

Abstract
In this study, we combined metabolic reconstruction, growth assays, and metabolome and transcriptome analyses to obtain a global view of the sulfur metabolic network and of the response to sulfur availability in Brevibacterium aurantiacum. In agreement with the growth of B. aurantiacum in the presence of sulfate and cystine, the metabolic reconstruction showed the presence of a sulfate assimilation pathway, thiolation pathways that produce cysteine (cysE and cysK) or homocysteine (metX and metY) from sulfide, at least one gene of the transsulfuration pathway (aecD), and genes encoding three MetE-type methionine synthases. We also compared the expression profiles of B. aurantiacum ATCC 9175 during sulfur starvation or in the presence of sulfate. Under sulfur starvation, 690 genes, including 21 genes involved in sulfur metabolism and 29 genes encoding amino acids and peptide transporters, were differentially expressed. We also investigated changes in pools of sulfur-containing metabolites and in expression profiles after growth in the presence of sulfate, cystine, or methionine plus cystine. The expression of genes involved in sulfate assimilation and cysteine synthesis was repressed in the presence of cystine, whereas the expression of metX, metY, metE1, metE2, and BL613, encoding a probable cystathionine-γ-synthase, decreased in the presence of methionine. We identified three ABC transporters: two operons encoding transporters were transcribed more strongly during cysteine limitation, and one was transcribed more strongly during methionine depletion. Finally, the expression of genes encoding a methionine γ-lyase (BL929) and a methionine transporter (metPS) was induced in the presence of methionine in conjunction with a significant increase in volatile sulfur compound production.
Brevibacteriaceae play a major role in the cheese microbial community (36, 40) and may constitute over 5% of the total bacterial counts of cheese surfaces (5). Brevibacterium linens and Brevibacterium aurantiacum strains have been used for a long time as ripening starters by the cheese industry. These bacteria are used for their ability to produce orange pigmentation and to release a variety of sulfur aromas during cheese ripening and for their inhibitory potential against food pathogens. B. linens and B. aurantiacum produce volatile sulfur compounds (VSCs), which are key aromas of cheese flavor. The metabolic pathways leading to VSC production have been investigated (1, 2, 14, 15). During the ripening process, amino acids such as methionine are gradually released due to the hydrolysis of casein by lactic acid bacteria (LAB) and yeasts (7, 12). In Brevibacteriaceae, VSCs arise from the degradation of methionine to methanethiol (MTL) by a methionine γ-lyase. MTL is then used as a common precursor for a wide variety of VSCs found in cheese, including dimethyl disulfide (DMDS), dimethyl trisulfide (DMTS), and S-methylthioesters (6). However, little is known about the central sulfur metabolism in Brevibacteriaceae (27).
Microorganisms frequently use sulfate for the synthesis of organic sulfur metabolites, mostly cysteine, methionine, and S-adenosylmethionine (SAM). The sulfate assimilation pathway involves uptake and activation of inorganic sulfate, followed by a stepwise reduction to sulfite. Sulfite could also be obtained from organic sulfonates via their oxidation by monooxygenases (48). Sulfite is then converted into sulfide by a sulfite reductase (48). An O-acetylserine-thiol-lyase, CysK, further catalyzes the reaction of sulfide and O-acetylserine (OAS) to generate cysteine (48). Homocysteine could be obtained from O-acylhomoserine and cysteine by the transsulfuration pathway via the intermediary formation of cystathionine or directly from O-acylhomoserine and sulfide by the thiolation pathway (24). Cystathionine γ-synthase and cystathionine β-lyase are required for transsulfuration, whereas O-acylhomoserine thiol-lyase is involved in the thiolation pathway. Homocysteine is then methylated by methionine synthases to form methionine (28). Methionine is also a sulfur source for several bacteria, indicating an efficient conversion of methionine into cysteine, usually through the SAM recycling pathway and then reverse transsulfuration (Fig. 1). This pathway, present in mammals, yeasts, Pseudomonas aeruginosa, and Bacillus subtilis, requires the sequential action of a cystathionine β-synthase and a cystathionine γ-lyase (48, 55). Sulfur-containing compounds are also efficiently taken up by specific transporters. Three main families of inorganic sulfate transporters are described (23, 29, 57). Moreover, ABC transporters and a symporter participate in the uptake of cystine in B. subtilis and Escherichia coli (9). Methionine can be taken up by high-affinity ABC transporters of the MUT family (48) or by a recently characterized MetPS symporter in Corynebacterium glutamicum (53). Finally, two different types of ABC transporters, the TauABCD and the SsuABC systems, are involved in sulfonate uptake (26, 48, 54).
FIG. 1.

Sulfur metabolism in B. aurantiacum. The BL numbers for B. aurantiacum genes correspond to those of Agmial (http://genome.jouy.inra.fr/∼vloux/publications/forquin08/) (8). A systematic search for E. coli, B. subtilis, C. glutamicum, and M. tuberculosis genes involved in sulfur metabolism was performed. The high-GC-content Gram-positive bacteria C. glutamicum and M. tuberculosis are phylogenetically related to Brevibacterium strains (33, 49). The genes were renamed according to B. subtilis and C. glutamicum orthologues. “?” indicates a step or a pathway for which the gene proposed remains to be more clearly identified. (A) Transport of sulfur compounds. Methionine transporters (MetNIQ, MetPS), cyst(e)ine transporters (BL1249 to BL1251, BL1293 to BL1296), sulfate transporters and sulfonate transporters are indicated. (B) Central sulfur metabolism. ATP sulfurylase, cysDN; APS reductase, cysH; sulfite reductase, cysJI; serine O-acetyltransferase, cysE; OAS thiol-lyase, cysK; homoserine dehydrogenase, hom; homoserine acetyl-transferase, metX; OAH thiol-lyase, metY; cystathionine β-lyase, aecD; cystathionine γ-synthase, metB; methionine synthase, metE1-3; methionine γ-lyase, mgl; SAM synthetase, metK; SAH hydrolase, sahH; alkanesulfonate monooxygenase, ssuD1-2; flavin mononucleotide (FMN) reductase, ssuI; FMNH2-dependent alkanesulfonate monooxygenase, seuA; electron transfer flavoprotein, fixA; glycosyltransferase, mshA; MSH deacetylase, mshB; MSH ligase, mshC; MSH synthase, mshD; MSH disulfide reductase, mtr; NAD/mycothiol-dependent formaldehyde dehydrogenase/nitrosothiol reductase, mscR; MSH-S-conjugate amidase, mca; Ac-CoA, acetyl coenzyme A; APS, adenylyl sulfate; PPi, diphosphate; OAS, O-acetylserine; OAH, O-acetylhomoserine; MSH, mycothiol; MSSM, mycothiol disulfide; GlcN-INS, glucoaminonylinositol; THF, tetrahydrofolate; SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; α-KB, α-ketobutyrate.
The unfinished genome sequence of the B. aurantiacum ATCC 9174 strain (20) has been recently released by the Joint Genome Institute (http://genome.jgi-psf.org/). In the present study, we performed a reconstruction of sulfur metabolism in B. aurantiacum. We further analyzed the metabolic and expression profiles of a strain of B. aurantiacum after growth under conditions of sulfur starvation or in the presence of various sulfur sources.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
B. aurantiacum strains ATCC 9174 and ATCC 9175 were used in the present study. These strains were grown in a synthetic medium (SM), adapted from the medium described by Mansour et al. (30), in 100-ml flasks containing 20 ml of medium at 25°C (180 rpm). In this medium, a residual growth in the absence of any added sulfur source was observed. To avoid this problem, a sulfur-depleted medium (SDM) was obtained by growing B. aurantiacum ATCC 9175 in a sulfur-free SM, followed by centrifugation (5,000 × g, 10 min, 4°C) and filtration of the supernatant. There was a complete absence of growth of B. aurantiacum strain ATCC 9175 in SDM without an added sulfur source. SDM was supplemented with a sulfur source as stated: 500 μM sulfate, 500 μM homocysteine, 500 μM sulfite, 500 μM S-methyl-l-cysteine, 500 μM thiosulfate, 500 μM cystine, or 10 mM methionine. In cow casein, methionine is ∼10-fold more abundant than cysteine (21). We then used SDM containing 500 μM l-cystine and 10 mM methionine to mimic the ratio present in casein.
Volatile sulfur compound extraction and analysis.
The volatile compounds were analyzed by a dynamic headspace analyzer (Purge and Trap HP 3547A; Agilent Technologies, Garches, France) fitted with a sorbent trap (Tenax) and a cryofocusing module. The concentrator was coupled to a gas chromatograph (GC G1530A; Agilent Technologies) connected to a mass spectrometer detector (MSD 5973; Agilent Technologies). Portions (5 ml) of nonfiltered culture were heated to 60°C and purged for 15 min with high-purity helium at 15 ml min−1. Volatile compounds were concentrated by adsorption to the Tenax trap maintained at 40°C. The trap was heated to 225°C for 2 min to desorb the volatile compounds, which were directly transferred at 150°C to the head of a capillary column and condensed by cryofocusing at −150°C. The condensed volatile compounds were then injected by heating (180°C) and analyzed as previously described (2). The concentrations of each volatile compound were quantified by using a standard curve of pure DMDS (Sigma-Aldrich, Saint Quentin Fallavier, France). Measurements were made in triplicate for each concentration in the noninoculated media, and the concentrations of volatile compounds were expressed as equivalents of DMDS in μg kg−1.
RNA extraction, quantitative real-time reverse transcription-PCR (qRT-PCR), and 5′RACE (rapid amplification of cDNA ends) analysis.
Exponentially growing cells (optical density at 600 nm of 5) were collected and broken by shaking in a FastPrep apparatus (Bio101; Qbiogene, Illkirch, France). Total RNA was extracted from cells of three independent cultures for each growth condition. The total RNA was then extracted by TRIzol treatment (Invitrogen, Carlsbad, CA), as described previously (22). RNA quality was assessed with an Agilent 2100 Bioanalyzer. After RNA extraction, the cDNAs were synthesized by using the SuperScript III first-strand synthesis system (Invitrogen) according to the manufacturer's recommendations. The primers used are listed in Table S1 in the supplemental material. In each sample, the quantity of cDNA for each gene was normalized to the quantity of the rpoB cDNA (35, 52), which is stably expressed in the transcriptome during growth in the presence of sulfate, cystine, and methionine plus cystine. The relative change in gene expression was recorded as the ratio of normalized target concentrations (ΔΔCT). The transcriptional start sites of the BL613, metY, BL2267, BL929, BL3001, cysK, and cysI genes were determined by using a 5′/3′RACE kit (Roche, Neuilly-sur-Seine, France) according to the manufacturer's recommendations. RNA preparations extracted from strain ATCC 9175 grown in the presence of various sulfur sources were used as a template. The primers used for 5′RACE analysis are listed in Table S1 in the supplemental material.
Transcriptome analysis.
The expression profiles of B. aurantiacum ATCC 9175 grown in the presence of various sulfur sources were analyzed by using DNA microarrays (Agilent Technologies). The DNA microarray was described in a previous study (20). RNAs were labeled with either Cy3 or Cy5 fluorescent dye (GE Healthcare, Little Chalfont, United Kingdom) by using a SuperScript Direct cDNA labeling system kit (Invitrogen) according to the manufacturer's recommendations. The levels of Cy3-dCTP and Cy5-dCTP incorporation were quantified by absorbance measurements. Hybridizations were then performed for 17 h at 65°C in dedicated microchambers with 150-pmol portions of the different samples. Array scanning was carried out with a GenePix 4000A dual-channel (635- and 532-nm) laser scanner (GenePix, Sunnyvale, CA) with a resolution of 10 nm per pixel. The laser power was set to 100, and the photomultiplier tension was adjusted to between 680 and 800 V according to the average intensity of the hybridization of each slide in order to optimize the dynamic range of measurements. The complete experimental data set was deposited in the GEO database under accession number GSE25422. The analyses were performed twice by swapping the fluorescent dyes, Cy3 and Cy5, to reduce the dye bias and the technical variance. The statistical analysis was based on two dye swaps. For each array, the raw data comprised the logarithm of median feature pixel intensity at wavelengths of 635 nm (red) and 532 nm (green). No background was subtracted. In the following description, log ratio refers to the differential expression between two conditions. It is either log2 (red/green) or log2 (green/red) according to the experiment design. An array-by-array normalization was performed to remove systematic biases. First, we excluded spots that were considered to be badly formed features. We then performed a global intensity-dependent normalization with the Loess procedure to correct the dye bias (58). Finally, for each block, the log-ratio median calculated over the values for the entire block was subtracted from each individual log-ratio value to correct print tip effects. To determine differentially expressed genes, we performed a paired t test on the log ratios implemented in the Anapuce R package (Tools for Microarray Data Analysis 2010, R package [http://www.R-project.org]; J. Aubert [INRA], Anapuce). The variance was divided between subgroups of genes with homogeneous variance (13). Statistical raw P values were adjusted for multiple comparisons by using the Benjamini-Hochberg procedure (4), which controls the false discovery rate after estimation of the proportion of differentially expressed genes by using the smoother method (50). The level of statistical significance was set at 0.05 or 0.1 for all of the comparisons. Each experiment was performed in three biological replicates and two technical replicates.
Extraction of intracellular metabolites and analysis by liquid chromatography coupled to mass spectrometry.
Cells of B. aurantiacum ATCC 9175 were grown in SDM with various sulfur sources. Exponentially growing cells (1010 bacteria) were harvested by centrifugation (5,000 × g, 2 min, 4°C). Cells were washed with 20 ml of Milli-Q water to prevent contamination with SDM, centrifuged again (5,000 × g, 2 min, 4°C), and then stored at −80°C. The cells were resuspended in 1 ml of formic acid 1%, incubated for 10 min at 95°C, and centrifuged (5,000 × g, 30 min, 4°C). The supernatant was evaporated to dryness, stored at −80°C, and then resuspended in water containing 0.1% formic acid prior to injection. Chromatographic separation was performed on a Discovery HS-F5 (2.1 by 250 mm, 5 μm) from Supelco Analytical (Interchim, Montluçon, France) by using a Surveyor LC system (Thermo Fisher Scientific, Courtaboeuf, France). Before sample injection (20 μl), the samples were resuspended with 400 μl of formic acid 1% and stored at 4°C in the tray of the autosampler. Separations were carried out using the following gradient at 200 μl min−1: 0 to 3 min, 0% solvent B; 3 to 20 min, from 0 to 100% solvent B; 20 to 25 min, 100% solvent B; and 25 to 45 min, 0% solvent B. Solvent A was water, and solvent B was acetonitrile; both contained 0.1% formic acid. The column temperature was set to 30°C. Mass spectrometric detection was performed with a LTQ/Orbitrap hybrid mass spectrometer (Thermo Fisher Scientific) fitted to an electrospray source operated in the positive and negative ionization modes. The detection was achieved from 75 to 1,000 U at the maximum resolving power of 30,000 (expressed as full width at half-maximum for an ion at 400 U). The mass spectrometer was operated with the capillary voltage at 4 kV and the capillary temperature at 275°C. The sheath gas pressure and the auxiliary gas pressure were set at 45 and 10 arbitrary units, respectively, with nitrogen gas.
Data processing and statistical analyses.
Data were processed by using the Xcalibur Qualbrowser module, version 2.0.7 (Thermo Fisher Scientifics), and its chemical formula generator was used to provide elemental formulas. Peak detection from liquid chromatography-mass spectrometry chromatograms was performed using XCMS software (47), version 1.14.1, run under R version 2.8.1. The matched filter algorithm was used, and the default values were set for all parameters except for the fwhm, step, steps, mzdiff, mzwid, bw, and snthresh parameters, which were set at 25, 0.01, 3, 0.1, 0.1, 5, and 3, respectively. The areas of the peaks detected were obtained for each sample, using a combination of the retention time and the m/z ratio as an identifier. An arbitrary number was assigned to each of these retention time-m/z pairs. The data were then combined into a single matrix by aligning peaks with the same mass-retention time pair from each data file in the data set. The XCMS datasets were annotated by using tools developed in-house: the spectral database of the laboratory that contains MS and MS/MS spectra of 300 metabolites and an informatics tool for automatic query of public metabolic and metabolomic databases (i.e., HMDB, KEGG, and METLIN) with the measured accurate masses.
The resulting data matrix was processed by the SIMCA P software (version12.0; Umetrics, Malmö, Sweden) for multivariate analyses (MVAs). The data were first mean centered and scaled either to unit variance or to Pareto variance and then subjected to principal component analysis (PCA) and projection on latent structure-discriminant analysis (PLS-DA). PLS-DA models were validated by using the cross-validation function of SIMCA P and were tested with permutation tests (k = 20). The relevance of the discriminant variables highlighted by MVAs was confirmed by one-way analysis of variance (ANOVA) performed on the areas of chromatographic peaks. The mean values of each group were then compared to a control group using Dunnett's test (16). Normality was checked prior to ANOVA. P values of <0.05 were considered significant. Calculations were done using GraphPad Prism version 5 software (GraphPad Software, Inc., La Jolla, CA).
RESULTS
Reconstruction of sulfur metabolism in B. aurantiacum.
We performed a systematic search (Fig. 1) in the B. aurantiacum genome of strain ATCC 9174 (formerly B. linens) for genes of E. coli, B. subtilis, C. glutamicum, and Mycobacterium tuberculosis involved in the assimilation pathways of sulfur-containing compounds. Growth of B. aurantiacum ATCC 9174 and ATCC 9175 in SDM with various sulfur sources supported the proposed reconstruction (Fig. 2). Most of the genes of ATCC 9174 involved in sulfur metabolism, with two exceptions, are present in ATCC 9175 (20). Both strains were able to grow on sulfate as the sole sulfur source, an observation in agreement with the presence of a sulfate assimilation pathway (CysIHDN-BL2146-BL2147-CysJ) in the B. aurantiacum genome. Interestingly, a gene encoding an APS kinase, CysC, is absent, indicating that PAPS is probably not produced and that CysH is an APS reductase rather than a PAPS reductase. This three-step pathway for sulfide synthesis has been proposed for C. glutamicum and M. tuberculosis (41, 45). Strain ATCC 9174 also grew in the presence of several organic sulfonates (taurine, methanesulfonate, and ethanesulfonate), in agreement with the presence of several operons possibly involved in sulfonate assimilation (Fig. 2B). However, sulfonates were poor sulfur sources, since the growth rate and the growth yield of ATCC 9174 in the presence of sulfonates remained low. Interestingly, B. aurantiacum ATCC 9175 cannot use taurine, methanesulfonate, or ethanesulfonate, whereas this strain grew similarly to ATCC 9174 with sulfate or cystine (Fig. 2B and data not shown). This result may be correlated with the deletion of the BL615-to-BL621 region containing genes similar to ssuD and ssuE in the genome of ATCC 9175, as observed by CGH analysis (20). B. aurantiacum further utilized sulfide for cysteine synthesis using the OAS thiol-lyase, CysK, and the serine acetyltransferase, CysE (Fig. 1B). Both strains grew in the presence of cysteine or cystine, indicating efficient methionine biosynthesis.
FIG. 2.

Growth of B. aurantiacum strains in the presence of various sulfur sources. (A) Growth of strain ATCC 9175 in SDM in the presence of various sulfur sources: 10 mM methionine-500 μM cystine (▪), 10 mM methionine (▴), or 500 μM cystine (•). The growth of strain ATCC 9175 in SDM in the presence of 500 μM sulfite, 500 μM homocysteine, 500 μM cystathionine, and 500 μM thiosulfate was similar to the growth in the presence of 500 μM cystine (data not shown). (B) Growth of strains ATCC 9174 (open symbols) and ATCC 9175 (closed symbols) in SDM in the presence of sulfate or sulfonates: 500 μM sulfate (□, ▪) or 500 μM taurine (▵, ▴). The growth of strains ATCC 9175 and ATCC 9174 in SDM in the presence of 500 μM methanesulfonate or ethanesulfonate was similar to the growth in the presence of 500 μM taurine (data not shown).
Two main pathways for homocysteine production could be involved: the thiolation pathway, involving direct synthesis of homocysteine from sulfide by an O-acetylhomoserine (OAH) thiol-lyase, and the transsulfuration pathway, requiring a cystathionine γ-synthase and a cystathionine β-lyase. Both pathways first required the synthesis of OAH by a homoserine acetyl-transferase encoded by metX (Fig. 1B). Cystathionine β-lyase, cystathionine γ-synthase, OAH thiol-lyase, methionine γ-lyase, and cystathionine γ-lyase belong to the same family of pyridoxal-P-dependent enzymes (34). Transcriptomic analyses will therefore be helpful in assigning functions to the four members of this family: BL56, BL541, BL613, and BL929. However, the BL56 protein (MetY) has similarities to the OAH thiol-lyases of C. glutamicum (54% identity) and M. tuberculosis (61% identity), and contains a 30-amino-acid insertion usually present in OAH thiol-lyases (10). A gene (aecD) encoding a cystathionine β-lyase of the PatC family is also present in the genome. Homocysteine is further methylated by MetE-type methionine synthases: BL2266, BL2839, and/or BL1340. Methionine can be converted into homocysteine by the SAM recycling pathway, which requires MetK, the SAM synthase, SAM-dependent methylases, and SahH, the S-adenosylhomocysteine (SAH) hydrolase (Fig. 1B). The ATCC 9174 and ATCC 9175 strains grew poorly in the presence of methionine as the sole sulfur source. The doubling time is 24 h in the presence of methionine compared to 3.5 h in the presence of sulfate or cystine (Fig. 2B). In contrast, the growth rate with 500 μM homocysteine was similar to that observed in the presence of cystine or sulfate (data not shown), indicating the existence of an efficient conversion of homocysteine to cysteine in B. aurantiacum.
Regulation of sulfur metabolism in strain ATCC 9175 in response to sulfur availability.
To analyze the global response of B. aurantiacum to sulfur starvation, we first compared the expression profiles of strain ATCC 9175 after growth in the presence of sulfate or after resuspension of the cells 2 h in the absence of any sulfur source (Table 1). We used strain ATCC 9175, a technological strain with a better cheese matrix colonization capability than the sequenced strain ATCC 9174. A total of 690 genes were differentially expressed at least 2-fold in the presence of sulfate compared to sulfur starvation. About 300 and 390 genes were more and less expressed during sulfur starvation, respectively. A total of 113 genes differentially expressed in the presence of sulfate or during sulfur starvation participated in central cellular processes such as transcription, translation, replication, and cell wall and macromolecule degradation or respiration. Fourteen genes related to stress response were differentially expressed. There were 19, 31, 1, 21, 56, and 28 genes involved in nucleotide, carbohydrate, secondary metabolite, lipid/energy, amino acid, and cofactor/vitamin metabolisms, respectively, that were also differentially expressed. In addition, 36 genes encoding regulators were identified: 9 were induced in the presence of sulfate, and 27 were induced during sulfur starvation. Such regulators may help B. aurantiacum to adapt to environmental changes.
TABLE 1.
Genes related to sulfur metabolism differentially expressed in B. aurantiacum ATCC 9175 grown in the presence of cystine, sulfate, or methionine plus cystine or in the absence of any sulfur source
| Gene | Function and/or similarity | Ratio and P valuea |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| S/no sulfur |
S/MC |
C/MC |
S/C |
||||||
| Ratio | P | Ratio | P | Ratio | P | Ratio | P | ||
| BL146 | MetK, SAM synthase | 0.34 | 4.5E-6 | -* | 2.8E-2 | ||||
| BL613 | MetB, putative cystathionine γ-synthase | 0.26 | 2.8E-7 | 7.55 | 1.8E-4 | ||||
| BL1112 | CysE, serine acetyltransferase | 0.16 | 1.1E-12 | 2.82 | 2.1E-3 | 2.37 | 1.7E-2 | ||
| BL1113 | CysK, OAS thiol-lyase | 0.32 | 2.5E-5 | 8.65 | 1.2E-6 | 3.39 | 1.8E-5 | ||
| BL2145 | CysJ, sulfite reductase, flavoprotein | 0.38 | 6.1E-6 | 3.50 | 4.5E-2 | 2.86 | 2.8E-2 | ||
| BL2147 | Uroporphyrin III C-methyltransferase, C terminal | 0.47 | 2.9E-3 | 4.51 | 1.7E-3 | 2.47 | 8.5E-5 | ||
| BL2148 | CysN, sulfate adenylyltransferase | 0.33 | 5.1E-6 | 14.21 | 4.3E-5 | 3.53 | 2.41E-5 | ||
| BL2149 | CysD, sulfate adenylyltransferase | 0.44 | 2.5E-3 | 16.95 | 1.4E-7 | 3.38 | 3.1E-3 | ||
| BL2150 | CysH, phosphoadenosine 5′-phosphosulfate sulfotransferase | 0.26 | 4.5E-7 | 7.04 | 1.5E-3 | 2.79 | 6.1E-3 | ||
| BL2151 | CysI, sulfite reductase, hemoprotein | 0.25 | 3.0E-6 | 10.39 | 3.1E-3 | 3.58 | 7.4E-6 | ||
| BL2839 | MetE2, cobalamin-independent methionine synthase | 0.30 | 4.9E-7 | 4.02 | 2.2E-2 | ||||
| BL2146 | Unknown function | 0.46 | 7.1E-4 | 4.51 | 1.7E-3 | ||||
| BL1340 | MetE3, cobalamin-independent methionine synthase | 0.31 | 1.1E-6 | ||||||
| BL2496 | Protein sharing similarity with part of MetH | 0.27 | 9.0E-9 | ||||||
| BL1114 | Formylmethionine deformylase | 0.39 | 1.2E-5 | ||||||
| BL3618 | AecD, cystathionine β-lyase | 0.21 | 3.4E-10 | ||||||
| BL2478 | Glutamate-cysteine ligase | 0.18 | 8.0E-8 | ||||||
| BL3579 | IscS, cysteine desulfurase | 3.85 | 1.1E-7 | ||||||
| BL36 | MetX, homoserine acetyltransferase | 0.50 | 9.6E-3 | 5.26 | 7.4E-3 | -† | -‡ | ||
| BL56 | MetY, OAH thiol-lyase | -** | 1.7E-3 | 18.77 | 1.9E-4 | 16.13 | 8.0E-2 | 2.2 | 5.1E-2 |
| BL2266 | MetE1, cobalamin-independent methionine synthase | 16.10 | 0.0E+0 | 11.84 | 5.5E-10 | 32.40 | 5.5E-4 | 0.45 | 4.0E-2 |
| BL2267 | Unknown function | 19.3 | 0.0E+0 | 13.8 | 1.9E-10 | 32.4 | 3.5E-2 | ||
| BL891 | MerR-like regulator | 0.10 | 5.9E-06 | 0.07 | 3.6E-2 | ||||
| BL929 | Mgl, methionine γ-lyase | 0.06 | 5.5E-10 | 0.07 | 8.2E-2 | ||||
Abbreviations: S, sulfate; MC, methionine plus cystine; C, cystine. A P value of ≤0.05 was obtained using a Benjamini-Hochberg (BH) test under the S/no sulfur condition, and a P value of ≤0.1 was obtained using the BH test under S/MC, S/C, and C/MC conditions, where the difference is >2 or <0.5. *, BL146 = 1.8 under this condition; **, BL56 = 0.52 under this condition; †, based on qRT-PCR, the C/MC ratio is 7.0; ‡, based on qRT-PCR, the S/C ratio is 1.6.
Finally, 21 genes involved in sulfur metabolism were identified in this analysis (Table 1). During sulfur starvation, we observed an increased expression of genes involved in sulfate assimilation (CysND, CysH, and CysJI), in cysteine biosynthesis from sulfide (CysE and CysK), in methionine synthesis via transsulfuration or thiolation (MetX, BL613, AecD, MetY, and two MetE-like enzymes [BL1340 and BL2839]), and in SAM synthesis (MetK) (Fig. 1B). The expression of BL2478, encoding a glutamate-cysteine ligase, also increased during sulfur depletion. In contrast, only three genes involved in sulfur metabolism were more transcribed in the presence of sulfate: BL3579, encoding a cysteine desulfurase that incorporates sulfur in iron-sulfur clusters using cysteine as a donor, as well as BL2266 and BL2267, encoding a MetE-type protein and a protein of unknown function, respectively.
Changes in the metabolome of strain ATCC 9175 grown in the presence of various sulfur sources.
To obtain new insights into regulation in response to sulfur availability, we compared the metabolome of strain ATCC 9175 after growth in the presence of sulfate (S), cystine (C), or methionine plus cystine (MC). We first focused our metabolomic analyses on the intermediary compounds of sulfur metabolism. The cysteine remained undetectable under the three growth conditions (Table 2). OAH and cystathionine were only detectable during growth with cystine or sulfate (Table 2). The methionine concentration increased 8- and 11-fold during growth in the presence of methionine plus cystine compared to cystine or sulfate, respectively. The homocysteine concentration similarly increased 5- and 1,000-fold during growth in the presence of methionine plus cystine.
TABLE 2.
Sulfur-containing compounds and amino acids detected with the metabolomic analysis
| Identificationa | m/z | Retention time (min) | Detection modeb | MS/MS | Ratioc |
||
|---|---|---|---|---|---|---|---|
| MC/S | C/S | MC/C | |||||
| Target analysis | |||||||
| Cysteine* | NDd | ND | ND | ND | ND | ND | ND |
| Cystine* | ND | ND | ND | ND | ND | ND | ND |
| Cystathionine* | 222.06797 | 3.6 | + | No | ND in MC | 2.2* | ND in MC |
| Homocysteine* | 135.03556 | 5.1 | + | No | 1,000* | 230* | 4.6* |
| Methionine* | 149.05126 | 9.1 | +/- | No | 11* | 1.4* | 8.0* |
| OAS | ND | ND | ND | ND | ND | ND | ND |
| OAH | 161.06901 | 6.3 | +/- | Yes | 0.003* | 1.0* | 0.003* |
| Global analysis | |||||||
| Glutamate* | 147.05341 | 3.7 | +/- | No | 0.6* | 0.8* | 0.8* |
| Isoleucine* | 131.09453 | 11.8 | +/- | No | 4.2* | 1.2* | 3.6* |
| Leucine* | 131.09453 | 12.5 | +/- | No | 1.2* | 1.5* | 0.8* |
| Proline* | 115.06344 | 4.8 | +/- | No | 1.7* | 1.8* | 0.9* |
| Aspartic acid* | 133.03754 | 3.6 | +/- | No | 1.3* | 1.8* | 0.7* |
| Phenylalanine* | 165.07909 | 13.6 | +/- | No | 1.0 | 1.2* | 0.8* |
| Tryptophan* | 204.08993 | 17.0 | +/- | No | 1.0 | 1.6* | 0.6* |
| Valine* | 117.07901 | 7.6 | +/- | No | 0.2 | 1.0* | 0.2 |
| Tyrosine* | 181.07383 | 11.8 | +/- | No | 0.8* | 1.9* | 0.4* |
| Ophthalmic acid† | 289.12827 | 5.3 | +/- | Yes | 0.03* | 0.2 | 0.2 |
| Glu-Met | 278.09199 | 12.3 | +/- | Yes | 8.3* | 2.2* | 3.7* |
| Glu-Met sulfoxide | 294.08895 | 5.2 | +/- | Yes | 58* | 4.7* | 13.0* |
| 5-Methylthioadenosine† | 297.08831 | 14.7 | +/- | Yes | 2.8 | 1.2* | 2.3 |
| Putative mycothiol† | 486.15231 | 5.2 | +/- | No | 0.3* | 1.2* | 0.2* |
| Putative Glu-Glu-Met or Met-Glu-Glu | 407.13559 | 14.6 | +/- | No | ND in S | ND in S | 7.3* |
| Putative N-acetylcystathionine | 264.07855 | 11.8 | +/- | No | 12* | 62* | 0.2* |
| Putative sulfur compound 1 | 378.10787 | 12.0 | +/- | No | ND in S* | ND in S* | 3.0* |
| Putative sulfur compound 2 | 343.09281 | 15.3 | - | No | 5.7* | 1.8* | 3.3* |
| Putative sulfur compound 3 | 266.15407 | 19.8 | - | No | 1.0* | 0.6* | 1.5* |
| Putative sulfur compound 4 | 256.05309 | 7.0 | - | No | 0.4* | 0.6* | 0.7* |
| Putative sulfur compound 5 | 424.13834 | 10.9 | + | No | 83* | 1.3* | 63* |
| Putative sulfur compound 6 | 345.11127 | 14.0 | + | No | 0.1* | 8.6* | 0.01* |
*, compounds identified on the basis of the similarity of the retention time, accurate mass, and mass spectrum compared to those of an authentic standard; †, compounds characterized on the basis of the elemental formula provided by accurate mass measurements, by the presence of sulfur in the molecule assessed by the isotopic pattern observed in the mass spectra, and by the interpretation of the fragmentation spectra (for MTA and mycothiol).
+, signal in positive (ESI+) ion detection mode; -, signal in negative (ESI−) ion detection mode.
MC, methionine plus cystine; C, cystine; S, sulfate. P values of <0.05 are indicated by an asterisk.
ND, not detected.
Furthermore, a global approach was performed using automatic peak detection and integration with XCMS software, leading to the detection of 4,335 and 1,225 signals in positive (ESI+) and negative (ESI−) ion detection mode, respectively. The score plot obtained from PCA exhibited a clear separation between our three conditions, and more particularly in the presence of methionine, suggesting that the sulfur source was the main source of variance. The metabolites of biological relevance are listed in Table 2. Some of them were identified on the basis of the similarity of their retention time, accurate mass, and multistage tandem mass spectrometry (MSn) spectra, compared to those of standards. The other metabolites were annotated according to the definition established by the Metabolomics Standard Initiative (51): the matching was based on the elemental formula obtained from accurate mass measurements or on the basis of the interpretation of fragmentation spectra. This analysis highlighted the presence of 23 putative dipeptides and tripeptides in B. aurantiacum cells (data not shown). Among them, we identified a Glu-Met dipeptide, its oxidized form, Glu-methionine sulfoxide, and a tripeptide (Glu-Glu-Met or Met-Glu-Glu). The global analysis also strongly suggested the presence of mycothiol, 5-methylthioadenosine (MTA) and ophthalmic acid, a tripeptide analogue of glutathione in which the cysteine group is replaced by L-2-aminobutyrate (Table 2 and see Fig. S1 in the supplemental material). The levels of mycothiol and ophthalmic acid decreased under MC conditions, whereas those of MTA increased. A pathway for the biosynthesis of mycothiol (mshABCD) and a gene encoding a mycothiol disulfide reductase (mtr) are found in the B. aurantiacum genome (Fig. 1B). The mycothiol present in Actinomycetales protects cells against oxidative stress. MTA might be a by-product of spermidine synthesis. However, a SpeE protein is present in the genome of B. aurantiacum ATCC 9174, but we were unable to identify a SpeD protein, a less-conserved protein (46). Finally, seven putative sulfur-containing compounds were detected on the basis of their isotopic patterns with the presence of the sulfur-34 ion (Table 2). Using metabolomic databases, we identified one of these compounds as N-acetylcystathionine. Its concentration increased 62- and 5-fold in the presence of cystine compared to sulfate or methionine plus cystine. We also observed an impact of the sulfur source on the concentrations of isoleucine, valine, or glutamate. The decreased levels of the latter under MC conditions could be explained by its use for the synthesis of glutamyl-containing peptides, which accumulated under these conditions (see Fig. S1 in the supplemental material).
Comparison of the expression profiles after the growth of strain ATCC 9175 in the presence of various sulfur sources.
After the growth of B. aurantiacum ATCC 9175 in the presence of sulfate, cystine, or cystine plus methionine, we observed major changes in the pools of sulfur-containing compounds that may influence the transcription of genes involved in sulfur metabolism. We then compared the expression profiles of strain ATCC 9175 grown under these conditions (Table 1 and data not shown). Totals of 15 and 30 genes were differentially expressed after the growth of strain ATCC 9175 in the presence of sulfate compared to growth in the presence of cystine or methionine plus cystine, respectively. The expression profiles of 15 genes also differed when we compared growth with cystine and growth with cystine plus methionine. Most of the genes differentially expressed participate in sulfur metabolism. However, the expression of genes encoding a peptidase (BL978), a putative carboxymuconolactone decarboxylase (BL1185), and a putative monooxygenase (BL1738), as well as proteins of unknown function, changed in S or MC conditions.
Sulfate assimilation and cysteine biosynthesis from sulfide are coordinately regulated.
A first set of genes was derepressed during sulfur starvation and was more expressed in strain ATCC 9175 grown in the presence of sulfate than in the presence of cystine or methionine-cystine (Table 1). The cysN, cysD, cysH, cysI, cysJ, cysE, and cysK genes were clearly transcribed more weakly in the presence of cystine. The metY gene encoding the OAH thiol-lyase that also uses sulfide as a substrate was similarly regulated. However, metY expression also responded to methionine availability, indicating a pattern of expression different from that of the cys genes. The genes involved in sulfate assimilation (cysIHDN-BL2146-BL2147-cysJ) and in cysteine biosynthesis (cysKE) are organized into two operons (Fig. 3A). We then determined the transcriptional start site of these operons by 5′RACE. The transcription of the sulfate assimilation operon is initiated 32 bp upstream of the cysI start codon, whereas that of the cysKE operon is initiated 117 bp upstream of the cysK start codon (Fig. 3A). Upstream of these transcriptional start sites, we did not identify the TATAAT and TTGACA sequences characteristic of promoters in E. coli and B. subtilis. A TAN3TG motif that shares similarities with part of the consensus of the −10 region of C. glutamicum [tgngnTA(c/t)aaTgg] (uppercase letters indicate highly conserved; lowercase indicates poorly conserved) (37) is present upstream of the transcriptional start site of cysK and cysJ (Fig. 3A).
FIG. 3.

Genetic organization of operons involved in sulfate assimilation or cysteine biosynthesis (A) and in methionine biosynthesis (B). A bent arrow indicates the experimental transcriptional start site. The potential −10 boxes for the promoters are underlined. The translational start codon is indicated in italics. The gray boxes correspond to conserved motifs. The terminators are represented by a loop. The positions of the primers used for the determination of the operon by RT-PCR are indicated by arrows.
Regulation of methionine biosynthesis and catabolism in response to methionine availability.
Comparison of the expression profiles of strain ATCC 9175 in the presence of cystine or cystine-methionine revealed a few genes specifically regulated in response to methionine availability (Table 1). These genes probably participated in either methionine biosynthesis or degradation. Only two adjacent genes, BL929 (encoding a putative methionine γ-lyase) and BL891 (encoding a regulator), were upregulated in the presence of methionine (Table 1). We demonstrated by RT-PCR the existence of a common transcript for these genes (data not shown). Surprisingly, the transcription of the BL929 operon is initiated at the ATG start codon of BL929 (Fig. 4A), as observed for several genes in actinomycetes (37). Accordingly, no RBS is present upstream of the ATG of BL929. Methionine is usually regarded as the VSC precursor. We thus measured the total production of VSCs after the growth of strain ATCC 9175 in the presence of various sulfur sources (Table 3). This strain did not produce VSCs in the presence of cystine or sulfate. Large amounts of VSCs were detected when methionine was added to the SDM containing cystine. DMDS and DMTS were the major VSCs produced under these conditions, with 643.5 ppb and 201.6 ppb, respectively.
FIG. 4.
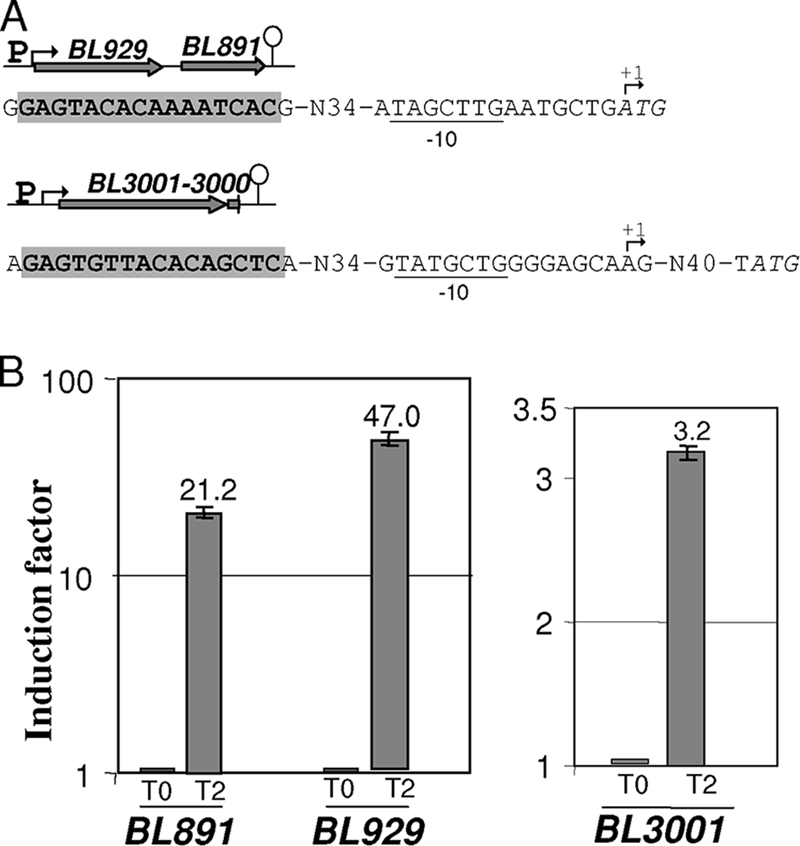
Operons induced in the presence of methionine. (A) Genetic organization of the BL929-to-BL891 and BL3000-BL3001 operons. A bent arrow indicates the transcriptional start site. The potential −10 boxes of the promoters are underlined. The start codons of BL929 and BL3001 are indicated in italics. The gray box corresponds to a conserved motif. The terminator is represented by a loop. (B) qRT-PCR analysis of BL929, BL891, and BL3001 expression after the addition of methionine. The 2−ΔΔCT method was applied to calculate the relative amount of cDNAs of interest. Methionine (10 mM) was added to a culture of strain ATCC 9175 in SDM containing 500 μM cystine, and the cells were then harvested before (T0) and 2 h (T2) after methionine addition (induction factor = 1 at T0). This experiment was performed in three biological replicates.
TABLE 3.
Specific production of volatile sulfur compounds by B. aurantiacum ATCC 9175 in SDM with different sulfur sourcesa
| Volatile sulfur compound | Mean sulfur compound concn (μg kg−1) ± SD |
||
|---|---|---|---|
| MC | C | S | |
| Methanethiol | 1.04 ± 0.40 | ND | ND |
| DMDS | 643.51 ± 136.35 | 0.70 ± 0.06 | 0.23 ± 0.05 |
| 2,4-Dithiapentane | 0.08 ± 0.05 | ND | ND |
| DMTS | 201.63 ± 50.45 | 0.05 ± 0.02 | ND |
| DMQS | 0.59 ± 0.31 | ND | ND |
ND, not detected. Abbreviations: MC, methionine plus cystine (10 mM l-methionine plus 500 μM l-cystine); C, cystine (500 μM l-cystine); S, sulfate (500 μM sulfate); DMDS, dimethyl disulfide; DMTS, dimethyl trisulfide; DMQS, dimethyl tetrasulfide. All experiments were performed in triplicate.
The expression of metY, encoding the OAH thiol-lyase, and of BL2266 (metE1), encoding a methionine synthase, was 16- and 32-fold repressed, respectively, when methionine was added to the medium (Table 1). This was also the case for BL2267, encoding a protein of unknown function, which is adjacent to metE1. Using RT-PCR, we demonstrated that BL2267 and BL2266, as well as metY and metX, form operons (Fig. 3B). The methionine-dependent repression of these operons suggested that MetY, MetX, and MetE1 play an important role in methionine biosynthesis in B. aurantiacum. The function of BL613 and BL541, which share similarities with MetY and BL929, remains unclear. The expression of BL613 was higher in the presence of sulfate than in the presence of methionine plus cystine. A 6-fold repression was also observed in qRT-PCR, 2 h after methionine addition to a medium containing cystine (see Table S2 in the supplemental material). Finally, BL613 expression decreased 30-fold in the presence of homocysteine compared to sulfate or cystine, suggesting that BL613 could participate in methionine synthesis via transsulfuration. In contrast, BL541 expression was not modulated under any of the conditions tested.
Regulation of transporter synthesis in response to sulfur, methionine, or cysteine starvation.
Genes encoding several transporters were differentially expressed in the presence of sulfate compared to sulfur limitation (Table 4). Eleven peptide transporters are present in B. aurantiacum. Among them, the expression of an operon encoding an oligopeptide ABC transporter (BL1734 to BL1737) increased 5-fold during sulfur depletion. In contrast, other genes encoding ABC transporters for oligopeptides (BL250 to BL253, BL634 to BL637, BL2377, and BL2733) or a dipeptide/tripeptide transporter (BL1264) were expressed more during growth with sulfate. Interestingly, the expression of five genes encoding proteases (AprE/BL4033, BL2348, and BL494) or peptidases (BL1078 and BL1007) also increased 2- to 4-fold in the presence of sulfate. These enzymes may contribute to the production or degradation of peptides.
TABLE 4.
Genes encoding oligopeptide or amino acid transporters differentially expressed in B. aurantiacum ATCC 9175 grown in the presence of sulfate, grown in the presence of methionine plus cystine, or grown during sulfur limitation
| Gene | Function and/or similarity | Ratio and P valuea |
|||
|---|---|---|---|---|---|
| S/no sulfur |
S/MC |
||||
| Ratio | P | Ratio | P | ||
| BL1734 | Oligopeptide ABC transporter, solute-binding protein | 0.14 | 5.4E-12 | ||
| BL1736 | Oligopeptide ABC transporter, permease | 0.22 | 7.9E-10 | ||
| BL1735 | Oligopeptide ABC transporter, permease | 0.46 | 6.3E-10 | ||
| BL1737b | Oligopeptide ABC transporter, ATP-binding protein | 0.19 | 5.3E-10 | ||
| BL380 | Probable glutamine ABC transporter, ATP-binding protein | 0.47 | 6.6E-3 | ||
| BL250 | Oligopeptide ABC transporter, solute-binding component | 2.15 | 2.7E-3 | ||
| BL251 | Oligopeptide ABC transporter, permease | 9.00 | 2.1E-14 | ||
| BL252 | Oligopeptide ABC transporter solute-binding protein | 14.23 | 2.1E-14 | ||
| BL253 | Oligopeptide ABC transporter ATP-binding protein | 12.30 | 2.1E-14 | ||
| BL634 | Oligopeptide ABC transporter ATP-binding protein | 2.95 | 5.6E-6 | ||
| BL635 | Oligopeptide ABC transporter, permease | 3.25 | 1.5E-6 | ||
| BL636 | Oligopeptide ABC transporter solute-binding protein | 3.40 | 4.8E-7 | ||
| BL637 | Oligopeptide ABC transporter solute-binding protein | 5.90 | 4.4E-5 | ||
| BL1103 | Oligopeptide ABC transporter, solute-binding protein | 9.95 | 0.0E+0 | ||
| BL1104 | Oligopeptide ABC transporter, ATP-binding protein | 2.25 | 8.1E-3 | ||
| BL2377 | Oligopeptide ABC transporter, permease | 2.96 | 5.2E-5 | ||
| BL2733 | Oligopeptide ABC transporter, permease | 2.50 | 6.7E-4 | ||
| BL1197 | Amino acid transporter | 2.05 | 2.0E-4 | ||
| BL1264 | Dipeptide/tripeptide permease | 5.16 | 2.7E-11 | ||
| BL1657 | Methionine ABC transporter, MetQ | 6.90 | 5.1E-4 | ||
| BL2207 | Lysine transporter | 2.14 | 1.4E-3 | ||
| BL3604 | LivF leucine/isoleucine/valine transporter, ATP-binding protein | 3.51 | 6.2E-7 | ||
| BL3605 | LivJ leucine/isoleucine/valine transporter, solute-binding protein | 2.93 | 4.2E-6 | ||
| BL3607 | LivM leucine/isoleucine/valine transporter, permease | 3.32 | 3.5E-7 | ||
| BL3608 | LivH leucine/isoleucine/valine transporter, permease | 2.24 | 7.3E-4 | ||
| BL1293 | Probable cystine ABC transporter ATP-binding protein | 8.27 | 0.0E+0 | 1.82 | 7.9E-2 |
| BL1294 | Probable cystine ABC transporter, permease | 10.74 | 2.1E-14 | 2.01 | 3.9E-2 |
| BL1295 | Probable cystine ABC transporter, permease | 6.01 | 6.2E-13 | 1.92 | 2.6E-2 |
| BL1296 | Probable cystine ABC transporter, solute-binding component | 2.14 | 2.1E-3 | ||
S, sulfate; MC, methionine plus cystine.
The expression of this gene increased 2-fold in the presence of sulfate compared to cystine.
Genes encoding two amino acid ABC transporters (BL1657 and BL1293-BL1296) were transcribed stronger during growth with sulfate (Table 4). BL1655 to BL1657 and BL1293 to BL1296 share similarities with MetNPQ, a high-affinity ABC transporter of B. subtilis (53, 35, and 36% identities) (48) and a cystine ABC transporter of E. coli (48, 31, 33, and 26% identities), respectively. Other putative cystine or methionine transporters are present in the B. aurantiacum genome. The BL1249-BL1250-BL1251 proteins correspond to an ABC transporter that shares similarities with BL1293 to BL1296. BL3001 and BL3000 are 48 and 30% identical to MetPS, a low-affinity methionine transporter of C. glutamicum (53). We tested the effect of cysteine or methionine limitation on their expression. Strain ATCC 9175 was first grown in the presence of methionine plus cystine and then resuspended either in the presence of cystine alone (methionine depletion) or in the presence of methionine alone (cysteine depletion). The expression of BL1250 and BL1296 increased 2.1- and 3.2-fold, respectively, during cysteine limitation (see Table S2 in the supplemental material). Both ABC transporters (BL1249 to BL1251 and BL1293 to BL1296) probably constitute uptake systems for cystine and/or cysteine (Fig. 1A).
During methionine limitation, BL1657 gene expression increased 25-fold in agreement with a key role of the BL1655-1657 ABC transporter for methionine supply. In contrast, the BL3001 expression did not change during methionine starvation but increased 3.2-fold when methionine was added. Interestingly, the expression of BL929 encoding a probable methionine γ-lyase also increased in MC medium. An additional qRT-PCR analysis was performed to analyze the induction of the BL929-to-BL891 and BL3001-BL3000 operons. The transcription of BL891, BL929, and BL3001 increased 21-, 47-, and 3-fold, respectively, 2 h after the addition of 10 mM methionine in SDM containing cystine (Fig. 4B), indicating a methionine-specific induction.
DISCUSSION
We combined metabolic reconstruction and growth assays with expression and metabolic profiling to obtain a global view of the sulfur metabolic network and of the response to sulfur availability in B. aurantiacum, a major VSC producer in cheese-ripening bacteria. The sulfate assimilation pathway encoded by the cysI operon and pathways required for direct formation of cysteine or homocysteine from sulfide are present in the B. aurantiacum genome. The expression of the cysI and cysK operons is high in the presence of sulfate and reduced with cystine, as expected for genes involved in cysteine production from sulfate. Using IMOMI software (38), we identified a conserved TTGTTGAGCAA motif upstream of cysK and cysI (Fig. 3A), suggesting a cysteine-dependent repression by a common regulator. Proteins sharing similarities with regulators controlling sulfate assimilation and/or cysteine biosynthesis in other Gram-positive bacteria, such as McbR and CysR of C. glutamicum (41, 43) or CymR of B. subtilis (17), are absent in B. aurantiacum, suggesting the involvement of a still-uncharacterized regulator. In addition, effectors of regulators of these pathways, such as cysteine, OAS, and OAH, are either undetectable or not differentially accumulated during growth with sulfate or cystine.
Two methionine biosynthesis pathways, direct thiolation and transsulfuration, are present in the genome of B. aurantiacum. C. glutamicum utilizes both pathways with equal efficiency (42). While the OAH-thiol-lyase, MetY, is clearly identified, the annotation of the enzymes for the transsulfuration pathway, the cystathionine γ-synthase (MetB) and the cystathionine β-lyases (MetC- or AecD-type), is less evident. BL613, which is more similar to MetB of C. glutamicum and M. tuberculosis than is BL541, is the probable cystathionine γ-synthase. Furthermore, homocysteine and, to a lesser extent, methionine repressed BL613 expression. The synthesis of AecD, a probable cystathionine β-lyase, is constitutive, as observed for AecD of C. glutamicum and PatB of B. subtilis (3). BL541, which shares similarities with Cg3086, could be either a second cystathionine β-lyase or a cystathionine γ-lyase (44). Three cobalamin-independent methionine synthases required for homocysteine methylation are found in the B. aurantiacum genome. MetE2 and MetE3 share 56 and 43% identity with MetE of C. glutamicum. Interestingly, MetE1 (BL2266) is highly similar to MetE from Arthrobacter, Pseudomonas, Xanthomonas, Psychrobacter, and Acinetobacter (78 to 82% identity), suggesting the existence of a horizontal gene transfer between gammaproteobacteria and Micrococcineae in common biotopes such as soils or foods. The metE1 and metE2 genes are repressed in the presence of methionine, as observed for BL613, metX, metY, and BL1657. A conserved motif, CATGACG-N4-GTGACA, is present ∼50 bp upstream of the metY and BL613 transcriptional start sites (Fig. 3B). A coordinated regulation of these genes by a still-uncharacterized regulator could be proposed. However, this motif is absent in the promoter region of the BL2267-metE1 and BL1655-to-BL1657 operons and metE2, suggesting the possible existence of several regulators for methionine biosynthesis, as observed in E. coli (56). In the presence of methionine, the pools of methionine and homocysteine increase whereas cystathionine and OAH are depleted. All of these compounds might be effectors of these regulators.
Surprisingly, the doubling time of ATCC 9175 on methionine (24 h) is very high compared to that observed with cystine or homocysteine (3.5 h). Its ability to grow on homocysteine indicates an efficient conversion of this compound into cysteine. Several pathways could be involved: the reverse transsulfuration pathway (48), the presence of an homocysteine γ-lyase, or the degradation of methionine to MTL and its further conversion into methanesulfonate (55). This latter possibility seems unlikely since strain ATCC 9175 grows poorly in the presence of methionine. Genes encoding proteins sharing only weak similarities with cystathionine β-synthases (BL781) or cystathionine γ-lyases (BL541) are present in the B. aurantiacum genome. Interestingly, the methionine γ-lyase of B. aurantiacum BL2 (formerly B. linens) also has a homocysteine γ-lyase activity (15). The expression of BL929 is induced in the presence of homocysteine (see Table S2 in the supplemental material). The BL929 enzyme could participate in cysteine production from homocysteine via the intermediary formation of sulfide (Fig. 1B). Two hypotheses for methionine being a poor sulfur source for B. aurantiacum can be proposed: (i) methionine is inefficiently taken up, and (ii) the SAM recycling pathway is limiting. Two methionine transporters are present in the B. aurantiacum genome: one, similar to the high-affinity transporter MetNPQ of B. subtilis (48), is induced during methionine limitation, and the second, which shares similarities with the low-affinity transporter MetPS of C. glutamicum (53), is induced in the presence of methionine. In addition, the intracellular concentration of methionine strongly increased when methionine was added to the medium, indicating an efficient uptake of this amino acid (Table 2).
The catabolism of methionine in Brevibacteriaceae has been the subject of several studies because this amino acid is the precursor of MTL, which is required for the production of VSCs (11, 14, 15). The methionine γ-lyase is the key enzyme for methionine degradation in Brevibacteriaceae and other bacteria (25, 32). The expression of genes encoding methionine γ-lyases is induced by methionine in Pseudomonas putida (25) and Citrobacter freundii (31). Methionine γ-lyase activity in B. antiquum CNRZ918 (formerly B. linens) also increases in the presence of methionine (18). BL929 is the only enzyme similar to methionine γ-lyase, whose synthesis is induced by methionine. This indicated that BL929 is the major methionine γ-lyase of B. aurantiacum ATCC 9175. In addition to MTL, methionine degradation leads to the production of α-ketobutyrate, a precursor of isoleucine (39). Interestingly, we observed an increase in isoleucine concentration in the presence of methionine (Table 2; see also Fig. S1 in the supplemental material). The BL3000-BL3001 transporter, similar to the MetPS system of C. glutamicum, probably corresponds to the Na+-stimulated l-methionine transporter identified in B. antiquum CNRZ918 (19). The expression of the BL3000-BL3001 operon, in contrast to that of metPS in C. glutamicum (53), is induced in the presence of 10 mM methionine. BL929 forms an operon with BL891 encoding a MerR-type regulator. This regulator is a good candidate to participate to the control of methionine γ-lyase and MetPS synthesis. Interestingly, a common motif, GAGTRYWMMAMAKCWC, is located 51 bp upstream of the transcriptional start site of the BL929 and BL3001 operons. The existence of a coordinated regulation of methionine transport and degradation could be an adaptation to the cheese matrix in which amino acids constitute a major substrate for growth and/or aroma production (7, 12). This work represents the first picture of sulfur metabolism in B. aurantiacum, a bacterium of major importance in cheese ripening. It also gives an overview of the global regulation of sulfur metabolism through transcriptomic and metabolomic approaches. This bacterium develops at the cheese surface and is then exposed to oxygen. The production and/or accumulation of reducing thiol molecules either extracellularly (sulfide, MTL) or intracellularly (mycothiol, homocysteine) could be an interesting adaptive feature to its biotope.
Supplementary Material
Acknowledgments
The sequence data of B. aurantiacum BL2 (formerly B. linens) were produced by the U.S. Department of Energy's Joint Genome Institute (http://genome.jgi-psf.org/).
We thank E. Guédon and F. Rul for helpful discussions and G. Wagman for correcting the English. We are also grateful to R. Tâche for her technical support.
This study was supported by the EcoMet program (ANR-06-PNRA-014) funded by the French National Research Agency (ANR). M.-P.F. and A.H. are grateful to the ANR for a Ph.D. scholarship.
Footnotes
Published ahead of print on 17 December 2010.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Amarita, F., et al. 2004. Identification and functional analysis of the gene encoding methionine-γ-lyase in Brevibacterium linens. Appl. Environ. Microbiol. 70:7348-7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arfi, K., F. Amarita, H. E. Spinnler, and P. Bonnarme. 2003. Catabolism of volatile sulfur compounds precursors by Brevibacterium linens and Geotrichum candidum, two microorganisms of the cheese ecosystem. J. Biotechnol. 105:245-253. [DOI] [PubMed] [Google Scholar]
- 3.Auger, S., M. P. Gomez, A. Danchin, and I. Martin-Verstraete. 2005. The PatB protein of Bacillus subtilis is a C-S-lyase. Biochimie 87:231-238. [DOI] [PubMed] [Google Scholar]
- 4.Benjamini, Y., and Y. Hochberg. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Stat. Methods 57:289-300. [Google Scholar]
- 5.Bockelmann, W. 2003. Smear ripened cheeses, p. 391-401. In H. Roginski, J. W. Fuquay, and P. F. Fox (ed.), Encyclopedia of dairy sciences. Academic Press, Inc., New York, NY.
- 6.Bonnarme, P., C. Lapadatescu, M. Yvon, and H. E. Spinnler. 2001. l-Methionine degradation potentialities of cheese-ripening microorganisms. J. Dairy Res. 68:663-674. [DOI] [PubMed] [Google Scholar]
- 7.Boutrou, R., and M. Guéguen. 2005. Interests in Geotrichum candidum for cheese technology. Int. J. Food Microbiol. 102:1-20. [DOI] [PubMed] [Google Scholar]
- 8.Bryson, K., et al. 2006. AGMIAL: implementing an annotation strategy for prokaryote genomes as a distributed system. Nucleic Acids Res. 34:3533-3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burguiere, P., S. Auger, M.-F. Hullo, A. Danchin, and I. Martin-Verstraete. 2004. Three different systems participate in l-cystine uptake in Bacillus subtilis. J. Bacteriol. 186:4875-4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cherest, H., D. Thomas, and Y. Surdin-Kerjan. 1993. Cysteine biosynthesis in Saccharomyces cerevisiae occurs through the transsulfuration pathway which has been built up by enzyme recruitment. J. Bacteriol. 175:5366-5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cholet, O., A. Hénaut, and P. Bonnarme. 2007. Transcriptional analysis of l-methionine catabolism in Brevibacterium linens ATCC 9175. Appl. Microbiol. Biotechnol. 74:1320-1332. [DOI] [PubMed] [Google Scholar]
- 12.Christensen, J., E. Dudley, J. Pederson, and J. Steele. 1999. Peptidases and amino acid catabolism in lactic acid bacteria. Antonie Van Leeuwenhoek 76:217-246. [PubMed] [Google Scholar]
- 13.Delmar, P., S. Robin, and J. J. Daudin. 2005. VarMixt: efficient variance modeling for the differential analysis of replicated gene expression data. Bioinformatics 21:502-508. [DOI] [PubMed] [Google Scholar]
- 14.Dias, B., and B. Weimer. 1998. Conversion of methionine to thiols by lactococci, lactobacilli, and brevibacteria. Appl. Environ. Microbiol. 64:3320-3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dias, B., and B. Weimer. 1998. Purification and characterization of l-methionine γ-lyase from Brevibacterium linens BL2. Appl. Environ. Microbiol. 64:3327-3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunnett, C. W. 1955. Multiple comparisons procedure for comparing several treatments with a control. Am. Stat. Assoc. 50:1096-1121. [Google Scholar]
- 17.Even, S., et al. 2006. Global control of cysteine metabolism by CymR in Bacillus subtilis. J. Bacteriol. 188:2184-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferchichi, M., D. Hemme, and M. Nardi. 1986. Induction of methanethiol production by Brevibacterium linens CNRZ 918. J. Gen. Microbiol. 132:3075-3082. [DOI] [PubMed] [Google Scholar]
- 19.Ferchichi, M., D. Hemme, and M. Nardi. 1987. Na+-stimulated transport of l-methionine in Brevibacterium linens CNRZ 918. Appl. Environ. Microbiol. 53:2159-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forquin, M. P., et al. 2009. Identification of Brevibacteriaceae by multilocus sequence typing and comparative genomic hybridization analyses. Appl. Environ. Microbiol. 75:6406-6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fox, P. F., P. L. H. McSweeney, J. Wallace, and T. P. Guinee. 2004. Cheese: chemistry, physics, and microbiology, 3rd ed. Elsevier Academic Press, Amsterdam, Netherlands.
- 22.Guillouard, I., et al. 2002. Identification of Bacillus subtilis CysL, a regulator of the cysJI operon, which encodes sulfite reductase. J. Bacteriol. 184:4681-4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hryniewicz, M., A. Sirko, A. Palucha, A. Bock, and D. Hulanicka. 1990. Sulfate and thiosulfate transport in Escherichia coli K-12: identification of a gene encoding a novel protein involved in thiosulfate binding. J. Bacteriol. 172:3358-3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hwang, B.-J., H.-J. Yeom, Y. Kim, and H.-S. Lee. 2002. Corynebacterium glutamicum utilizes both transsulfuration and direct sulfhydrylation pathways for methionine biosynthesis. J. Bacteriol. 184:1277-1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inoue, H., et al. 1997. Molecular characterization of the mde operon involved in l-methionine catabolism of Pseudomonas putida. J. Bacteriol. 179:3956-3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koch, D. J., et al. 2005. Role of the ssu and seu genes of Corynebacterium glutamicum ATCC 13032 in the utilization of sulfonates and sulfonate esters as sulfur sources. Appl. Environ. Microbiol. 71:6104-6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landaud, S., S. Helinck, and P. Bonnarme. 2008. Formation of volatile sulfur compounds and metabolism of methionine and other sulfur compounds in fermented food. Appl. Microbiol. Biotechnol. 77:1191-1205. [DOI] [PubMed] [Google Scholar]
- 28.Ludwig, M. L., and R. G. Matthews. 1997. Structure-based perspectives on B12-dependent enzymes. Annu. Rev. Biochem. 66:269-313. [DOI] [PubMed] [Google Scholar]
- 29.Mansilla, M. C., and D. De Mendoza. 2000. The Bacillus subtilis cysP gene encodes a novel sulfate permease related to the inorganic phosphate transporter (Pit) family. Microbiology 146:815-821. [DOI] [PubMed] [Google Scholar]
- 30.Mansour, S., J. M. Beckerich, and P. Bonnarme. 2008. Lactate and amino acid catabolism in the cheese-ripening yeast Yarrowia lipolytica. Appl. Environ. Microbiol. 74:6505-6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manukhov, I. V., et al. 2006. l-Methionine γ-lyase from Citrobacter freundii: cloning of the gene and kinetic parameters of the enzyme. Biochemistry Mosc. 71:361-369. [DOI] [PubMed] [Google Scholar]
- 32.Manukhov, I. V., et al. 2005. A gene encoding l-methionine γ-lyase is present in Enterobacteriaceae family genomes: identification and characterization of Citrobacter freundii l-methionine γ-lyase. J. Bacteriol. 187:3889-3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martın, J. F., C. Barreiro, E. Gonzalez-Lavado, and M. Barriuso. 2003. Ribosomal RNA and ribosomal proteins in corynebacteria. J. Biotechnol. 104:41-53. [DOI] [PubMed] [Google Scholar]
- 34.Mehta, P. K., and P. Christen. 2000. The molecular evolution of pyridoxal-5′-phosphate-dependent enzymes. Adv. Enzymol. Relat. Areas Mol. Biol. 74:129-184. [DOI] [PubMed] [Google Scholar]
- 35.Milohanic, E., et al. 2003. Transcriptome analysis of Listeria monocytogenes identifies three groups of genes differently regulated by PrfA. Mol. Microbiol. 47:1613-1625. [DOI] [PubMed] [Google Scholar]
- 36.Mounier, J., et al. 2005. Surface microflora of four smear-ripened cheeses. Appl. Environ. Microbiol. 71:6489-6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patek, M., J. Nesvera, A. Guyonvarch, O. Reyes, and G. Leblon. 2003. Promoters of Corynebacterium glutamicum. J. Biotechnol. 104:311-323. [DOI] [PubMed] [Google Scholar]
- 38.Pons, N., J. M. Batto, S. D. Ehrlich, and P. Renault. 2008. Development of software facilities to characterize regulatory binding motifs and application to Streptococcaceae. J. Mol. Microbiol. Biotechnol. 14:67-73. [DOI] [PubMed] [Google Scholar]
- 39.Ramakrishnan, T., and E. A. Adelberg. 1965. Regulatory mechanisms in the biosynthesis of isoleucine and valine. II. Identification of two operator genes. J. Bacteriol. 89:654-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rea, M. C., et al. 2007. Stability of the biodiversity of the surface consortia of Gubbeen, a red-smear cheese. J. Dairy Sci. 90:2200-2210. [DOI] [PubMed] [Google Scholar]
- 41.Rey, D. A., et al. 2005. The McbR repressor modulated by the effector substance S-adenosylhomocysteine controls directly the transcription of a regulon involved in sulfur metabolism of Corynebacterium glutamicum ATCC 13032. Mol. Microbiol. 56:871-887. [DOI] [PubMed] [Google Scholar]
- 42.Rückert, C., et al. 2005. Functional genomics and expression analysis of the Corynebacterium glutamicum fpr2-cysIXHDNYZ gene cluster involved in assimilatory sulfate reduction. BMC Genomics 6:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rückert, C., et al. 2008. The dual transcriptional regulator CysR in Corynebacterium glutamicum ATCC 13032 controls a subset of genes of the McbR regulon in response to the availability of sulfide acceptor molecules. BMC Genomics 9:483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rückert, C., A. Pühler, and J. Kalinowski. 2003. Genome-wide analysis of the l-methionine biosynthetic pathway in Corynebacterium glutamicum by targeted gene deletion and homologous complementation. J. Biotechnol. 104:213-228. [DOI] [PubMed] [Google Scholar]
- 45.Schelle, M. W., and C. R. Bertozzi. 2006. Sulfate metabolism in mycobacteria. Chembiochem 7:1516-1524. [DOI] [PubMed] [Google Scholar]
- 46.Sekowska, A., J. Y. Coppée, J. P. Le Caer, I. Martin-Verstraete, and A. Danchin. 2000. S-Adenosylmethionine decarboxylase of Bacillus subtilis is closely related to archaebacterial counterparts. Mol. Microbiol. 36:1135-1147. [DOI] [PubMed] [Google Scholar]
- 47.Smith, C. A., E. J. Want, G. O'Maille, R. Abagyan, and G. Siuzdak. 2006. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78:779-787. [DOI] [PubMed] [Google Scholar]
- 48.Soutourina, O., and I. Martin-Verstraete. 2007. Global regulatory network of sulfur metabolism in Bacillus subtilis, p. 111-141. In Y. Fujita (ed.), Global regulatory networks in Bacillus subtilis. Transworld Research Network, Kerala, India.
- 49.Stackebrandt, E., F. A. Rainey, and N. L. Ward-Rainey. 1997. Proposal for a new hierarchic classification system, Actinobacteria classis nov. Int. J. Syst. Bacteriol. 47:479-491. [Google Scholar]
- 50.Storey, J. D., and R. Tibshirani. 2003. Statistical significance for genome-wide experiments. Proc. Natl. Acad. Sci. U. S. A. 100:9440-9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sumner, L. W., E. Urbanczyk-Wochniak, and C. D. Broeckling. 2007. Metabolomics data analysis, visualization, and integration. Methods Mol. Biol. 406:409-436. [DOI] [PubMed] [Google Scholar]
- 52.Tasara, T., and R. Stephan. 2007. Evaluation of housekeeping genes in Listeria monocytogenes as potential internal control references for normalizing mRNA expression levels in stress adaptation models using real-time PCR. FEMS Microbiol. Rev. 269:265-272. [DOI] [PubMed] [Google Scholar]
- 53.Trötschel, C., et al. 2008. Methionine uptake in Corynebacterium glutamicum by MetQNI and by MetPS, a novel methionine and alanine importer of the NSS neurotransmitter transporter family. Biochemistry 47:12698-12709. [DOI] [PubMed] [Google Scholar]
- 54.van der Ploeg, J. R., et al. 1996. Identification of sulfate starvation-regulated genes in Escherichia coli: a gene cluster involved in the utilization of taurine as a sulfur source. J. Bacteriol. 178:5438-5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vermeij, P., and M. A. Kertesz. 1999. Pathways of assimilative sulfur metabolism in Pseudomonas putida. J. Bacteriol. 181:5833-5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weissbach, H., and N. Brotn. 1991. Regulation of methionine synthesis in Escherichia coli. Mol. Microbiol. 5:1593-1597. [DOI] [PubMed] [Google Scholar]
- 57.Wooff, E., et al. 2002. Functional genomics reveals the sole sulfate transporter of the Mycobacterium tuberculosis complex and its relevance to the acquisition of sulfur in vivo. Mol. Microbiol. 43:653-663. [DOI] [PubMed] [Google Scholar]
- 58.Yang, Y. H., et al. 2002. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 30:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.