

Abstract
Borrelia burgdorferi, the causative agent of Lyme disease in North America, is an invasive pathogen that causes persistent multiorgan manifestations in humans and other mammals. Genetic studies of this bacterium are complicated by the presence of multiple plasmid replicons, many of which are readily lost during in vitro culture. The analysis of B. burgdorferi plasmid content by plasmid-specific PCR and agarose gel electrophoresis or other existing techniques is informative, but these techniques are cumbersome and challenging to perform in a high-throughput manner. In this study, a PCR-based Luminex assay was developed for determination of the plasmid content of the strain B. burgdorferi B31. This multiplex, high-throughput method allows simultaneous detection of the plasmid contents of many B. burgdorferi strains in a 96-well format. The procedure was used to evaluate the occurrence of plasmid loss in 44 low-passage B. burgdorferi B31 clones and in a library of over 4,000 signature-tagged mutagenesis (STM) transposon mutant clones. This analysis indicated that only 40% of the clones contained all plasmids, with (in order of decreasing frequency) lp5, lp56, lp28-1, lp25, cp9, lp28-4, lp28-2, and lp21 being the most commonly missing plasmids. These results further emphasize the need for careful plasmid analysis in Lyme disease Borrelia studies. Adaptations of this approach may also be useful in the evaluation of plasmid content and chromosomal gene variations in additional Lyme disease Borrelia strains and other organisms with variable genomes and in the correlation of these genetic differences with pathogenesis and other biological properties.
Lyme borreliosis is caused by a group of closely related spirochetes, including Borrelia afzelii and Borrelia garinii in Eurasia and Borrelia burgdorferi in North America and regions of Eurasia (3, 42, 46). These bacteria are transmitted to humans and other mammals by hard-bodied ticks of the genus Ixodes and are capable of causing persistent infection. The initial stage of infection often presents as an expanding erythematous rash (erythema migrans) at the site of the tick bite and may be accompanied by constitutional symptoms, including fatigue, malaise, low-grade fever, headache, and muscle and joint pain. Spirochetes can often be isolated from the erythema migrans lesion, and dissemination is known to occur even during this early, “localized” stage of infection. Neurologic, cardiac, and ophthalmic symptoms may occur as a result of disseminated disease, and persistent infection lasting for months to years may manifest as repeated episodes of arthritis, neurologic symptoms, or a skin condition called acroderma chronicum atrophicans (ACA).
B. burgdorferi sensu lato has an unusual segmented genome composed of a linear chromosome and a large number of linear and circular plasmids (reviewed in reference 11). The chromosome is ∼911 kb, and its gene content and order are highly conserved across all Borrelia species examined to date (8, 18-20, 35, 44). The B. burgdorferi strain B31-MI contains 12 linear and 9 circular plasmids, ranging in size from 5 kb to 56 kb. The extrachromosomal replicons comprise 612 kb, or about 47% of the genome, and some contain essential genetic elements. Comparison of the Lyme disease Borrelia genomes sequenced to date indicates that plasmid DNA rearrangements and sequence differences are common (9, 11, 44).
Repeated passage in culture (2, 22, 45) or genetic manipulation (15) of B. burgdorferi can result in the loss of many plasmids. However, some extrachromosomal elements appear to be essential for survival in vitro and are always present. For example, the circular plasmid cp26 is necessary for growth both in vitro and during the infection cycle. The cp26-encoded telomere resolvase ResT (30) and the products of two genes of unknown and apparently redundant functions, BBB26 and BBB27, are required for in vitro multiplication (27). At least some members of the cp32 plasmids are always present, and isolates lacking lp54 have only rarely been isolated (7, 11). Some plasmids are not required for growth but are essential for virulence in mice. Three plasmids, lp25, lp28-1, and lp36, are required for full infectivity in mice but not for growth in vitro (28, 32, 33, 41, 54). A number of genes on lp54 are likely to be important in both tick colonization (e.g., OspA and OspB genes) and mammalian infection (4, 16, 24, 25, 31, 53). OspC, encoded by cp26, fulfills a critical role in the early stages of mammalian infection (23). Loss of lp28-4 or lp25 is also associated with reduced tick colonization (49). Therefore, it is critical to ascertain the plasmid content of B. burgdorferi following in vitro growth or genetic manipulation in assessing pathogenesis, growth characteristics, and other biological properties.
Previously described methods for determining the plasmid content of B. burgdorferi include pulsed-field agarose gel electrophoresis (PFGE), electron microscopy, two-dimensional agarose gel electrophoresis, and Southern blotting (2, 38, 52, 54). The assembly of the plasmid sequences of low-passage, infectious B. burgdorferi B31-MI defined its plasmid complement (18) and permitted the design of primer pairs that amplified unique portions of each plasmid for use in determining the plasmid contents of individual B. burgdorferi clones (14, 41). Xu et al. (51) used a microarray method to detect plasmid content.
We have developed a rapid plasmid analysis assay for B. burgdorferi by using Luminex xMAP technology. The Luminex system is a liquid-flow multiplex assay designed to allow 100 or more bioassays to be run in a single well of a 96-well plate. The system uses 5.6-μm polystyrene microspheres that are internally labeled with fluorescent dyes and emit distinct fluorescence signatures (addresses). One hundred xTAG microspheres are available that are precoupled with unique oligonucleotide sequences. We used multiplex PCR, an asymmetric primer extension (ASPE) labeling step, and detection of labeled product via binding to xTAG microspheres to identify the B. burgdorferi plasmids. This high-throughput assay permits determination of the plasmid contents of several hundred B. burgdorferi clones or strains in a single day. Plasmid analyses can be done directly from B. burgdorferi cultures or frozen stocks and yield identical results to those obtained by more time-consuming traditional methods. The Luminex plasmid analysis assay (LPAA) was used to examine the plasmid contents of 44 B31 clones and 4,464 transposon mutants in a signature-tagged mutagenesis (STM) library.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The uncloned, infectious B. burgdorferi strain B31 was obtained from A. G. Barbour (University of California at Irvine College of Medicine). B31 clones (B31-5A1 through B31-5A19 and B31-2591 through B31-2621) were isolated by the subsurface agarose plating method as described previously (12, 40). B31 high-passage clones B312, B313, and B314 (43) and the B. afzelii ACA-1 and B. garinii Ip90 strains were also provided by A. G. Barbour. As part of an ongoing STM project (T. Lin, L. Gao, C. Zhang, E. Odeh, L. Coutte, and S. J. Norris, unpublished data), plasmid analysis was also performed on 4,464 clones derived from B. burgdorferi B31 5A18NP1 (lp56− lp28-4− Δbbe02::flgBP-Kanr) transformed with signature-tagged versions of the Himar1-based transposon vector pGKT (48).
The following low-passage B. burgdorferi strains were kindly provided by the indicated investigators: 297 (J. D. Radolf, University of Connecticut Health Science Center, Farmington, CT), N40 and p32.2 (P. A. Rosa, NIAID Rocky Mountain Laboratories, Hamilton, MT), 1989 Wisconsin human skin biopsy isolate B. burgdorferi NCH-1 (26) (K. J. Cann, Stoke Mandeville Hospital, Aylesbury, United Kingdom), human blood isolate B247-101 (I. Schwartz, New York Medical College, Valhalla, NY), and California wood rat isolate C57 (R. S. Lane, University of California, Berkeley, CA). Spirochetes were cultured as previously described (1, 40). Multiplex PCR was performed directly from fresh cultures (titration and mixing experiments, STM clones, reproducibility experiments, and B. burgdorferi strains not derived from B31), from purified DNA (B31-5A1 to -5A19), or from scrapings of frozen stocks (B31-2597 to -2621 and B312 to B314).
Identification of unique regions and design of PCR primers.
Candidate genomic regions for primer pairs that would unambiguously identify each replicon were selected using data from the work of Purser and Norris (41) and information shared by E. Fikrig and Y. Li (Yale University School of Medicine) (51). Primer pairs in unique regions were designed for the chromosome and each of the 21 plasmids by using Vector NTI Advance10 software from Invitrogen (Carlsbad, CA). Primers were chosen to possess melting temperatures (Tms) of ≥65°C and to produce amplicons of between 250 and 737 bp. Each primer sequence was examined for sequence identity with other B. burgdorferi genomic regions by using the NCBI nBLAST utility (http://blast.ncbi.nlm.nih.gov/). Primer sequences are presented in Table S1 in the supplemental material. ASPE primers contained a sequence specific for an individual amplicon at the 3′ end and a unique xTAG sequence at the 5′ end (see Table S2 in the supplemental material). The xTAG sequence is complementary to the unique antitag oligonucleotide attached to a specific Luminex xTAG microsphere. Thus, an ASPE primer provides a link between a particular amplicon and a specific bead type. ASPE primers had no sequence in common with the forward or reverse primers and had a melting temperature of ≥60°C. All oligonucleotides were made by IDT, Coralville, IA.
Recombinant plasmid pool (RPP) clones corresponding to each amplicon were made to serve as positive controls. A Qiagen multiplex PCR kit (Qiagen, Valencia, CA) or NEB Taq 2× mix (New England BioLabs, Ipswich, MA) was used to make each PCR product, which was cloned into the pCR2.1 vector by use of an Invitrogen TOP10 PCR cloning kit. Plasmid DNAs were purified, and pools of the recombinant plasmids were made by mixing equimolar amounts of each plasmid.
Multiplex PCR.
Genomic DNA, purified from B. burgdorferi by use of a Qiagen DNeasy kit or by phenol-chloroform extraction, was used as a template to optimize the procedure and to serve as a positive control for the PCRs. For routine analysis, bacterial cultures were diluted 1:50 in water. Qiagen multiplex PCR kits were used for all multiplex reactions. Plasmid-specific primers were mixed together in 3 pools, with each pool containing primer pairs for 8 different amplicons at a concentration of 2 μM for each primer. Multiplex reaction mixtures contained 25 μl 2× Qiagen reaction mix, 5 μl of one primer pool, 2 μl template DNA or culture, and 18 μl water, for a final volume of 50 μl. PCRs were performed in an Eppendorf Mastercycler thermocycler (Foster City, CA), using the following conditions: 95°C for 15 min followed by 30 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 90 s, and extension at 72°C for 60 s, with a final extension at 72°C for 10 min.
ASPE.
Prior to ASPE, each multiplex PCR mixture was treated with ExoSapIT (USB, Cleveland, OH) to degrade excess primers and dephosphorylate leftover deoxynucleoside triphosphates (dNTPs). A 1:1 dilution of ExoSapIT (2 μl) was added to 5 μl of the multiplex PCR product, incubated at 37°C for 30 min, and heat inactivated at 80°C for 15 min. The ASPE reaction mixture consisted of 10.1 μl of nuclease-free water, 2 μl of 10× PCR buffer (20 mM Tris-HCl, pH 8.4, 50 mM KCl), 1 μl of pooled tag-ASPE primers (8 primers per pool; 500 nM [each]), 1 μl of 20× dNTPs (without dCTP; 1 μM [each]), 0.5 μl 50 mM MgCl2, 0.25 μl 0.4 mM biotin-dCTP (Invitrogen), 0.15 μl of Platinum GenoTYPE Tsp DNA polymerase (Invitrogen), and 5 μl of ExoSapIT-treated PCR product. The reaction mixture was incubated at 96°C for 2 min and then subjected to 30 cycles at 94°C for 30 s, 55°C for 1 min, and 74°C for 2 min. A final extension was performed at 72°C for 1 min.
Hybridization with microspheres and detection.
Each B. burgdorferi amplicon was assigned to a specific xTAG bead type (see Table S2 in the supplemental material). Beads were mixed at a concentration of 2.5 × 105/ml of each bead to create xTAG bead pools that corresponded to the ASPE primer tags used in the multiplex PCRs. Bead pools were mixed before each use by vortexing for 20 s and then were diluted with 2× hybridization solution (0.4 M NaCl, 0.2 M Tris, 0.16% Triton X-100, pH 8.0) to yield a final concentration of 2,500 of each bead type per 25 μl. After being vortexed again for 20 s, 25 μl of the bead-hybridization buffer mixture was aliquoted into 96-well plates (Fisher). Eight microliters of ASPE product and 17 μl of nuclease-free water were added, for a final volume of 50 μl. Control wells (beads only) contained 25 μl of distilled H2O. Reaction mixtures were denatured at 96°C for 90 s and then incubated at 38°C for 30 min to overnight. After hybridization, the reactions were transferred to a Millipore filter plate (Millipore, Temecula, CA) and washed with 1× hybridization solution (0.2 M NaCl, 0.1 M Tris, 0.08% Triton X-100, pH 8.0) according to the Millipore apparatus procedure. PhycoLink streptavidin-R-phycoerythrin (Prozyme, Hayward, CA) was diluted to 2 μg/ml in 1× hybridization solution and added to each reaction mix in the filter plate at a volume of 100 μl. The plates were incubated at 38°C for 15 min on a microplate rotator at 250 rpm/min. Plates were analyzed using a Luminex 100/200 system according to the standard protocol (100 beads minimum per bead type). Results are recorded as the median fluorescence intensity (MFI) of the phycoerythrin output associated with each bead type. In each plasmid analysis experiment, normalization was performed by dividing the MFI value for a given isolate and plasmid by the MFI value for the corresponding RPP amplicon, e.g., (isolate 1 MFIlp54)/(RPP MFIlp54). For lp5, the RPP MFI value was generally much greater than the highest MFI values obtained with B. burgdorferi clones. Therefore, the median of the highest 5 MFI values in a given experiment was used for lp5 normalization. The same approach was utilized in rare experiments where, for a particular plasmid or plate, the highest normalized MFI values were either >1.5 or <0.5. The cutoff for plasmid positivity was determined empirically for each primer pair. An intermediate range was also established for some plasmids when there was not a clear distinction between positive and negative samples.
RESULTS
Overview of LPAA.
The Luminex system is designed to run multiple assays simultaneously in a single well of a 96-well plate, permitting high-throughput analysis. Polystyrene microspheres (beads) with unique spectral properties and oligonucleotide antitags link each assay product to a specific fluorescence signature. Detection of the multiplexed results takes place in a proprietary analyzer. This instrument is based on the principles of flow cytometry. Suspended microspheres pass in a single-file stream through a detection chamber. A red laser excites the red and infrared dyes, and sensors detect the unique fluorescence signature of each microsphere. A green laser excites orange fluorescence associated with the labeled product of the assay bound to the microsphere, yielding an MFI value that indicates the quantity of bound ligand reporter.
Our early attempts at B. burgdorferi plasmid detection with Luminex-based technology focused on the direct capture of unamplified Borrelia plasmid DNA by using plasmid-specific oligonucleotides bound to microspheres followed by detection with a second biotin-labeled oligonucleotide and a fluorophore-coupled streptavidin. However, this approach was found to lack sufficient sensitivity, even with the use of dendrimers to increase the amount of fluorescent signal. Therefore, we proceeded with the development of an assay that used amplification of plasmid-specific sequences followed by an allele-specific biotin labeling reaction to permit detection of the products.
An outline of the LPAA for determining the plasmid content of B. burgdorferi is shown in Fig. 1. We designed 24 primer sets to amplify unique regions of each of the 21 B. burgdorferi B31 plasmids and the chromosome, including two sets each for lp25 and lp28-1. Replicon detection of each B. burgdorferi sample was performed in 3 multiplex reactions. The multiplex reactions used genomic DNA or cultures of Borrelia as templates and were run in a 96-well plate format, allowing analysis of 30 B. burgdorferi clones per plate. Multiplex reaction products were used as templates for ASPE reactions in the presence of biotin-dCTP. The 3′ ends of the ASPE primers contained sequences that corresponded to the B. burgdorferi amplicons, while the 5′ ends of the primers contained xTAG sequences that annealed to specific xTAG microspheres (see Table S2 in the supplemental material). The labeled DNA was hybridized to xTAG microspheres, and microsphere-biotinylated DNA complexes were detected with fluorescently labeled streptavidin and quantitated in a Luminex 100/200 analyzer. The analyzer measures the number of each type of bead detected and the MFI for each bead type. Thus, the phycoerythrin signal on a specific microsphere indicates the presence of a specific B. burgdorferi replicon.
FIG. 1.

Overview of Luminex procedure for determining plasmid content. (1) Plasmid-specific regions are amplified by multiplex PCR, using genomic DNA or B. burgdorferi culture as the template. Unincorporated primers and dNTPs in amplified PCR products are removed by treatment with exonuclease I and alkaline phosphatase. (2) Primers that contain an xTAG sequence are utilized in an asymmetric PCR that incorporates biotin-dCTP. (3) Biotinylated products are hybridized with xTAG microspheres that are coupled to antitag sequences and detected by binding of streptavidin-R-phycoerythrin. MFI, mean fluorescence intensity.
Several controls were included in each 96-well plate. Wells containing only microspheres provided a positive control for the integrity of the bead pool and a negative control for background phycoerythrin fluorescence (data not shown). Other negative controls included a multiplex PCR with no borrelial DNA and an ASPE reaction with no multiplex PCR template. Recombinant plasmids containing the multiplex DNA amplicons and a previously characterized B. burgdorferi strain served as positive controls.
Assay for B. burgdorferi replicons.
An RPP containing equimolar amounts of each of the 24 B. burgdorferi plasmid-specific amplicons was used to examine the relative MFI produced in the multiplex Luminex assay. As shown in Fig. 2A, each plasmid sequence consistently produced a robust MFI signal, but the values varied considerably for each replicon. The relative signal produced did not correlate with the number of cytosines in the ASPE-amplified strand (and hence the number of biotin-dCTP molecules incorporated [data not shown]), the lengths of the amplicons, the Tms of primers (see Tables S1 and S2 in the supplemental material), or the overall GC content of the amplified region. We expect that the observed differences reflect disparity in the relative amplification efficiencies of the PCRs, as is commonly observed in comparing the PCR efficiencies of different sequences. Therefore, we routinely normalized the MFI value obtained for each B. burgdorferi replicon by dividing the MFI by the corresponding RPP MFI value obtained in the same assay.
FIG. 2.

Reproducibility of Luminex plasmid assay. (A) Mean MFI values obtained using RPP templates. The MFI values for each B. burgdorferi replicon in 10 independent experiments were averaged, and standard errors (SE) were calculated. (B) A culture of the B. burgdorferi strain B31-5A2 was assayed 10 times. The mean MFI values and standard errors are presented. (C) The MFI values were normalized by dividing the MFI value for the B. burgdorferi sample by the MFI value for the RPP from the same experiment. The A and B designations for plasmids lp25 and lp28-1 represent the two different primer sets that were used for these two plasmids.
To establish the reproducibility of the LPAA, we repeated the assay 10 times on a single sample, a bacterial culture of the previously characterized strain B31-5A2 (41). As shown in Fig. 2B, the observed MFI varied very little from assay to assay for any given sample, and each replicon tested as consistently present or absent. In addition, the maximal MFI values were similar for the RPP DNA templates and for B. burgdorferi culture templates (compare Fig. 2A and B), yielding normalized MFI ratios near 1.0 (Fig. 2C). When a B. burgdorferi replicon was absent, the normalized value was typically below 0.05; thus, there was a clear difference between positive and negative results in most cases.
To more clearly define criteria for establishing whether a replicon was present or absent, we performed a detailed analysis of the LPAA results obtained for 4,464 B. burgdorferi transformants and 44 B. burgdorferi B31 clones. Histograms of the normalized MFI values for each replicon were generated (see Fig. S1 and S2 in the supplemental material). Most replicon-primer pair combinations generated bell-shaped curve histograms, and we chose a normalized MFI value as a cutoff, below which a call of negative was generated (see Table S3 in the supplemental material). We found that for most replicon-primer pair combinations, a normalized MFI of >0.2 was consistent with the plasmid being present, whereas a normalized MFI of ≤0.2 was indicative of plasmid absence. For several replicons (cp32-6, cp32-7, and lp21), a lower cutoff of 0.15 was chosen due to low normalized MFI values at the histogram peak.
For 10 replicons, there was a region in which the normalized MFI could not be identified unambiguously as positive or negative. In these cases, an additional range was designated intermediate (see Table S3 in the supplemental material). For example, for cp26, a normalized MFI value of >0.2 but <0.5 was designated intermediate. The histogram of the normalized MFI values for cp26 formed a tight peak centered at 1.0. Values in the intermediate range were rarely seen (5 of 4,464 transformants). The intermediate designation indicates that those clones should be reexamined by the LPAA or other means before a final call of positive or negative is made for that replicon. Using the same reasoning, intermediate designations were also chosen for replicons cp9, cp32-8, cp32-9, lp17, lp28-3, lp36, and lp38.
The histograms derived for both sets of lp28-1 primers indicated that many transformants fell into an intermediate normalized MFI range (101 of 4,464 transformants). Given the importance of this replicon for infectivity and the relatively high rate of replicon loss (7% [see below]), a wide range of normalized MFI values were chosen as intermediate (0.2 to 0.55). In addition, examination of the data generated for the 4,464 transformants indicated that the lp28-1B primers were less effective at generating normalized MFI values near 1.0 than the lp28-1A primers (see Fig. S1 and S2 in the supplemental material). The histogram peak for primer pair B is centered over a value of 0.5 and trails into very low values, making this primer pair of limited value in calling the replicon positive or negative. Thus, we have discontinued the use of primer pair B and use the lp28-1A primers to evaluate the presence of lp28-1. Similarly, the lp25B primer set provided a clearer distinction between positive and negative values than the lp25A set. We also subjected 44 B. burgdorferi B31 clones to the plasmid analysis assay (see Table S4 and Fig. S2 in the supplemental material). The clones were obtained by colony formation after 5 in vitro passages (40, 41) of the uncloned B31 isolate. Nineteen of these clones, B31-5A1 through B31-5A19, had been analyzed for plasmid content previously (41). In addition, three noninfectious high-passage B31 clones (43) were subjected to the Luminex plasmid analysis assay. For each of the 19 previously characterized low-passage clones, the plasmid complement detected in the Luminex assay was identical to that previously characterized by PCR and agarose gel electrophoresis (41) (see Table S4). Figure 3 shows the normalized MFI values for four representative strains. For example, panel A shows the normalized MFI values for B31-5A3 (41), which lacks plasmids cp32-3, lp28-2, and lp56. The high-passage strains B312, B313, and B314 all retained cp26, as previously described (43), as well as cp32-1, cp32-3, and cp32-4, but no other plasmids were consistently retained (Fig. 3D; see Table S4 and Fig. S3).
FIG. 3.

Plasmid content analysis of B. burgdorferi B31 clones by using LPAA. Diluted liquid cultures or scrapings of frozen B. burgdorferi stocks were used as templates. The MFI for each B. burgdorferi replicon was normalized by dividing culture MFI values by RPP MFI values. (A) B31-5A3 lacks replicons cp32-3, lp28-2, and lp56. (B) B31-5A8 lacks replicon lp28-1. (C) B31-5A9 lacks replicons lp5 and lp56. (D) B312 is a high-passage strain that contains only the chromosome (BB147), cp26, cp32-1, cp32-3, cp32-4, and lp54.
Sensitivity of Luminex plasmid analysis assay.
To examine the sensitivity of the LPAA, we tested the procedure using different amounts of template DNA. The RPP control pool was serially diluted to provide templates with known concentrations, ranging from 1 million molecules to ∼1 molecule, for the multiplex PCRs. Figure 4A shows normalized MFI values obtained using the RPP preparations for pool 1 target sequences. The normalized MFI for most amplicons showed a clearly detectable signal at 100 copies of each DNA template and reached near maximum at about 1,000 DNA copies (Fig. 4A). Raw MFI values could not be increased by further increasing the amount of template. Similar results were obtained for multiplex pools 2 and 3 (data not shown). The data also demonstrated that the MFI levels obtained with each template-primer combination had different maximal asymptotic levels. For example, lp28-3 had the lowest maximal level, whereas cp26 exhibited the highest maximal level (data not shown). These results indicated that the differences in signal observed could not be altered by increasing template concentrations, reinforcing the concept that the observed differences were due to varied PCR or labeling efficiency. Thus, the LPAA provides excellent sensitivity and is relatively unaffected, above an input threshold, by differences in the amount of input DNA.
FIG. 4.

Sensitivity of B. burgdorferi Luminex plasmid analysis assay. (A) The RPP containing the PCR amplicons was diluted serially and used as a template in the Luminex assay. The MFIs were normalized by dividing culture MFI values by RPP MFI values. (B) Cultures of B. burgdorferi strain B31-5A4, which contains all replicons except for lp5, were used as templates. Spirochete concentrations of the cultures were determined, and cultures were serially diluted to yield the indicated number of organisms in each reaction. MFIs were normalized by dividing culture MFI values by RPP MFI values.
The sensitivity of the assay was then examined by using cultured organisms as a template. Cultures of B. burgdorferi with known plasmid contents were grown to mid-log phase. Concentrations were determined by light microscopy, and serial dilutions in culture medium were made such that a known number of organisms could be used for each Luminex reaction. Total organism counts ranged from 10,000 organisms per reaction to 0.01 organism per reaction (Fig. 4B). At counts of less than 12 organisms per reaction, little significant fluorescence was detected, whereas at 25 organisms per reaction, most replicons provided a detectable signal. The number of spirochetes required for a replicon to be scored as present ranged from 12 to 50 organisms (Table 1). The MFI values became asymptotic at 100 or more organisms per reaction. With 2 μl of diluted culture added per multiplex reaction, 100 organisms represented a culture concentration of 5 × 104 B. burgdorferi organisms per ml, a concentration lower than that normally used for plasmid analysis. Therefore, the assay in this format provides robust plasmid detection that is relatively independent of the B. burgdorferi concentration. In addition, the assay can be performed without any purification or pretreatment of the organisms.
TABLE 1.
Number of B. burgdorferi organisms per reaction required for positive results in the Luminex plasmid analysis assay
| No. of organisms | LPAA result for replicona |
||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BB147 | cp32-1 | cp32-3 | cp32-7 | lp28-3 | lp28-4 | lp36 | lp56 | cp32-6 | cp32-9 | cp9 | lp21 | lp28-1 | lp38 | lp54 | cp26 | cp32-4 | cp32-8 | lp17 | lp25 | lp28-2 | |
| 10,000 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 1,000 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 100 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 50 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 25 | + | + | + | + | + | + | + | + | − | Int | Int | + | − | Int | + | Int | + | Int | + | + | + |
| 12 | − | − | − | − | − | − | − | − | − | − | Int | + | − | − | + | Int | − | − | Int | − | − |
| 6 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| 3 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| 1 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| 0.5 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| 0.25 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
Int, intermediate (see the text for more details).
It was surprising that the MFI values were asymptotic at ∼100 B. burgdorferi organisms per reaction but were below asymptotic levels at an estimated 100 copies of DNA when the recombinant plasmid pool was used as the source of template DNA (Fig. 4). This result may reflect variations in PCR efficiency due to differences in size and shape of the DNA target.
Replicon detection in mixed cultures.
B. burgdorferi readily loses plasmids during in vitro culture (2, 39, 40). To determine whether the Luminex-based assay could discriminate a mixed population of cells with varied degrees of plasmid loss, we performed plasmid analysis of mixtures of cultures of two B. burgdorferi B31 clones with different plasmid contents. The two B. burgdorferi strains were mixed together as indicated in Fig. 5 and were used as a template for the Luminex assay. B31-5A2 (which lacks lp56, lp28-1, and cp9) was mixed with various amounts of B31-5A4, which contains the replicons absent in B31-5A2. Each multiplex reaction mixture contained a total of 1,000 organisms. The normalized MFIs obtained with lp36 (present in both strains) and the three plasmids not present in B31-5A2 are depicted in Fig. 5. As the number of B31-5A4 spirochetes increased, the normalized MFI values for the additional plasmids it contained also increased. Very few organisms were required to reach the normalized MFI maximum for cp9: as few as 50 organisms containing this replicon (in a pool of 1,000) were sufficient for the corresponding normalized MFI ratios to exceed the cutoff for a positive call (Table 2). The normalized MFI values for the lp56 and lp28-1 primer pairs exceeded this threshold when 250 B31-5A4 organisms were present. In addition, the normalized MFI values increased in a relatively linear fashion over a range of 10 to 500 organisms for these replicons. Similar data were obtained when B31-2599 (which also contains all plasmids) was used as the spiking strain in place of B31-5A4 (data not shown). These results indicate that a replicon may be detected if it is present in 25% of cells or fewer but that the sensitivity may vary depending on the replicon and the primer set used.
FIG. 5.
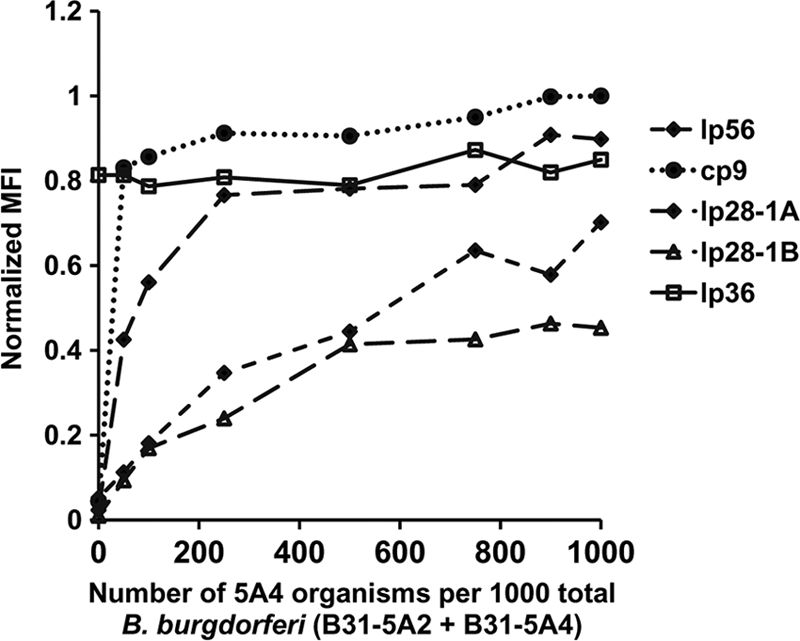
Detection of B. burgdorferi replicons in mixed cultures. B. burgdorferi strain B31-5A2, which lacks cp9, lp56, cp28-1, and lp28-1, was mixed with strain B31-5A4, which contains those replicons. Each sample contained 1,000 organisms, with different proportions of B31-5A2 and -5A4. The mixed cultures were used as templates for the LPAA. Normalized MFI values from these cultures were graphed against the number of organisms from strain B31-5A4 in each sample.
TABLE 2.
Number of B31-5A4 organisms (containing all plasmids) in a mixed population with B31-5A2 (cp9− lp56− lp28-1−) required to yield positive results in the Luminex plasmid analysis assay
| No. of contaminated organisms | LPAA result for replicona |
|||
|---|---|---|---|---|
| lp56 | cp9 | lp28-1A | lp28-1B | |
| 1,000 | + | + | + | + |
| 900 | + | + | + | + |
| 750 | + | + | + | + |
| 500 | + | + | + | + |
| 250 | + | + | + | + |
| 100 | − | + | Int | − |
| 50 | − | + | Int | − |
| 0 | − | − | − | − |
Int, intermediate (see the text for more details).
Frequency of plasmid loss.
Although the occurrence of plasmid loss in Borrelia species was reported several years ago, little information is available regarding the relative stability of individual plasmids. The LPAA allowed us to conduct a high-throughput examination of the frequency of plasmid loss. We analyzed the 44 clones obtained by passage of the uncloned B31 strain 5 times in vitro and isolation of clones by colony formation (Fig. 6A; see Table S4 in the supplemental material). Also, we examined the 4,464 individual transformants isolated by colony formation during the STM study (Fig. 6B). In the STM study, the parental strain used for transposon mutagenesis was B. burgdorferi 5A18NP1, which lacks lp56 and lp28-4 and contains a kanamycin resistance cassette inserted in the lp25-carried putative restriction enzyme gene bbe02 (29). The lp56 gene bbq67 and bbe02 have been implicated in the exceedingly low transformation rates of low-passage infectious B. burgdorferi strains, and 5A18NP1 retains infectivity but has a much higher transformation rate (29, 34). Both gentamicin (to select for transposon insertion) and kanamycin (to select for the Δbbe02::flgBP-Kanr locus in lp25 and hence preserve the required virulence determinant pncA) were used in the isolation of STM clones. Therefore, only a single lp25− clone (presumably a spontaneous Kanr mutant) was isolated during the creation of the STM library. An additional consideration in the evaluation of the STM library results was the possible occurrence of false-negative results due to transposon insertions in the LPAA target sites. Because the transposon insertion sites had also been mapped precisely by sequencing, we were able to determine that 50 clones had a transposition event that was within the LPAA target region for that plasmid. Only 5 of the clones had insertions within the forward and reverse primer sequences used for the initial amplification or within the ASPE primer sequence. However, the MFI obtained for the affected target sequence was reduced in 36 of these clones, in many cases to background levels. The plasmid content data were corrected for this occurrence.
FIG. 6.

Frequency of plasmid loss. (A) Samples of 44 clones obtained from subcloning an uncloned passage 5 B. burgdorferi B31 culture were analyzed using the Luminex plasmid content assay. The percentage of clones that had lost each replicon is presented. (B) A total of 4,464 transformants obtained from an STM screen were analyzed using the Luminex plasmid content assay. The percentage of clones that had lost each replicon is presented.
Similar results were obtained with the low-passage B31 clones and the STM library populations, with the exception of the absence of lp56 and lp28-4 and the nearly universal presence of lp25 in the STM study (Fig. 6). lp5 was the most frequently lost plasmid and was absent in 39% of the B31 clones and 43% of the STM transformants. lp28-1, which carries the vls antigenic variation system and is required for full infectivity in mice, was lost in 27% of clones obtained by passage and cloning, whereas it was lost less frequently in the 4,464 transformants analyzed (7%). lp25, another plasmid required for infectivity (21, 41), was absent in 23% of the passage 5 B31 clones. Other replicons that were frequently lost included cp9 (absent in 14% and 13% of B31 clones and transformants, respectively), lp56 (absent in 36% of B31 clones), lp28-4 (absent in 11% of B31 clones), and lp21 (lacking in 5% and 7% of B31 clones and transformants, respectively). As expected from previous work by others (10, 27, 55), low levels of replicon loss were observed for the plasmids cp32-1, cp32-3, and cp32-4 (0.1%, 0.8%, and 0.5%, respectively), and cp26 and lp54 were consistently retained by all isolates. The remaining plasmids were absent in a small percentage of clones among the 4,000+ clones in the STM library, although several were consistently present in the 44 B31 clones.
We further analyzed the average number of plasmids lost in each clone (Fig. 7). Because lp5 was lost at a high frequency and has no apparent effect on infectivity (data not shown), the results including and excluding the lp5 data are shown. Surprisingly, only 14% of the clones in the B31 population and 40% of the STM transformants retained all of the plasmids present in the parent strain; these proportions increased to 27% and 69%, respectively, when the lp5 data were excluded. A high percentage of clones had lost 1 or 2 plasmids, but some were missing as many as 4 or 5 plasmids. The mean (± standard deviation [SD]) number of plasmids lost in the B31 clone population was 1.64 ± 1.06 (1.25 ± 0.99 if excluding lp5 data). For the STM library, the mean value was 0.81 ± 0.81 (0.38 ± 0.62 if excluding lp5 data).
FIG. 7.

Number of plasmids lost per B. burgdorferi clone. The histograms represent the percentages of clones that had lost the indicated numbers of replicons. (A) Number of replicons lost from 44 clones obtained by subcloning passage 5 B. burgdorferi B31. (B) Results obtained with 4,464 transformants from an STM library.
The high-passage, noninfectious strains B312, B313, and B314 were notable for the few replicons that remained (see Table S4 and Fig. S3 in the supplemental material). All clones retained the chromosome (BB147), cp26, cp32-1, cp32-3, and cp32-4, indicating that these replicons may be required for in vitro growth, as reported previously (11, 27, 30, 55). Each clone contained only one or two other replicons.
Application of LPAA to other species and strains.
We tested B. garinii Ip90 and B. afzelii ACA-1 in the LPAA. The chromosome and two plasmids (B31 plasmids lp54 and cp26) are colinear between the three sequenced genospecies. In addition, other groups of plasmids share large colinear sequences. We found that few replicons were detected (BB147, cp26, and lp38 in the case of B. garinii and BB147 and lp54 in the case of B. afzelii). These data were consistent with the poor conservation of the B31-derived primer sequences in the available B. garinii and B. afzelii sequences, as determined by BLAST analysis. We also tested B. burgdorferi strains 297, C57, N-40, B247, and NCH-1 in the LPAA (data not shown). These strains yielded positive results for between 8 and 12 replicons. Every strain was positive for the chromosome, lp54, cp26, and lp17. These results indicate, not surprisingly, that adaptation of the LPAA to other strains would require modification of the primer sequences to match the plasmid DNA sequences of each strain.
Cost analysis.
In an effort to reduce the cost of each plasmid content determination, titrations of costly assay reagents were performed. We found that the volumes of the multiplex PCR and ASPE reaction mixtures could be reduced 2-fold with comparable assay results (data not shown). In addition, the concentrations of both the ExoSapIT nuclease and the xTAG microspheres could be reduced 4-fold, with no change to the assay results (data not shown). These modifications reduced the estimated reagent cost for the LPAA from $17.85 to $9.00 per sample. While these reagent costs are considerably higher than those for plasmid analysis using PCR and agarose gel electrophoresis, the time per sample, and hence the labor costs, are substantially lower.
DISCUSSION
Borrelia species have the largest complement of extrachromosomal replicons of any characterized bacterium (9, 18). Genes required for in vitro growth and/or the infectious cycle are located across both the chromosome and the plasmids, and loss of plasmids during in vitro manipulation can complicate the analysis of the roles of borrelial genes in infection and other biological functions. Here we describe a high-throughput method for determining the plasmid content of B. burgdorferi by utilizing multiplex PCR with replicon-specific primers followed by detection using Luminex xTAG technology.
The primer set utilized in this study was based on the B. burgdorferi B31-MI genome sequence and thus permits identification of the plasmids present in that sequence. Two additional 32-kb replicons (cp32-2 and cp32-5) and an additional cp9 replicon (cp9-2) have been described for B31-derived clones (8, 55). Sequences corresponding to these plasmids were not detected in the B31-MI genome sequence, most likely because those replicons were not present in that B31 derivative. Limited sequences are available for cp32-2, cp32-5, and cp9-2 (10, 37), so in future iterations of the LPAA we will attempt to include the detection of these plasmids. The results we obtained with other Lyme disease Borrelia strains verified that the LPAA in its current format would not be suitable for assessing plasmid content in strains other than B31; primers need to be chosen carefully for each B. burgdorferi strain to obtain an accurate assessment of replicon content. Our analysis of the plasmid content of a large set of B. burgdorferi clones provided precise statistics on the frequency of plasmid loss. The B31 clones were obtained after five in vitro passages of the uncloned B31 strain, originally cultured from a tick from Shelter Island, NY (5). The consistency of genomic sequencing results (18, 54) suggests that B31 represents the progeny of a single B. burgdorferi lineage. It is unclear, however, to what degree the observed plasmid content differences and other genetic heterogeneity (such as OspB truncation [15]) were present in the initial culture inoculum from the tick midgut. In the case of our study of B31 5A18NP1 transposon transformants, 5A18NP1 was a single clone isolated by colony formation after targeted mutagenesis of bbe02 with a suicide vector (29). Therefore, the observed plasmid content variation occurred at some point following the initial outgrowth of the colony. In both the B31 clones and the STM library, the clones had undergone five in vitro passages and ∼10 doublings per passage, or an estimated 50 generations, prior to testing for plasmid content. Given that 1 or 2 plasmids had been lost in both populations, on average, plasmid loss must be a high-frequency event occurring at a rate of roughly 0.02 plasmid lost per cell-generation.
As observed previously (15, 32, 41, 52), plasmids vary widely in stability. The smallest plasmid, lp5, appears to derive from one or more fragments of lp21, along with sequences homologous to lp56. Its high sequence similarity to other plasmids makes effective PCR primer design challenging, so the presence of lp5 was not examined in most prior plasmid content analyses. In this report, lp5 was the plasmid that was lost with the highest frequency (39% of B31 clones and 43% of STM transformants); a high frequency of lp5 loss was also observed in a previous study by Elias et al. (15). Loss of lp5 has no apparent effect on mouse infectivity (unpublished results), but its role in tick colonization or transmission has not been reported.
lp25 and lp28-1 are readily lost during in vitro culture but are required for full infectivity of B. burgdorferi B31 (15, 21, 32, 33, 41, 52). Consistent with this prior work, we found that lp25 was absent from 23% of the B31 clones and that lp28-1 was missing from 27% and 7% of the B31 clone and STM library populations, respectively. lp56, cp9, lp28-4, lp28-2, and lp21 were also lost at relatively high rates (Fig. 6). The absence of these plasmids does not result in any apparent growth defect, so they do not appear to be required for in vitro culture in BSK II medium. The remaining plasmids (the seven cp32 plasmids, lp28-3, lp36, and lp38) were each absent in small numbers of clones (ranging from 4 to 135 clones) in the STM library, which because of the large number of clones provided a more sensitive measure of low rates of plasmid loss.
These findings reinforce previously described results indicating that cp26 and lp54 are extremely stable (8, 11, 27, 30, 43). cp32 plasmids are also retained in high-passage B31 clones (Fig. 3; also see Fig. S3 in the supplemental material); B313 has also been reported to contain cp32-2 (8), which was not assayed in our study. cp32s are thought to represent closely related prophages of the lysogenic bacteriophage φBB-1 (11, 13, 47). Packaging and transduction of cp32 DNA have been confirmed (13), but thus far, lytic infection has not been demonstrated reproducibly. The long-term retention of cp32s indicates that these plasmids are required for in vitro growth, are unusually stable, or (potentially) are maintained in the bacterial population by transduction. Because the clones B312, B313, and B314 were all derived from the same parent culture, it is not known whether cp32-1, cp32-3, and cp32-4 are preferentially retained in comparison to the other cp32s; it is possible that a particular subset of plasmids was retained by chance in the high-passage culture (10).
In a comprehensive article, Grimm et al. (22) determined the effects of in vitro passage and cryogenic storage on plasmid retention in two low-passage, infectious B. burgdorferi B31 clones in which 21 plasmids were initially detected. The borreliae were cultured at either 35°C or 23°C and were examined at 3 passage (∼27 generations) intervals for up to 25 passages. The plasmids that were absent following culture at 35°C were (in order of decreasing frequency) cp9, lp28-4, lp25, lp21, lp17, and lp28-1. These results are similar to those reported for our B31 clone population (Fig. 6A), although our clones were derived from a passage 5, uncloned B31 culture rather than from a single isogenic progenitor. We additionally observed a high rate of lp5 and lp56 loss and a higher frequency of lp28-1 loss (Fig. 6A).
The histogram in Fig. 7 underscores the instability of B. burgdorferi plasmids during in vitro culture. Only 14% of the B31 clones and 40% of the STM library had retained all plasmids. The apparently lower rate of plasmid loss for the STM group was due in part to the lack of lp28-4 and lp56 in the 5A18NP1 parent clone (and hence the exclusion of these plasmids from the calculation) as well as to positive selection for lp25 via kanamycin resistance. The difference in plasmid loss between the two groups was not statistically significant. It is a positive sign that over 91% of the STM library clones contained lp25, lp28-1, lp36, and lp54, plasmids known to be required for the infectious cycle. Nevertheless, the results emphasize the need for close scrutiny of plasmid content.
The primer pairs used here were designed to maximize specificity for replicon identification. However, it would be possible to develop primer sets with different criteria, for example, to assay for virulence factors or to identify B. burgdorferi species in clinical samples. A number of studies have indicated that different B. burgdorferi species and strains produce distinct clinical manifestations (6, 50) and that coinfections with more than one strain can occur (17, 36, 50). A Luminex-based assay may be useful in rapid strain identification.
Luminex-based approaches should prove useful in evaluating the plasmid loss and genetic content of Lyme disease Borrelia and other organisms with unstable genomes, as well as the relationship between these genetic changes and pathogenesis and other biological properties.
Supplementary Material
Acknowledgments
We thank James Jacobson, Keld Sorenson, Sherry Dunbar, Michaela Hoffmeyer, and Jennifer Svoboda of the Luminex Corporation, Austin, TX, for providing equipment, supplies, and invaluable advice for this study. We also thank Erol Fikrig and Xin Li for sharing primer sequences and Kerry Gunning of IDT for helpful discussions on primer design.
This work was supported by grants RO1 AI37277 and RO1 AI 59048 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Published ahead of print on 17 December 2010.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Barbour, A. G. 1984. Isolation and cultivation of Lyme disease spirochetes. Yale J. Biol. Med. 57:521-525. [PMC free article] [PubMed] [Google Scholar]
- 2.Barbour, A. G. 1988. Plasmid analysis of Borrelia burgdorferi, the Lyme disease agent. J. Clin. Microbiol. 26:475-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbour, A. G., and D. Fish. 1993. The biological and social phenomenon of Lyme disease. Science 260:1610-1616. [DOI] [PubMed] [Google Scholar]
- 4.Bestor, A., et al. 2010. Use of the Cre-lox recombination system to investigate the lp54 gene requirement in the infectious cycle of Borrelia burgdorferi. Infect. Immun. 78:2397-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burgdorfer, W., et al. 1982. Lyme disease, a tick-borne spirochetosis? Science 216:1317-1319. [DOI] [PubMed] [Google Scholar]
- 6.Canica, M. M., et al. 1993. Monoclonal antibodies for identification of Borrelia afzelii sp. nov. associated with late cutaneous manifestations of Lyme borreliosis. Scand. J. Infect. Dis. 25:441-448. [DOI] [PubMed] [Google Scholar]
- 7.Casjens, S. 1999. Evolution of the linear DNA replicons of the Borrelia spirochetes. Curr. Opin. Microbiol. 2:529-534. [DOI] [PubMed] [Google Scholar]
- 8.Casjens, S., M. Delange, H. L. Ley III, P. Rosa, and W. M. Huang. 1995. Linear chromosomes of Lyme disease agent spirochetes: genetic diversity and conservation of gene order. J. Bacteriol. 177:2769-2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casjens, S., et al. 2000. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol. 35:490-516. [DOI] [PubMed] [Google Scholar]
- 10.Casjens, S., R. van Vugt, K. Tilly, P. A. Rosa, and B. Stevenson. 1997. Homology throughout the multiple 32-kilobase circular plasmids present in Lyme disease spirochetes. J. Bacteriol. 179:217-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casjens, S. R., C. H. Eggers, and I. Schwartz. 2010. Borrelia genomics: chromosome, plasmids, bacteriophages and genetic variation, p. 27-53. In D. S. Samuels and J. D. Radolf (ed.), Borrelia: molecular biology, host interaction and pathogenesis. Caister Academic Press, Hethersett, Norwich, United Kingdom.
- 12.Dever, L. L., J. H. Jorgensen, and A. G. Barbour. 1992. In vitro antimicrobial susceptibility testing of Borrelia burgdorferi: a microdilution MIC method and time-kill studies. J. Clin. Microbiol. 30:2692-2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eggers, C. H., et al. 2001. Transduction by phiBB-1, a bacteriophage of Borrelia burgdorferi. J. Bacteriol. 183:4771-4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elias, A. F., J. Schmutzhard, P. E. Stewart, T. G. Schwan, and P. Rosa. 2002. Population dynamics of a heterogeneous Borrelia burgdorferi B31 strain in an experimental mouse-tick infectious cycle. Wien. Klin. Wochenschr. 114:557-561. [PubMed] [Google Scholar]
- 15.Elias, A. F., et al. 2002. Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect. Immun. 70:2139-2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer, J. R., N. Parveen, L. Magoun, and J. M. Leong. 2003. Decorin-binding proteins A and B confer distinct mammalian cell type-specific attachment by Borrelia burgdorferi, the Lyme disease spirochete. Proc. Natl. Acad. Sci. U. S. A. 100:7307-7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Floris, R., et al. 2007. Evaluation of a genotyping method based on the ospA gene to detect Borrelia burgdorferi sensu lato in multiple samples of Lyme borreliosis patients. New Microbiol. 30:399-410. [PubMed] [Google Scholar]
- 18.Fraser, C. M., et al. 1997. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390:580-586. [DOI] [PubMed] [Google Scholar]
- 19.Glöckner, G., et al. 2004. Comparative analysis of the Borrelia garinii genome. Nucleic Acids Res. 32:6038-6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glöckner, G., et al. 2006. Comparative genome analysis: selection pressure on the Borrelia vls cassettes is essential for infectivity. BMC Genomics 7:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grimm, D., et al. 2004. Experimental assessment of the roles of linear plasmids lp25 and lp28-1 of Borrelia burgdorferi throughout the infectious cycle. Infect. Immun. 72:5938-5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grimm, D., A. F. Elias, K. Tilly, and P. A. Rosa. 2003. Plasmid stability during in vitro propagation of Borrelia burgdorferi assessed at a clonal level. Infect. Immun. 71:3138-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grimm, D., et al. 2004. Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc. Natl. Acad. Sci. U. S. A. 101:3142-3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo, B. P., E. L. Brown, D. W. Dorward, L. C. Rosenberg, and M. Hook. 1998. Decorin-binding adhesins from Borrelia burgdorferi. Mol. Microbiol. 30:711-723. [DOI] [PubMed] [Google Scholar]
- 25.Guo, B. P., S. J. Norris, L. C. Rosenberg, and M. Hook. 1995. Adherence of Borrelia burgdorferi to the proteoglycan decorin. Infect. Immun. 63:3467-3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hughes, C. A., C. B. Kodner, and R. C. Johnson. 1992. DNA analysis of Borrelia burgdorferi NCH-1, the first north-central U.S. human Lyme disease isolate. J. Clin. Microbiol. 30:698-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jewett, M. W., et al. 2007. Genetic basis for retention of a critical virulence plasmid of Borrelia burgdorferi. Mol. Microbiol. 66:975-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jewett, M. W., et al. 2007. The critical role of the linear plasmid lp36 in the infectious cycle of Borrelia burgdorferi. Mol. Microbiol. 64:1358-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawabata, H., S. J. Norris, and H. Watanabe. 2004. BBE02 disruption mutants of Borrelia burgdorferi B31 have a highly transformable, infectious phenotype. Infect. Immun. 72:7147-7154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobryn, K., and G. Chaconas. 2002. ResT, a telomere resolvase encoded by the Lyme disease spirochete. Mol. Cell 9:195-201. [DOI] [PubMed] [Google Scholar]
- 31.Kraiczy, P., et al. 2004. Immunological characterization of the complement regulator factor H-binding CRASP and Erp proteins of Borrelia burgdorferi. Int. J. Med. Microbiol. 293(Suppl. 37):152-157. [DOI] [PubMed] [Google Scholar]
- 32.Labandeira-Rey, M., E. Baker, and J. Skare. 2001. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect. Immun. 69:446-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Labandeira-Rey, M., J. Seshu, and J. T. Skare. 2003. The absence of linear plasmid 25 or 28-1 of Borrelia burgdorferi dramatically alters the kinetics of experimental infection via distinct mechanisms. Infect. Immun. 71:4608-4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawrenz, M. B., H. Kawabata, J. E. Purser, and S. J. Norris. 2002. Decreased electroporation efficiency in Borrelia burgdorferi containing linear plasmids lp25 and lp56: impact on transformation of infectious B. burgdorferi. Infect. Immun. 70:4798-4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lescot, M., et al. 2008. The genome of Borrelia recurrentis, the agent of deadly louse-borne relapsing fever, is a degraded subset of tick-borne Borrelia duttonii. PLoS Genet. 4:e1000185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liveris, D., et al. 1999. Genetic diversity of Borrelia burgdorferi in Lyme disease patients as determined by culture versus direct PCR with clinical specimens. J. Clin. Microbiol. 37:565-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller, J. C., et al. 2000. A second allele of eppA in Borrelia burgdorferi strain B31 is located on the previously undetected circular plasmid cp9-2. J. Bacteriol. 182:6254-6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moody, K. D., S. W. Barthold, and G. A. Terwilliger. 1990. Lyme borreliosis in laboratory animals: effect of host species and in vitro passage of Borrelia burgdorferi. Am. J. Trop. Med. Hyg. 43:87-92. [DOI] [PubMed] [Google Scholar]
- 39.Norris, S. J., C. J. Carter, J. K. Howell, and A. G. Barbour. 1992. Low-passage-associated proteins of Borrelia burgdorferi B31: characterization and molecular cloning of OspD, a surface-exposed, plasmid-encoded lipoprotein. Infect. Immun. 60:4662-4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Norris, S. J., J. K. Howell, S. A. Garza, M. S. Ferdows, and A. G. Barbour. 1995. High- and low-infectivity phenotypes of clonal populations of in vitro-cultured Borrelia burgdorferi. Infect. Immun. 63:2206-2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Purser, J. E., and S. J. Norris. 2000. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc. Natl. Acad. Sci. U. S. A. 97:13865-13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radolf, J. D., J. C. Salazar, and R. J. Dattwyler. 2010. Lyme disease in humans, p. 487-533. In D. S. Samuels and J. D. Radolf (ed.), Borrelia: molecular biology, host interaction and pathogenesis. Caister Academic Press, Hethersett, Norwich, United Kingdom.
- 43.Sadziene, A., B. Wilske, M. S. Ferdows, and A. G. Barbour. 1993. The cryptic ospC gene of Borrelia burgdorferi B31 is located on a circular plasmid. Infect. Immun. 61:2192-2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schutzer, S. E., et al. 8 October 2010. Whole genome sequences of thirteen isolates of Borrelia burgdorferi. J. Bacteriol. doi: 10.1128/JB.01158-10. [DOI] [PMC free article] [PubMed]
- 45.Simpson, W. J., C. F. Garon, and T. G. Schwan. 1990. Analysis of supercoiled circular plasmids in infectious and non-infectious Borrelia burgdorferi. Microb. Pathog. 8:109-118. [DOI] [PubMed] [Google Scholar]
- 46.Steere, A. C. 2001. Lyme disease. N. Engl. J. Med. 345:115-125. [DOI] [PubMed] [Google Scholar]
- 47.Stevenson, B., W. R. Zuckert, and D. R. Akins. 2000. Repetition, conservation, and variation: the multiple cp32 plasmids of Borrelia species. J. Mol. Microbiol. Biotechnol. 2:411-422. [PubMed] [Google Scholar]
- 48.Stewart, P. E., and P. A. Rosa. 2008. Transposon mutagenesis of the Lyme disease agent Borrelia burgdorferi. Methods Mol. Biol. 431:85-95. [DOI] [PubMed] [Google Scholar]
- 49.Strother, K. O., A. Broadwater, and A. De Silva. 2005. Plasmid requirements for infection of ticks by Borrelia burgdorferi. Vector Borne Zoonotic Dis. 5:237-245. [DOI] [PubMed] [Google Scholar]
- 50.van Dam, A. P., et al. 1993. Different genospecies of Borrelia burgdorferi are associated with distinct clinical manifestations of Lyme borreliosis. Clin. Infect. Dis. 17:708-717. [DOI] [PubMed] [Google Scholar]
- 51.Xu, Q., et al. 2005. Association of linear plasmid 28-1 with an arthritic phenotype of Borrelia burgdorferi. Infect. Immun. 73:7208-7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu, Y., C. Kodner, L. Coleman, and R. C. Johnson. 1996. Correlation of plasmids with infectivity of Borrelia burgdorferi sensu stricto type strain B31. Infect. Immun. 64:3870-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang, X. F., U. Pal, S. M. Alani, E. Fikrig, and M. V. Norgard. 2004. Essential role for OspA/B in the life cycle of the Lyme disease spirochete. J. Exp. Med. 199:641-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang, J. R., J. M. Hardham, A. G. Barbour, and S. J. Norris. 1997. Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell 89:275-285. [DOI] [PubMed] [Google Scholar]
- 55.Zückert, W. R., J. Meyer, and A. G. Barbour. 1999. Comparative analysis and immunological characterization of the Borrelia Bdr protein family. Infect. Immun. 67:3257-3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.