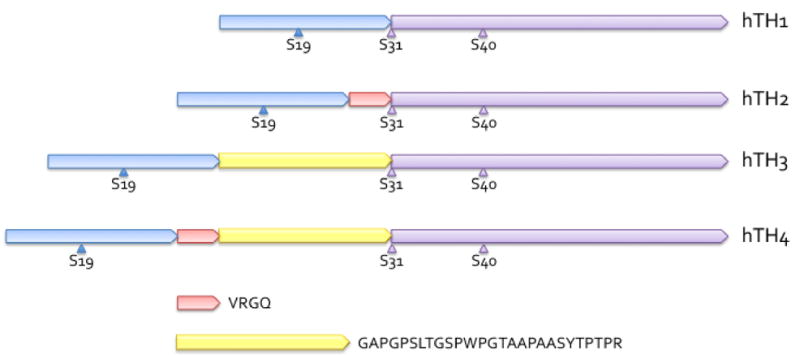
Drawing of the R domains of the human isoforms of TyrH illustrating their structural differences. Amino termini are to the left. hTH1 is identical in length to rat TyrH, and appears first, with the regulatory serines shown. hTH2 has four additional amino acids after met30, and those amino acids are shown as a short pink segment of the R domain. hTH3 has 27 additional amino acids, shown as a yellow segment, included after met30. hTH4 has both additional segments so has 31 amino acids more than hTH1. These additional amino acids are included via alternative mRNA splicing. Since the additional amino acids come immediately before ser31, the serine residues homologous to ser31 and ser40 have different numbers in hTH’s 2, 3, and 4, but are rarely referred to by these numbers.
Summary Section 2.3: R domain differences between rat and human TyrH
Human TyrH comes in four isoforms; isoform 1 is very similar to rat TyrH. Isoforms 2–4 contain additional amino acids in the very region of the R domain where the regulatory serines are positioned. The human isoforms have slightly different affinities for DOPA and dopamine than rat TyrH, and slightly different affinities from each other as well. To be discussed in section 2, there may be some differences in binding affinity for other proteins.
3. Protein complexes
3.1 14-3-3 proteins
In 1987 Ichimura et al. reported that TyrH forms complexes with the 14-3-3 protein after phosphorylation by CaMKII, upon which TyrH activity is increased (23); others have reported that 14-3-3 binding does not affect TyrHpser19 activity (13). The 14-3-3 proteins are highly conserved regulatory proteins involved in a variety of signaling pathways due to their affinity for phosphorylated proteins. For some time after their discovery very few binding partners for the 14-3-3 proteins were known. Since then literally hundreds of proteins have been found to bind to these phosphoprotein chaperones (56).
There are seven isoforms of 14-3-3 in mammals. The 14-3-3s are homodimers of subunits with a molecular weight of 28,000. They are designated by Greek letters: β, ε, γ, η, τ (sometimes called θ), σ, and ζ. Figure 11 contains a three-dimensional representation of 14-3-3ζ with bound phosphopeptide; a strong case has been made for the zeta isoform being a major binding partner for TyrH (57). The 14-3-3 proteins have a preference, somewhat flexible, for phosphorylated residues that are three or four positions after an arginine residue and two before a proline residue (58). CaMKII phosphorylates TyrH at both ser19 and ser40 in vitro, so it is uncertain whether only ser19 or perhaps both serines must be phosphorylated for 14-3-3 binding. The 14-3-3 proteins have no effect on the activity of TyrH which has been phosphorylated only on ser40 (59). Binding of 14-3-3 proteins is reported to decrease the rate of dephosphorylation of ser19 and ser40 by PP2A (59). Cyclin-dependent kinase 11p110 and casein kinase 2 also compete with the 14-3-3 proteins for binding to TyrH (60), so one possibility for the role of 14-3-3 is to prevent access by kinases and phosphatases.
Figure 11.
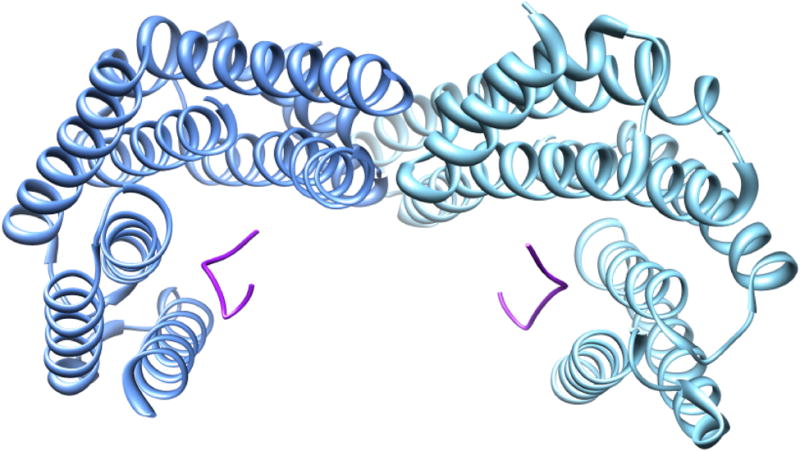
Crystal structure of the dimer of 14-3-3ζ protein with phosphopeptides bound (1QJA).
Neither ser19 nor ser40 lie within sequences similar to the ones described as 14-3-3-favored sequences. It has been suggested that for phosphoserines that are not located in a preferred 14-3-3 protein binding motif, two phosphorylated residues are required for the tightest binding (61). That CaMKII phosphorylates two serines suggests that the 14-3-3 proteins bind TyrH only after both serines are phosphorylated. Other kinases besides CaMKII phosphorylate ser19 and ser40, such as MAPKAPK-2 (25); perhaps all phosphorylation strategies that result in two serine residues being phosphorylated enhance 14-3-3 binding. Kleppe et al. studied the binding of 14-3-3 to full-length human TyrH isoforms using surface plasmon resonance and measured binding affinities (59). Presumably due to limited availability of 14-3-3 proteins at the time of publication, they tested a varied and incomplete library of them, ranging from a yeast homolog (BMH1) to bovine and sheep forms. The results from the yeast 14-3-3 protein may be of limited interest since BMH1 is only 62% identical to human 14-3-3ζ, and bovine 14-3-3ζ is 99+% identical to human 14-3-3ζ. Therefore binding by BMH1 will not be discussed here. No binding was seen between hTH1pser40 and bovine 14-3-3ζ. These data argue that phosphorylation at ser40 is not enough to enable binding of 14-3-3 protein. Experiments also tested whether double phosphorylation of hTH1 affected 14-3-3 affinity; between hTH1pser19pser40 and bovine 14-3-3ζ the Kd value was 10 nM. A subsequent report from the same laboratory described similar measurements with singly-labeled hTH1pser19, phosphorylated by PRAK; in this case, binding between this TyrH form and bovine 14-3-3ζ was fitted to a two-site model, with the tighter-binding site having a Kd value of about 2 nM and the looser site having an affinity of about 50 nM (62). The use of a two-site model in only the second publication makes direct comparisons between the two difficult. Using the Kd values for the tighter site, hTH1pser19 binds more tightly than hTH1pser40 to 14-3-3ζ, and hTH1pser19pser40 binds 14-3-3 ζ more tightly than hTH1pser19. That is, binding between TyrH and 14-3-3 is tighter for pser19 than pser40, and affinity increases with multiple phosphorylation of TyrH. Without knowing the in vivo concentrations of the 14-3-3 proteins, it is difficult to know how important a difference between Kd values of 0.2 nM and 50 nM is. However, since the 14-3-3 concentrations could be in this range, it is probably wise for now to consider that these affinities are physiologically pertinent.
The above data were obtained using hTH1. Data for isoforms 3 and 4 were also collected. Because the distance between ser19 and the homolog to ser40 differs among the four human isoforms due to the insertion of 4, 27, or 31 amino acids between position 30 and 31, 14-3-3 binding may differ among the isoforms. The data did confirm this hypothesis; hTyrH3 and hTyrH4, when phosphorylated at both ser19 and the homolog of ser40, bound 14-3-3ζ more tightly than hTyrH1. These data are represented in Figure 12.
Figure 12.

Dissociation constants measured for the binding of bovine 14-3-3 ζ protein to human TyrH isoforms 1, 3, and 4 phosphorylated at ser19 and ser40.
Obsilova et al. (63) studied the isolated R domain of human TyrH 1 (hTH1R), unphosphorylated, singly phosphorylated at ser40 or phosphorylated at both ser19 and ser40 (dphTH1R) and found a difference in 14-3-3ζ affinity for the three forms. The binding constant for dphTH1R and 14-3-3ζ was 421 nM, measured using native polyacrylamide gel electrophoresis to detect the complex between the two proteins. This number is higher than the ones obtained using surface plasmon resonance (62). Obsilova et al. also compared proteolysis rates of hTH1R and dphTH1R in the presence of 14-3-3ζ; 14-3-3ζ protected the R domain from proteolysis to a greater degree when the R domain was phosphorylated at both ser19 and ser40. Furthermore, they used hTH1R with tryptophan residues at position 14, 34, and 73, and relying on the fluorescence lifetime of those tryptophan residues, concluded that binding of 14-3-3ζ causes a structural change in the areas of position 13, 34, and especially position 73. The homologous position to 74 in PheH is not far away from the active site opening, so it is quite plausible that the 14-3-3 protein would cover this portion of the TyrH R domain upon binding. 14-3-3ζ has been shown to co-precipitate with TyrH from the dopaminergic cell MN9D under conditions in which 14-3-3η did not have an effect (57). Halskau et al. (64) have expanded studies of TyrH binding to the 14-3-3 proteins by considering the possibility that TyrH or complexes including TyrH are membrane-bound. They measured binding affinities between 14-3-3ζ or η and short polypeptides of TyrH consisting of amino acids 1-43, in the presence of membranes of different composition. They recorded much tighter binding with the ζ isoform than the η isoform (S0.5 = 0.5 μM vs. 7 μM, respectively), and also 10-fold tighter binding to membranes for TyrHpser19 vs. unphosphorylated TyrH. These results are surprising in light of the assumption for many years that TyrH is a mainly cytosolic enzyme. Purification of TyrH has been successful from eukaryotic cells and from bacteria with no effort to extract proteins from membranes (20, 42).
A model of TyrH docked into position within a dimer of 14-3-3ζ is shown in Figure 13. Alignment of ser19 and ser40 with the phosphoserine binding sites of 14-3-3 is not possible since this R domain structure is that of PheH. The reader is reminded that there are approximately 30 more amino acids in the flexible region of the TyrH R domain than the homologous region of PheH, and that there are another 4, 27, or 31 amino acids (depending on the isoform) in hTH isoforms 2, 3, and 4. Even faced with these caveats it does seem that the R domain of the hydroxylases is geometrically suited for binding to 14-3-3 proteins.
Figure 13.
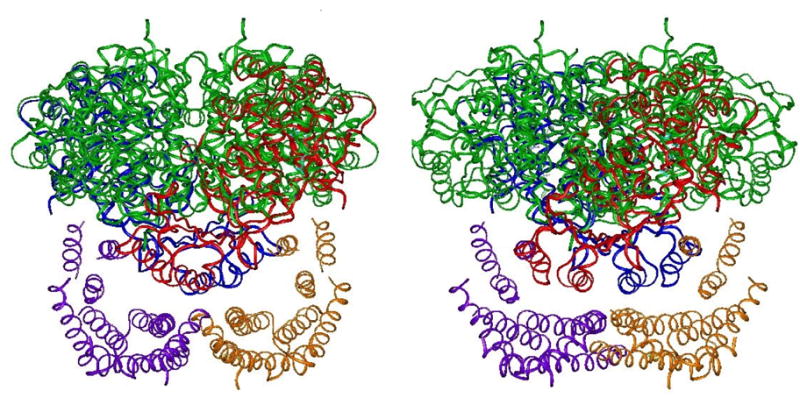
Two views of possible orientations of 14-3-3 protein and the R domain of TyrH. The figure was composed using the structure of PheH (1PHZ) since no TyrH R domain structure is available. The green ribbons are a tetramer of TyrH (1TOH). The blue and red ribbons are 2 monomers of PheH superimposed upon the TyrH tetramer. One monomer of a 14-3-3ζ dimer (1QJA) is colored purple and the other is gold. The views represent the model at two positions rotated 90° on the x-axis.
Summary Section 3.1: Complexes with 14-3-3 proteins
TyrH is activated by phosphorylation of ser19 but only in the presence of 14-3-3 proteins (23). Singly-phosphorylated (at ser40 or ser19) TyrH binds 14-3-3 proteins but affinity is increased if TyrH is phosphorylated at both ser19 and ser40. The result of 14-3-3 binding is not definitively known but may include stabilization, exclusion of PP2A or other proteins, and localization to organelles or other proteins (further covered below).
3.2 Alpha-synuclein
Recently another protein, α-synuclein (α-syn), has been implicated in the regulation of TyrH. Alpha-synuclein is an abundant presynaptic protein implicated in neuronal plasticity and neurodegenerative diseases. An association between α-syn and dopamine leads to the selective death of neuronal cells and the accumulation of misfolded α-syn (65). Misfolded α-syn is a major component of the Lewy body, the diagnostic histological feature of Parkinson disease. Patients with Parkinson disease suffer from a low level of dopamine due to the deterioration of the substantia nigra, the region of the brain where TyrH is most abundant. The function of α-syn is uncertain but it has been shown to bind to TyrH and to diminish TyrH phosphorylation and DOPA production (66). Alpha-synuclein has some limited similarity to the 14-3-3 proteins (67), which are also found in large quantities in the Lewy body. One study has found that mouse dopaminergic cells overexpressing α-syn contain normal amounts of TyrH but that the TyrH is more phosphorylated at ser40 (66); this effect has since been explained as an inhibitory effect of α-syn on PP2A. The same phenomenon has been found for TyrHpser19 (68). Alpha-synuclein is itself altered by phosphorylation; when its ser129 is modified by PLK2 (polo-like kinase) its affinity for TyrH is decreased, freeing TyrH for inactivation by PP2A (68). These studies include clear microscopic and immunochemical data showing co-localization and binding between α-syn and TyrH.
Summary Section 3.2: Complex with α-synuclein
TyrH forms complexes with α-syn with the result of continued TyrH activation due to the exclusion of PP2A. Binding between TyrH and α-syn may play a role in cellular localization of a complex associated with dopamine production and oxidative stress in the neuron.
3.3 PP2A, AADC, GTP cyclohydrolase, VMAT, DJ-1
Studies also provide evidence for localization of TyrH and its effector proteins near neuron vesicles and mitochondria, along with PP2A and aromatic amino acid decarboxylase (AADC) (57, 66). Further evidence for this near-vesicle association and perhaps a dopaminergic protein complex comes from immunoprecipitation experiments that co-localize TyrH with the vesicular monoamine transporter-2 (VMAT), along with AADC (69). This protein complex, associated at the vesicles that contain the enzyme dopamine β-hydroxylase, would ensure that dopamine is promptly packaged into secretory vesicle rather than remaining free in the cytosol. Other studies have found that the enzyme GTP cyclohydrolase (GTPCH) (70, 71), the rate-limiting enzyme of tetrahydrobiopterin synthesis, is in direct contact with TyrH. This may be a strategy to ensure that TyrH can obtain enough tetrahydrobiopterin to displace the dopamine that is abundant due to the proximity of AADC. These studies have relied heavily on in situ techniques. While the present review is focused mainly on biochemical studies, TyrH association with these neuronal proteins has not yet reached the level of in vitro study. Halskau et al. have shown an association between TyrH, 14-3-3, and negatively charged membranes (64) using purified components and surface plasmon resonance, in part corroborating the in situ work. It will be the task of the next review on TyrH to cover the biophysical experiments with purified TyrH, associated proteins, and other effectors, that yield the relative binding constants and Michaelis constants telling us when binding is occurring, when it is not, and what effect binding has on TyrH activity.
One last protein to be mentioned as a possible large-molecule effector is DJ-1. DJ-1 is a transcriptional regulator and plays a role in circumventing oxidative stress; variants of DJ-1 are identified in patients with Parkinson disease (72). With respect to TyrH it was first identified as a repressor binding to the TyrH gene promoter, but Ishikawa et al. report that it binds directly to TyrH itself and also to aromatic amino acid decarboxylase (AADC), activating both enzymes. DJ-1 has three cysteine residues, at positions 46, 53, and 106; DJ-1cys106ser is not an activator of TyrH (73), suggesting that the oxidation state of DJ-1 may regulate TyrH and DOPA levels. Because only the sulfinic acid form of DJ-1cys106 is effective in preventing fibrillation of α-syn, it has been postulated to act as an α-syn chaperone (74). It remains to be determined whether the three activities of DJ-1, i.e., activation of TyrH, activation of AADC, and chaperone for α-syn, are interdependent.
Summary Section 3.3: Complexes with other proteins
Considerable data are accumulating that suggest that dopamine production involves a complex of proteins. The complex may serve to stabilize tetrahydrobiopterin, dopamine, phosphorylated proteins, or to localize proteins near secretory vesicles or mitochondria. TyrH has been detected in complex with PP2A, AADC, GTPCH, VMAT and DJ-1.
4 Nitration/S-thiolation
It is appropriate at this point, having just introduced the importance of the oxidation state of cysteine residues of particular neuronal proteins, to turn to the phenomenon of nitration and thiolation of TyrH. Peroxynitrite, which is formed by the reaction of nitric oxide and superoxide, generates an intermediate that nitrates tyrosine residues in proteins (75), and tyrosine nitration in proteins is currently considered a functionally significant post-translational modification that indicates the cellular level of NO-mediated oxidative reactions (76). Reactivity of nitrating species is emerging as a mode of regulation in many tissues (76). Parkinson Disease is characterized by the deterioration of dopamine-producing neurons in the brain, believed to result from exposing neurons to oxidative and nitrosative stress (77, 78). Tyrosine hydroxylase activity and dopamine levels are decreased in the Parkinson brain more than would be expected simply from the loss of the dopaminergic neurons (79, 80). Therefore, chemical modifications to TyrH consistent with etiology of Parkinson disease are of great interest. Because certain amphetamines that damage dopaminergic nerve endings also inhibit TyrH, the enzyme was considered a possible target for peroxynitrite. When neurons are exposed to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), an amphetamine that produces a Parkinson-like dystonia, or when neurons are exposed to peroxynitrite, TyrH is nitrated and inactivated (81). Since this discovery by Ara et al. in 1998, considerable in vitro work has been carried out to understand the reaction and to determine whether it is reversible and a possible mode of enzyme regulation.
Experiments to document the nitration of TyrH, the loss of activity in nitrated TyrH, the specificity of the nitration, and the possible reversibility of the reaction, have shown TyrH to be nitrated by peroxynitrite in vitro. Freshly prepared peroxynitrite inactivates purified TyrH in a concentration-dependent manner with an IC50 of ~160 μM (82). Reduced pterins can protect TyrH from nitration, suggesting that the reaction takes place at the active site (83, 84). Reaction of TyrH and peroxynitrite followed by proteolysis and mass spec determination of the modified peptides showed that TyrH is nitrated at three tyrosine residues, tyr423, tyr428, and tyr432 (85). These three tyrosine residues are located on a flexible loop that lies at the entrance to the active site (50). The position and arrangement of these tyrosine residues on the outer surface of the opening to the active site are shown in Figure 14. Kuhn et al. made variants of TyrH with these tyrosine residues singly, doubly, or triply substituted with alanine. To see if nitration of these tyrosines caused inactivation, each variant lacking the tyrosine(s) was nitrated. For each variant, the enzyme was not as extensively nitrated by peroxynitrite, as expected if the target for nitration is removed, but all the variants were inactivated to some extent by the treatment, even the one containing none of the tyrosines (85). That is, even though the sites of nitration were removed by the mutagenesis, the mutants were still as susceptible to inactivation by peroxynitrite as wild-type TyrH (85). The finding that removal of all the nitrated tyrosine residues did not prevent inactivation of TyrH by peroxynitrite suggested that in addition to nitration, some other modification was also taking place.
Figure 14.
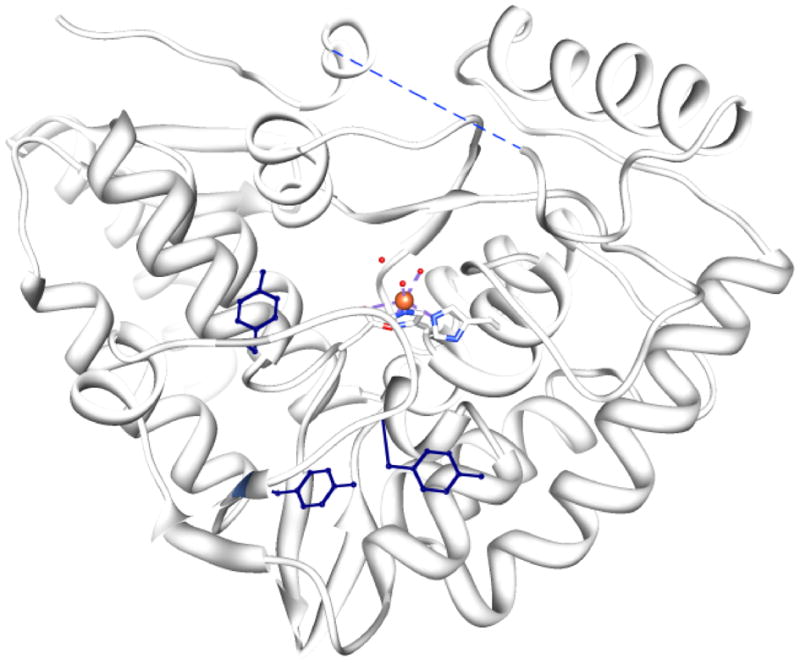
Locations of the three tyrosine residues of TyrH that become nitrated as a result of reaction with peroxyitrate. The three tyrosine residues are at positions 423, 428, and 432 and are shown in dark blue. The active site it identifiable by the bound iron and the ligands that surround it. The figure illustrates that each tyrosine residue is very exposed to solvent.
Further experiments by the Kuhn lab pointed to S-thiolation of cysteine residues as the main reason for loss of activity after exposure to peroxynitrite (86). S-thiolated proteins contain mixed disulfides between cysteine residues of the protein and small molecular weight thiols such as glutathione. These adducts are the result of oxidative injury sustained in vivo. Peroxynitrite, besides reaction at tyrosine residues, also reacts at the thiol groups of cysteine, causing reactive species that result in disulfide bond formation (86).
TyrH has no disulfide bonds but does have 7 cysteine residues (17). The positions of these residues and their surface exposure are illustrated in Figure 15. Reaction of TyrH with peroxynitrite resulted in loss of TyrH activity concurrently with loss of reactivity of 6 cysteine residues with DTNB (82). Treatment of TyrH with the thiol-reactive compounds labeled TyrH much less if the enzyme had already reacted with peroxynitrite, suggesting that peroxynitrite had oxidized cysteine residues of TyrH. TyrH exposed to the sulfhydryl oxidant diamide lost activity; when glutathione was included in the reaction, the loss of activity was greater. Proteolysis and peptide mapping of TyrH that had undergone this treatment showed that all of the cysteine residues of TyrH but one (cys311) had become glutathionylated (87). Glutaredoxin and DTT were able to release glutathione from TyrH and partially reverse the loss of TyrH activity, a requisite characteristic if glutathionylation is to be considered a mode of TyrH regulation. The possibility that TyrH is regulated in part by cysteine oxidation when exposed to NO is demonstrated by these experiments. However, it should be noted that in the Ischiropoulos lab, where TyrH nitration was first studied, glutathionylation at cysteine residues in TyrH does not occur (88). The Ischiropoulos laboratory suggests that differences in purification schemes may make TyrH less prone to unfolding in their hands, so that cysteine residues are more protected (88). Figure 15 illustrates that few of the cysteine residues of TyrH are exposed to solvent. Reduced biopterin protected TyrH from nitration with peroxynitrite, but it did not protect TyrH from inactivation after peroxynitrite treatment or from inactivation by S-thiolation (77), also suggesting that the cysteine residues may be reacting in a non-specific way.
Figure 15.

Locations of the seven cysteine residues of TyrH. At left is the structure of TyrH overlaid on the structure of PheH; PheH is included to keep in mind where the R domain would be. The cysteine residues are shown as spheres and the sulfur atoms are yellow. At right is the same structure but TyrH is represented as a partially transparent surface rendering to illustrate the relative exposure of the cysteine residues.
Summary Section 4: Nitration/thiolation of TyrH
In the presence of NO and superoxide TyrH is nitrated at three tyrosine residues with resulting loss of activity. Nitration may be a mode of regulation of TyrH activity in the cell. Oxidizing conditions also result in the modification of 6 of the 7 cysteine residues of TyrH by thiolation. It remains to be proven that these modifications are physiologically relevant, but the importance of biological oxidation in the brain, and of dopamine levels, suggest that any changes in TyrH associated with biological oxidations such as these should be thoroughly investigated.
CONCLUSION
This review is written at a time of very active research on levels of dopamine and its effects. TyrH as the rate-limiting enzyme of dopamine synthesis has to be considered one of the major molecular agents in determining dopamine levels. Some of the work on TyrH activity is sophisticated; biophysical measurements of complicated binding equilibria between purified components are being made. Other studies are still at the comparatively early level of qualitative immunochemical assessments. The sites of phosphorylation of TyrH and the kinases involved are known, but the molecular effects are largely unknown. Some of the macromolecular binding partners of TyrH are known, but few effects are known. The involvement of membranes and organelles are only beginning to be studied. As more knowledge is gathered on oxidative neurochemistry, TyrH may seem even more central to mental health. The field of TyrH regulation, while mature in some respects, is in its infancy in others.
Abbreviations
- AAAHs
aromatic amino acid hydroxylases
- AADC
aromatic amino acid decarboxylase
- BMHI
a 14-3-3 protein homolog from yeast
- CaMKII
Ca++/calmodulin-dependent protein kinase II
- cAMP
cyclic AMP
- C domain
catalytic domain
- CdK5
cyclin-dependent kinase 5
- DOPA
dihydroxyphenylalanine
- ERK
extracellularly-regulated kinase
- hTH1
human tyrosine hydroxylase isoform 1, etc
- MAPKAPKII
mitogen-activated protein kinase-activating protein kinase II
- MSKI
mitogen- and stress-activated protein kinase
- NO
nitric oxide
- PheH
phenylalanine hydroxylase
- PKA
cyclic AMP-dependent protein kinase
- PKC
protein kinase C
- PRAK
p38-regulated/activated protein kinase
- R domain
regulatory domain
- rTyrH
rat tyrosine hydroxylase
- TrpH
tryptophan hydroxylase
- TyrH
tyrosine hydroxylase
- TyrHpser19,-pser31, and pser40, etc
TyrH phosphorylated at serine19, serine31, or serine40, etc
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Molinoff PB, Axelrod J. Biochemistry of catecholamines. Annual Review of Biochemistry. 1971;40:465–500. doi: 10.1146/annurev.bi.40.070171.002341. [DOI] [PubMed] [Google Scholar]
- 2.Weiner N. Tyrosine-3-monooxygenase (tyrosine hydroxylase) In: Youdim MBH, editor. Aromatic amino acid hydroxylases and mental disease. John Wiley & Sons, Ltd; New York: 1979. pp. 141–190. [Google Scholar]
- 3.Tripp G, Wickens JR. Neurobiology of adhd. Neuropharmacology. 2009;57:579–589. doi: 10.1016/j.neuropharm.2009.07.026. [DOI] [PubMed] [Google Scholar]
- 4.Arnsten AFT. Catecholamine regulation of the prefrontal cortex. J Psychopharmacol. 1997;11:151–162. doi: 10.1177/026988119701100208. [DOI] [PubMed] [Google Scholar]
- 5.Assadi SM, Yücel M, Pantelis C. Dopamine modulates neural networks involved in effort-based decision-making. Neuroscience & Biobehavioral Reviews. 2009;33:383–393. doi: 10.1016/j.neubiorev.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 6.Anisman H, Zacharko RM. Behavioral and neurochemical consequences associated with stressors. Annals of the New York Academy of Sciences. 1986;467:205–225. doi: 10.1111/j.1749-6632.1986.tb14630.x. [DOI] [PubMed] [Google Scholar]
- 7.David J, Brooks PP. Imaging in Parkinson’s disease: The role of monoamines in behavior. Biological Psychiatry. 2006;59:908–918. doi: 10.1016/j.biopsych.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 8.Weiner N. Regulation of norepinephrine biosynthesis. Annual Review of Pharmacology. 1970;10:273–290. doi: 10.1146/annurev.pa.10.040170.001421. [DOI] [PubMed] [Google Scholar]
- 9.Cousins DA, Butts K, Young AH. The role of dopamine in bipolar disorder. Bipolar Disorders. 2009;11:787–806. doi: 10.1111/j.1399-5618.2009.00760.x. [DOI] [PubMed] [Google Scholar]
- 10.Grobecker H, Saavedra JM, McCarty R, Weise VK, Kopin IJ. Role of noradrenergic nerves and adrenal medulla during the development of genetic and experimental hypertension in rats. In: Usdin E, Kopin IJ, Barchas J, editors. Catecholamines: Basic and clinical frontiers, proc Int Catecholamine symp. 4. Pergamon Press; New York: 1978. pp. 906–908. [Google Scholar]
- 11.Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2009;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Döskeland AP, Flatmark T. Ubiquitination of soluble and membrane-bound tyrosine hydroxylase and degradation of the soluble form. European Journal of Biochemistry. 2002;269:1561–1569. doi: 10.1046/j.1432-1033.2002.02808.x. [DOI] [PubMed] [Google Scholar]
- 13.Dunkley PR, Bobrovskaya L, Graham ME, Nagy-Felsobuki EIV, Dickson PW. Tyrosine hydroxylase phosphorylation: Regulation and consequences. Journal of Neurochemistry. 2004;91:1025–1043. doi: 10.1111/j.1471-4159.2004.02797.x. [DOI] [PubMed] [Google Scholar]
- 14.Fitzpatrick PF. The aromatic amino acid hydroxylases. In: Purich DL, editor. Advances in enzymology and related areas of molecular biology. John Wiley & Sons, Inc; 2000. pp. 235–294. [DOI] [PubMed] [Google Scholar]
- 15.Hufton SE, Jennings IG, Cotton RGH. Structure and function of the aromatic amino acid hydroxylases. Biochem J. 1995;311:353–366. doi: 10.1042/bj3110353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ledley FD, DiLella AG, Kwok SCM, Woo SLC. Homology between phenylalanine and tyrosine hydroxylases reveals common structural and functional domains. Biochemistry. 1985;24:3389–3394. doi: 10.1021/bi00335a001. [DOI] [PubMed] [Google Scholar]
- 17.Goodwill KE, Sabatier C, Marks C, Raag R, Fitzpatrick PF, Stevens RC. Crystal structure of tyrosine hydroxylase at 2.3 angstrom and its implications for inherited neurodegenerative diseases. Nature Structural Biology. 1997;4:578–585. doi: 10.1038/nsb0797-578. [DOI] [PubMed] [Google Scholar]
- 18.Kobe G, Jennings IG, House CM, Feil SC, Michell BJ, Tiganis T, Parker MW, Cotton RGH, Kemp BE. Regulation and crystallization of phosphorylated and dephosphorylated forms of truncated dimeric phenylalanine hydroxylase. Protein Science. 1997;6:1352–1357. doi: 10.1002/pro.5560060626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fitzpatrick PF. The tetrahydropterin-dependent amino acid hydroxylases. Ann Rev Biochem. 1999;68:355–381. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- 20.Daubner S, Lauriano C, Haycock J, Fitzpatrick P. Site-directed mutagenesis of serine 40 of rat tyrosine hydroxylase. Effects of dopamine and cAMP-dependent phosphorylation on enzyme activity. J Biol Chem. 1992;267:12639–12646. [PubMed] [Google Scholar]
- 21.Doskeland AP, Martinez A, Knappskog PM, Flatmark T. Phosphorylation of recombinant human phenylalanine hydroxylase: Effect on catalytic activity, substrate activation and protection against non-specific cleavage of the fusion protein by restriction protease. Biochem J. 1996;313:409–414. doi: 10.1042/bj3130409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC. The structural basis for 14-3-3: phosphopeptide binding specificity. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 23.Ichimura T, Isobe T, Okuyama T, Yamauchi T, Fujisawa H. Brain 14-3-3 protein is an activator protein that activates tryptophan 5-monooxygenase and tyrosine 3-monooxygenase in the presence of Ca2+, calmodulin-dependent protein kinase II. FEBS Letts. 1987;219:79–82. doi: 10.1016/0014-5793(87)81194-8. [DOI] [PubMed] [Google Scholar]
- 24.Kuhn DM, Sakowski SA, Sadidi M, Geddes TJ. Nitrotyrosine as a marker for peroxynitrite-induced neurotoxicity: The beginning or the end of the end of dopamine neurons? J Neurochem. 2004;89:529–536. doi: 10.1111/j.1471-4159.2004.02346.x. [DOI] [PubMed] [Google Scholar]
- 25.Dunkley PR, Bobrovskaya L, Graham ME, von Nagy-Felsobuki EI, Dickson PW. Tyrosine hydroxylase phosphorylation: Regulation and consequences. J Neurochem. 2004;91:1025–1043. doi: 10.1111/j.1471-4159.2004.02797.x. [DOI] [PubMed] [Google Scholar]
- 26.Saraf A, Oberg EA, Strack S. Molecular determinants for PP2A substrate specificity: Charged residues mediate dephosphorylation of tyrosine hydroxylase by the PP2A/B′ regulatory subunit. Biochemistry. 2009;49:986–995. doi: 10.1021/bi902160t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bobrovskaya L, Dunkley PR, Dickson PW. Phosphorylation of ser19 increases both ser40 phosphorylation and enzyme activity of tyrosine hydroxylase in intact cells. J Neurochem. 2004;90:857–864. doi: 10.1111/j.1471-4159.2004.02550.x. [DOI] [PubMed] [Google Scholar]
- 28.Bevilaqua LRM, Graham ME, Dunkley PR, von Nagy-Felsobuki EI, Dickson PW. Phosphorylation of ser19 alters the conformation of tyrosine hydroxylase to increase the rate of phosphorylation of ser40. J Biol Chem. 2001;276:40411–40416. doi: 10.1074/jbc.M105280200. [DOI] [PubMed] [Google Scholar]
- 29.Royo M, Fitzpatrick PF, Daubner SC. Mutation of regulatory serines of rat tyrosine hydroxylase to glutamate: Effects on enzyme stability and activity. Arch Biochem Biophys. 2005;434:266–274. doi: 10.1016/j.abb.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Ramsey AJ, Fitzpatrick PF. Effects of phosphorylation on binding of catecholamines to tyrosine hydroxylase: Specificity and thermodynamics. Biochemistry. 2000;39:773–778. doi: 10.1021/bi991901r. [DOI] [PubMed] [Google Scholar]
- 31.McCulloch RI, Daubner SC, Fitzpatrick PF. Effects of substitution at serine 40 of tyrosine hydroxylase on catecholamine binding. Biochemistry. 2001;40:7273–7278. doi: 10.1021/bi010546d. [DOI] [PubMed] [Google Scholar]
- 32.Sutherland C, Alterio J, Campbell DG, Le Bourdelles B, Mallet J, Haavik J, Cohen P. Phosphorylation and activation of human tyrosine hydroxylase in vitro by mitogen-activated protein (MAP) kinase and MAP-kinase-activated kinases 1 and 2. Eur J Biochem. 1993;217:715–722. doi: 10.1111/j.1432-1033.1993.tb18297.x. [DOI] [PubMed] [Google Scholar]
- 33.Vulliet PR, Woodgett JR, Ferrari S, Hardie DG. Characterization of the sites phosphorylated on tyrosine hydroxylase by Ca2+ and phospholipid-dependent protein kinase, calmodulin-dependent multiprotein kinase and cyclic AMP-dependent protein kinase. FEBS Letts. 1985;182:335–339. doi: 10.1016/0014-5793(85)80328-8. [DOI] [PubMed] [Google Scholar]
- 34.Gahn LG, Roskoski R., Jr Thermal stability and cd analysis of rat tyrosine hydroxylase. Biochemistry. 1995;34:252–256. doi: 10.1021/bi00001a030. [DOI] [PubMed] [Google Scholar]
- 35.Martinez A, Haavik J, Flatmark T, Arrondo JL, Muga A. Conformational properties and stability of tyrosine hydroxylase studied by infrared spectroscopy: Effect of iron/catecholamine binding and phosphorylation. J Biol Chem. 1996;271:19737–19742. doi: 10.1074/jbc.271.33.19737. [DOI] [PubMed] [Google Scholar]
- 36.Williams JW, Northrup DB. Substrate specificity and structure-activity relationships of gentamicin acetyltransferase i. The dependence of antibiotic resistance upon substrate Vmax/KM values. J Biol Chem. 1978;253:5908–5914. [PubMed] [Google Scholar]
- 37.Royo M, Colette Daubner S. Kinetics of regulatory serine variants of tyrosine hydroxylase with cyclic AMP-dependent protein kinase and extracellular signal-regulated protein kinase 2. Biochimica et Biophysica Acta (BBA) - Proteins & Proteomics. 2006;1764:786–792. doi: 10.1016/j.bbapap.2006.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vigny A, Henry J-P. Bovine adrenal tyrosine hydroxylase: Comparative study of native and proteolyzed enzyme, and their interaction with anions. J Neurochem. 1981;36:483–489. doi: 10.1111/j.1471-4159.1981.tb01618.x. [DOI] [PubMed] [Google Scholar]
- 39.Phillips RS, Iwaki M, Kaufman S. Ligand effects on the limited proteolysis of phenylalanine hydroxylase: Evidence for multiple conformational states. Biochem Biophys Res Commun. 1983;110:919–925. doi: 10.1016/0006-291x(83)91050-1. [DOI] [PubMed] [Google Scholar]
- 40.McCulloch RI, Fitzpatrick PF. Limited proteolysis of tyrosine hydroxylase identifies residues 33–50 as conformationally sensitive to phosphorylation state and dopamine binding. Arch Biochem Biophys. 1999;367:143–145. doi: 10.1006/abbi.1999.1259. [DOI] [PubMed] [Google Scholar]
- 41.Ramsey AJ, Fitzpatrick PF. Effects of phosphorylation of serine 40 of tyrosine hydroxylase on binding of catecholamines: Evidence for a novel regulatory mechanism. Biochemistry. 1998;37:8980–8986. doi: 10.1021/bi980582l. [DOI] [PubMed] [Google Scholar]
- 42.Fitzpatrick PF. The pH dependency of binding of inhibitors to bovine adrenal tyrosine hydroxylase. J Biol Chem. 1988;263:16058–16062. [PubMed] [Google Scholar]
- 43.Gordon S, Webb J, Shehadeh J, Dunkley P, Dickson P. The low affinity dopamine binding site on tyrosine hydroxylase: The role of the N-terminus and in situ regulation of enzyme activity. Neurochemical Research. 2009;34:1830–1837. doi: 10.1007/s11064-009-9989-5. [DOI] [PubMed] [Google Scholar]
- 44.Gordon SL, Quinsey NS, Dunkley PR, Dickson PW. Tyrosine hydroxylase activity is regulated by two distinct dopamine-binding sites. J Neurochem. 2008;106:1614–1623. doi: 10.1111/j.1471-4159.2008.05509.x. [DOI] [PubMed] [Google Scholar]
- 45.Erlandsen H, Flatmark T, Stevens RC, Hough E. Crystallographic analysis of the human phenylalanine hydroxylase catalytic domain with bound catechol inhibitors at 2.0 å resolution. Biochemistry. 1998;37:15638–15646. doi: 10.1021/bi9815290. [DOI] [PubMed] [Google Scholar]
- 46.Andersen OA, Flatmark T, Hough E. Crystal structure of the ternary complex of the catalytic domain of human phenylalanine hydroxylase with tetrahydrobiopterin and 3-(2-thienyl)-l-alanine, and its implications for the mechanism of catalysis and substrate activation. J Mol Biol. 2002;320:1095–1108. doi: 10.1016/s0022-2836(02)00560-0. [DOI] [PubMed] [Google Scholar]
- 47.Nakashima A, Hayashi N, Kaneko Y, Mori K, Sabban E, Nagatsu T, Ota A. Role of N-terminus of tyrosine hydroxylase in the biosynthesis of catecholamines. Journal of Neural Transmission. 2009;116:1355–1362. doi: 10.1007/s00702-009-0227-8. [DOI] [PubMed] [Google Scholar]
- 48.Daubner SC, Piper MM. Deletion mutants of tyrosine hydroxylase identify a region critical for heparin binding. Protein Expression and Purification. 1995;4:538–541. doi: 10.1002/pro.5560040320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alieva IN, Mustafayeva NN, Gojayev NM. Conformational analysis of the N-terminal sequence met1-val60 of the tyrosine hydroxylase. Journal of Molecular Structure. 2006;785:76–84. [Google Scholar]
- 50.Wang S, Sura GR, Dangott LJ, Fitzpatrick PF. Identification by hydrogen/deuterium exchange of structural changes in tyrosine hydroxylase associated with regulation. Biochemistry. 2009;48:4972–4979. doi: 10.1021/bi9004254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Busenlehner LS, Armstrong RN. Insights into enzyme structure and dynamics elucidated by amide H/D exchange mass spectrometry. Arch Biochem Biophys. 2005;433:34–46. doi: 10.1016/j.abb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 52.Lewis DA, Melchitzky DS, Haycock JW. Four isoforms of tyrosine hydroxylase are expressed in human brain. Neuroscience. 1993;54:477–492. doi: 10.1016/0306-4522(93)90267-j. [DOI] [PubMed] [Google Scholar]
- 53.Haycock J. Species differences in the expression of multiple tyrosine hydroxylase protein isoforms. J Neurochem. 2002;81:947–953. doi: 10.1046/j.1471-4159.2002.00881.x. [DOI] [PubMed] [Google Scholar]
- 54.Sura G, Daubner SC, Fitzpatrick PF. Effects of phosphorylation by protein kinase a on binding of catecholamines to the human tyrosine hydroxylase isoforms. J Neurochem. 2004;90:970–978. doi: 10.1111/j.1471-4159.2004.02566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gordon SL, Bobrovskaya L, Dunkley PR, Dickson PW. Differential regulation of human tyrosine hydroxylase isoforms 1 and 2 in situ: Isoform 2 is not phosphorylated at ser35. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2009;1793:1860–1867. doi: 10.1016/j.bbamcr.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 56.Pozuelo Rubio M, Geraghty KM, Wong BHC, Wood NT, Campbell DG, Morrice N, Mackintosh C. 14-3-3-affinity purification of over 200 human phosphoproteins reveals new links to regulation of cellular metabolism, proliferation and trafficking. Biochem J. 2004;379:395–408. doi: 10.1042/BJ20031797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang J, Lou H, Pedersen CJ, Smith AD, Perez RG. 14-3-3ζ contributes to tyrosine hydroxylase activity in MN9D cells: Localization of dopamine regulatory proteins to mitochondria. J Biol Chem. 2009;284:14011–14019. doi: 10.1074/jbc.M901310200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rittinger K, Budman J, Xu J, Volinia S, Cantley LC, Smerdon SJ, Gamblin SJ, Yaffee MB. Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Molecular Cell. 1999;4:143–166. doi: 10.1016/s1097-2765(00)80363-9. [DOI] [PubMed] [Google Scholar]
- 59.Kleppe R, Toska K, Haavik J. Interaction of phosphorylated tyrosine hydroxylase with 14-3-3 proteins: Evidence for a phosphoserine 40-dependent association. J Neurochem. 2001;77:1097–1107. doi: 10.1046/j.1471-4159.2001.00318.x. [DOI] [PubMed] [Google Scholar]
- 60.Sachs NA, Vaillancourt RR. Cyclin-dependent kinase 11p110 and casein kinase 2 (CK2) inhibit the interaction between tyrosine hydroxylase and 14-3-3. J Neurochem. 2004;88:51–62. doi: 10.1046/j.1471-4159.2003.02119.x. [DOI] [PubMed] [Google Scholar]
- 61.Tzivion G, Avruch J. 14-3-3 proteins: Active cofactors in cellular regulation by serine/threonine phosphorylation. J Biol Chem. 2002;277:3061–3064. doi: 10.1074/jbc.R100059200. [DOI] [PubMed] [Google Scholar]
- 62.Toska K, Kleppe R, Armstrong CG, Morrice NA, Cohen P, Haavik J. Regulation of tyrosine hydroxylase by stress-activated protein kinases. J Neurochem. 2002;83:775–783. doi: 10.1046/j.1471-4159.2002.01172.x. [DOI] [PubMed] [Google Scholar]
- 63.Obsilova V, Nedbalkova E, Silhan J, Boura E, Herman P, Vecer J, Sulc M, Teisinger J, Dyda F, Obsil T. The 14-3-3 protein affects the conformation of the regulatory domain of human tyrosine hydroxylase. Biochemistry. 2008;47:1768–1777. doi: 10.1021/bi7019468. [DOI] [PubMed] [Google Scholar]
- 64.Halskau O, Jr, Ying M, Baumann A, Kleppe R, Rodriguez-Larrea D, Almås B, Haavik J, Martinez A. Three-way interaction between 14-3-3 proteins, the N-terminal region of tyrosine hydroxylase, and negatively charged membranes. J Biol Chem. 2009;284:32758. doi: 10.1074/jbc.M109.027706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leong S, Cappai R, Barnham K, Pham C. Modulation of α-synuclein aggregation by dopamine: A review. Neurochemical Research. 2009;34:1838–1846. doi: 10.1007/s11064-009-9986-8. [DOI] [PubMed] [Google Scholar]
- 66.Perez RG, Waymire JC, Lin E, Liu JJ, Guo F, Zigmond MJ. A role for α-synuclein in the regulation of dopamine biosynthesis. J Neurosci. 2002;22:3090–3099. doi: 10.1523/JNEUROSCI.22-08-03090.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ostrerova N, Petrucelli L, Farrer M, Mehta N, Choi P, Hardy J, Wolozin B. α-synuclein shares physical and functional homology with 14-3-3 proteins. J Neurosci. 1999;19:5782–5791. doi: 10.1523/JNEUROSCI.19-14-05782.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lou HMSE, Alerte TN, Wang J, Wu J, Peng XM, Hong CS, Friedrich EE, Mader SA, Pedersen CJ, Marcus BS, McCormack AL, Di Monte DA, Daubner SC, Perez RG. Serine 129 phosphorylation reduces α–synuclein’s ability to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J Biol Chem. 2010;285:17648–11766. doi: 10.1074/jbc.M110.100867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cartier EA, Parra LA, Baust TB, Quiroz M, Salazar G, Faundez V, Egaña L, Torres GE. A biochemical and functional protein complex involving dopamine synthesis and transport into synaptic vesicles. J Biol Chem. 2010;285:1957–1966. doi: 10.1074/jbc.M109.054510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hirayama K, Kapatos G. Nigrostriatal dopamine neurons express low levels of GTP cyclohydrolase I protein. J Neurochem. 1998;70:164–170. doi: 10.1046/j.1471-4159.1998.70010164.x. [DOI] [PubMed] [Google Scholar]
- 71.Bowling KM, Huang Z, Xu D, Ferdousy F, Funderburk CD, Karnik N, Neckameyer W, O’Donnell JM. Direct binding of GTP cyclohydrolase and tyrosine hydroxylase: Regulatory interactions between key enzymes in dopamine biosynthesis. J Biol Chem. 2008;283:31449–31459. doi: 10.1074/jbc.M802552200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang YX, Wood Nicholas W, Latchman David S. Molecular basis of Parkinson’s disease. NeuroReport. 2008;20:150–156. doi: 10.1097/WNR.0b013e32831c50df. [DOI] [PubMed] [Google Scholar]
- 73.Ishikawa STT, Niki T, Takahashi-Niki K, Maita C, Maita H, Ariga H, Iguchi-Ariga SM. Oxidative status of DJ-1-dependent activation of dopamine synthesis through interaction of tyrosine hydroxylase and 4-dihydroxy-l-phenylalanine (L-DOPA) decarboxylase with DJ-1. J Biol Chem. 2009;284:28832–28844. doi: 10.1074/jbc.M109.019950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou W, Zhu M, Wilson MA, Petsko GA, Fink AL. The oxidation state of DJ-1 regulates its chaperone activity toward α-synuclein. Journal of Molecular Biology. 2006;356:1036–1048. doi: 10.1016/j.jmb.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 75.Ischiropoulos H. Biological selectivity and functional aspects of protein tyrosine nitration. Biochemical and Biophysical Research Communications. 2003;305:776–783. doi: 10.1016/s0006-291x(03)00814-3. [DOI] [PubMed] [Google Scholar]
- 76.Schopfer FJ, Baker PRS, Freeman BA. NO-dependent protein nitration: A cell signaling event or an oxidative inflammatory response? Trends in Biochemical Sciences. 2003;28:646–654. doi: 10.1016/j.tibs.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 77.Kuhn DM, Geddes TM. Tetrahydrobiopterin prevents nitration of tyrosine hydroxylase by peroxynitrite and nitrogen dioxide. Molecular Pharmacology. 2003;64:946–953. doi: 10.1124/mol.64.4.946. [DOI] [PubMed] [Google Scholar]
- 78.Tsang AHK, Chung KKK. Oxidative and nitrosative stress in Parkinson’s disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2009;1792:643–650. doi: 10.1016/j.bbadis.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 79.Pakkenberg B, Møller A, Gundersen HJ, Mouritzen Dam A, Pakkenberg H. The absolute number of nerve cells in substantia nigra in normal subjects and in patients with Parkinson’s disease estimated with an unbiased stereological method. J Neurol Neurosurg Psychiatry. 1991;54:30–33. doi: 10.1136/jnnp.54.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hornykiewicz O. Biochemical pathophysiology of Parkinson’s disease. Advances in neurology. 1987;45:19–34. [PubMed] [Google Scholar]
- 81.Ara J, Prezedborski S, Naini AB, Jackson-Lewis V, Trifletti RR, Horwitz J, Ischiropoulos H. Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Proc Natl Acad Sci USA. 1998;95:7659–7663. doi: 10.1073/pnas.95.13.7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kuhn DM, Aretha CW, Geddes TJ. Peroxynitrite inactivation of tyrosine hydroxylase: Mediation by sulfhydryl oxidation, not tyrosine nitration. J Neurosci. 1999;19:10289–10294. doi: 10.1523/JNEUROSCI.19-23-10289.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kuhn D. Tetrahydrobiopterin prevents nitration of tyrosine hydroxylase by peroxynitrite and nitrogen dioxide. Molecular Pharmacology. 2003;64:946. doi: 10.1124/mol.64.4.946. [DOI] [PubMed] [Google Scholar]
- 84.Kuhn DM, Geddes TJ. Reduced nicotinamide nucleotides prevent nitration of tyrosine hydroxylase by peroxynitrite. Brain Research. 2002;933:85–89. doi: 10.1016/s0006-8993(02)02307-7. [DOI] [PubMed] [Google Scholar]
- 85.Kuhn DM, Sadidi M, Liu X, Kreipke C, Geddes T, Borges C, Watson JT. Peroxynitrite-induced nitration of tyrosine hydroxylase: Identification of tyrosines 423, 428, and 432 as sites of modification by matrix-assisted laser desorption ionization time-of-flight mass spectrometry and tyrosine-scanning mutagenesis. J Biol Chem. 2002;277:14336–14342. doi: 10.1074/jbc.M200290200. [DOI] [PubMed] [Google Scholar]
- 86.Quijano C, Alvarez B, Gatti RM, Augusto O, Radi R. Pathways of peroxynitrite oxidation of thiol groups. Biochem J. 1997;322:167–173. doi: 10.1042/bj3220167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Borges CR, Geddes T, Watson JT, Kuhn DM. Dopamine biosynthesis is regulated by S-glutathionylation. Potential mechanism of tyrosine hydroxylase inhibition during oxidative stress. J Biol Chem. 2002;277:48295–48302. doi: 10.1074/jbc.M209042200. [DOI] [PubMed] [Google Scholar]
- 88.Blanchard-Fillion B, Souza JM, Friel T, Jiang GCT, Vrana K, Sharov V, Barron L, Schoneich C, Quijano C, Alvarez B, Radi R, Przedborski S, Fernando GS, Horwitz J, Ischiropoulos H. Nitration and inactivation of tyrosine hydroxylase by peroxynitrite. J Biol Chem. 2001;278:46017–46023. doi: 10.1074/jbc.M105564200. [DOI] [PubMed] [Google Scholar]