

Abstract
Regulation of transcription factor Nrf2 (NF-E2-related factor 2) involves redox-sensitive proteasomal degradation via the E3 ubiquitin ligase Keap1/Cul3. However, Nrf2 is controlled by other mechanisms that have not yet been elucidated. We now show that glycogen synthase kinase 3 (GSK-3) phosphorylates a group of Ser residues in the Neh6 domain of mouse Nrf2 that overlap with an SCF/β-TrCP destruction motif (DSGIS, residues 334 to 338) and promotes its degradation in a Keap1-independent manner. Nrf2 was stabilized by GSK-3 inhibitors in Keap1-null mouse embryo fibroblasts. Similarly, an Nrf2ΔETGE mutant, which cannot be degraded via Keap1, accumulated when GSK-3 activity was blocked. Phosphorylation of a Ser cluster in the Neh6 domain of Nrf2 stimulated its degradation because a mutant Nrf2ΔETGE 6S/6A protein, lacking these Ser residues, exhibited a longer half-life than Nrf2ΔETGE. Moreover, Nrf2ΔETGE 6S/6A was insensitive to β-TrCP regulation and exhibited lower levels of ubiquitination than Nrf2ΔETGE. GSK-3β enhanced ubiquitination of Nrf2ΔETGE but not that of Nrf2ΔETGE 6S/6A. The Nrf2ΔETGE protein but not Nrf2ΔETGE 6S/6A coimmunoprecipitated with β-TrCP, and this association was enhanced by GSK-3β. Our results show for the first time that Nrf2 is targeted by GSK-3 for SCF/β-TrCP-dependent degradation. We propose a “dual degradation” model to describe the regulation of Nrf2 under different pathophysiological conditions.
A disadvantage of aerobic life is the constant generation of potentially damaging reactive oxygen species (ROS). The intracellular levels of such species need to be tightly controlled in order to avoid oxidative stress. Transcription factor Nrf2 (NF-E2-related factor 2) plays a critical role in redox homeostasis since it increases the expression of many antioxidant and drug-metabolizing genes, including those encoding heme oxygenase 1 (HO-1), NADPH:quinone oxidoreductase 1, glutathione S-transferases, glutamate-cysteine ligase, and glutathione peroxidases, in response to oxidative and electrophile stressors (13). These genes all contain a common promoter enhancer called the antioxidant response element (ARE) and are transactivated by Nrf2. Because ROS play a role as intracellular signaling molecules for many physiological processes, Nrf2 can have an impact on numerous cell functions, ranging from differentiation and development to proliferation and inflammation. Therefore, Nrf2 activity influences neurodegenerative disease, cardiovascular disease, and cancer (3, 4, 14, 16, 17, 49, 53).
While increased Nrf2 transcriptional activity enhances cellular antioxidant defenses and increases the capacity to detoxify drugs, it may also lead to unwanted side effects. For instance, in tumors, high levels of Nrf2 activity have been correlated with a poor prognosis (41). Indeed, high Nrf2 activity has not been favored during evolution (25), but its levels are restricted via both redox-dependent and redox-independent pathways in normal healthy cells (29).
In normal cells, Keap1 (Kelch-like ECH-associated protein 1), an E3 ubiquitin ligase substrate adaptor, regulates the level of Nrf2 protein in a redox-dependent fashion (5, 20, 51). The interaction between Nrf2 and Keap1 occurs via a “two-site tethering” process, otherwise called the “hinge and latch” mechanism. In this model, two motifs, a high-affinity ETGE motif and a low-affinity DLG motif, within the N-terminal Neh2 domain of Nrf2 each interact with a separate Kelch repeat domain present in the Keap1 homodimer (40). Both the ETGE motif and the DLG motif are required for the transcription factor to be repressed by Keap1 (28). In addition to its interaction with Nrf2, Keap1 also binds Cullin 3 (Cul3), which forms a core E3 ubiquitin ligase complex through an association with Ring-box1 protein (Rbx1, also called Roc1) (5, 10, 20, 51). The Keap1-Cul3-Rbx1 complex is able to ubiquitinate Nrf2 and target it for proteasomal degradation only under normal redox conditions, and upon exposure to oxidants or electrophiles, Cys-151, Cys-273, and Cys-288 in Keap1 become modified, leading to disturbance of the interaction between Nrf2 and Keap1 (8, 21, 49, 50). Failure of Nrf2 to dock simultaneously onto both Kelch repeat domains enables it to escape ubiquitination by Cul3-Rbx1 (21, 32, 47, 50). Thus, stress-related modification of Keap1 results in Nrf2 stabilization, accumulation of the transcription factor in the nucleus, and upregulation of ARE-driven genes. Perturbation of the Nrf2-Keap1 complex by oxidants and electrophiles is considered the principal mechanism by which Nrf2 accumulates and induces the ARE-gene battery. However, other regulatory mechanisms must exist in order to explain the following: (i) how Nrf2 contributes to the basal expression of certain ARE-driven genes under normal homeostatic conditions, (ii) how Nrf2 activity returns to its low basal levels after the intracellular redox balance has been restored, and (iii) how Nrf2 activity is limited during oxidative and electrophile stress.
Conventional cell signaling studies have suggested that Nrf2 might be regulated by protein phosphorylation (2, 6, 15, 18, 36, 44). Previously, we presented data suggesting that GSK-3β (glycogen synthase kinase 3β) influences the nuclear exclusion and inactivation of Nrf2 (37-39). However, the mechanistic connection between GSK-3 and Nrf2 remains largely unexplored. A number of studies have demonstrated that GSK-3 directs the ubiquitination and proteasomal degradation of various transcription factors and other proteins by SCF/β-TrCP; these include Snail (54), β-catenin (1, 22, 34), Gli2 and Gli3 (33, 48), Xom (55), Cdc 25a (19), FGD1 and -3 (11, 12), Mcl-1 (7), securin (24), prolactin receptor (46), and the phosphatase PHLPP1 (23). In these instances, GSK-3 phosphorylates a cluster of Ser/Thr residues in target proteins, which are then recognized by SCF/β-TrCP. In turn, the complex formed by SCF/β-TrCP binds the Cullin-1 (Cul1) scaffold protein to form a complete E3 ligase by association with a linker protein called Skp1 and with Rbx1. Therefore, β-TrCP is an adapter protein that contains a Skp1-binding site called F-box and a WD recognition domain for phosphorylated substrates in the consensus motif DpSGX(1-4)pS (9, 42).
To date, the existence of a phosphodegron in Nrf2 has not been explored. In the present article, we report that Nrf2 is destabilized as a consequence of its phosphorylation by GSK-3 and subsequent ubiquitination by SCF/β-TrCP. This pathway represents an alternative mechanism to the Keap1-dependent degradation of Nrf2 and provides a means by which this transcription factor can be regulated in a redox-independent manner.
MATERIALS AND METHODS
Cell culture and reagents.
Human embryonic kidney (HEK) 293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 80 mg/ml gentamicin. Mouse embryo fibroblasts (MEFs) from Keap1 knockout mice and wild-type littermates (kindly provided by Ken Itoh, Center for Advanced Medical Research, Hirosaki University Graduate School of Medicine, Japan) were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 0.5 U/ml penicillin, and 0.5 μg/ml streptomycin. Transient transfection of HEK293T cells was performed with calcium phosphate, using reagents from Sigma-Aldrich (Madrid, Spain). SB216763 and MG132 were from Sigma-Aldrich (Madrid, Spain). Cycloheximide (CHX) was purchased from Boehringer Mannheim (Germany).
Plasmids.
The expression vectors pcDNA3.1/V5HisB-mNrf2ΔETGE, pcDNA3.1/V5-mNrf2ΔETGEΔNeh6, pHis-Ub, and pET-mNrf2 have been described previously (29). The vectors pCGN-HA-GSK-3βΔ9 and pCGN-HA-GSK-3βY216F were provided by Akira Kikuchi (Department of Biochemistry, Faculty of Medicine, Hiroshima University), and GSK-3βS9A (pcDNA3-GSK-3βS9A-HA) was a kind gift of Richard Jope (Department of Psychiatry, University of Alabama at Birmingham, Birmingham, AL). A plasmid encoding β-TrCP2ΔFbox (pcDNA3β-TrCP2ΔFbox-HA) was provided by Serge Y. Fuchs (Department of Animal Biology, University of Pennsylvania, Philadelphia, PA). pcDNA3-Flag-β-TrCP2 was provided by Tomoki Chiba (Department of Molecular Biology, University of Tsukuba, Japan). The expression construct pcDNA3.1/V5HisB-mNrf2ΔETGE 6S/6A, with point mutations S335A, S338A, S342A, S347A, S351A, and S355A, was created using the GeneTailor site-directed mutagenesis system (Invitrogen) and the following primers: 5′ TGGAATTCAATGACTCTGACGCTGGCATTGCACTGAAGACGGCTCCCAGCCGAGCGCCCCAGA 3′ and 5′ GTCAGAGTCATTGAATTCCATTGTGCCTTCAGCGTGCTTC 3′ (mutation sites are underlined) using pcDNA3.1/V5 HisB-mNrf2ΔETGE 3S/3A (point mutations at S347A, S351A, and S355A) as a template in two sequential PCR amplifications with the following primers: forward (PCR1), 5′-GAGCGGCCCCAGAGCATGCCGTGGAGTCTGCCATTTACGG-3′, and reverse (PCR1), 5′-CGATCTCGAGGCCACTGTGCTGGAT-3′; forward (PCR2), 5′-CGATCATATGATGGACTTGGAGTTG-3′, and reverse (PCR2), 5′-CCGTAAATGGCAGACTCCACGGCATGCTCTGGGGCCGCTC-3′. The NdeI/XhoI fragment from pcDNA3.1/V5 HisB-mNrf2ΔETGE 6S/6A was cloned into pET-15b to generate the plasmid pET-mNrf26S/6A. All sequences were verified by automated sequencing. For the in vivo ubiquitination assays, the polyhistidine tag was removed from pcDNA3.1/V5HisB-mNrf2ΔETGE and pcDNA3.1/V5HisB-mNrf2ΔETGE 6S/6A by GeneTailor site-directed mutagenesis with the following pair of primers, which introduced a STOP codon in front of the 6-histidine-coding sequence: forward, 5′-TCGATTCTACGCGTACCGGTTAACATCACCATC-3′, and reverse, 5′-ACCGGTACGCGTAGAATCGAGACCGAGGAG-3′.
Luciferase assays.
Transient transfections of HEK293T cells were performed with the expression vectors for renilla (Promega, Madison, CA) and 3×ARE-Luc (a gift of J. Alam, Department of Molecular Genetics, Ochsner Clinic Foundation) as described previously (26). Cells were seeded on 24-well plates (100,000 cells per well), cultured for 16 h, and transfected using calcium phosphate. After 24 h of recovery from transfection, the cells were lysed and assayed for luciferase activity with a dual-luciferase assay system (Promega) according to the manufacturer's instructions. Relative light units were measured in a GloMax 96 microplate luminometer with dual injectors (Promega).
Immunoblotting.
The primary antibodies used were anti-V5 (Invitrogen, Carlsbad, CA), antihemagglutinin (anti-HA) (Covance, Berkeley, CA), anti-Flag (Sigma Aldrich, Madrid, Spain), anti-Nrf2 (generated by the Hayes laboratory), anti-glucose 6-phosphate dehydrogenase (G6PDH), and anti-β-actin and anti-lamin B (Santa Cruz Biotechnology, Santa Cruz, CA). Cell lysates were resolved by SDS-PAGE and transferred to Immobilon-P membranes (Millipore, Billerica, MA). These membranes were analyzed using the appropriate primary antibodies and peroxidase-conjugated secondary antibodies. Proteins were detected by enhanced chemiluminescence (Amersham Biosciences).
Coimmunoprecipitation (co-IP).
A monoclonal antibody from Invitrogen (catalog no. 37-3400) was used to immunoprecipitate β-TrCP, whereas “in-house” polyclonal antibodies were used to immunoprecipitate Nrf2. Cells were washed once with cold phosphate-buffered saline (PBS) and harvested by centrifugation at 1,100 rpm for 10 min. The cell pellet was resuspended in 0.45 ml of ice-cold lysis buffer (50 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin). Five microliters of the anti-Flag antibody (Sigma) or anti-V5 (Invitrogen) was added per lysate, and after incubation for 2 h at 4°C in a rotating wheel, gamma-bind Sepharose-protein G was added (Amersham Biosciences), followed by incubation for 1 h at 4°C. A lysate from nontransfected cells was incubated only with G protein to control for nonspecific binding (data not shown). The complexes were harvested by centrifugation and washed first with lysis buffer (washing buffer 1), second with washing buffer 2 (50 mM Tris-HCl [pH 7.6], 500 mM NaCl, 1% NP-40, 0.05% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride, and 1 μg/ml leupeptin), and finally with washing buffer 3 (50 mM Tris-HCl [pH 7.6], 1% NP-40, 0.05% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin). The samples were boiled, resolved by SDS-PAGE, and immunoblotted. Mouse IgG TrueBlot (eBiosciences) was used as a peroxidase-conjugated secondary antibody (1:10,000 dilution) because it reduces interference by the 55-kDa heavy and 23-kDa light chains of the immunoprecipitating antibody. In control experiments (not shown), it was established that anti-V5 antibodies did not recognize Flag-tagged proteins and that anti-Flag antibodies did not recognize V5-tagged proteins.
In vitro kinase assays.
In vitro phosphorylation was performed using as a substrate bacterially expressed His-tagged Nrf2, isolated using the ProBond purification system (Invitrogen). GSK-3β kinases were obtained by HA immunoprecipitation of whole-cell lysates of HEK293T cells that had been transfected with HA-GSK-3β Y216F, HA-GSK-3βΔ9, or HA-GSK-3βS9A. For in vitro phosphorylation studies, the substrate (0.5 μg) was incubated with the kinase and 5 μCi of [γ-32P]ATP in 25 μl of reaction buffer (10 mM MgCl2, 100 μM ATP in 40 mM morpholinepropanesulfonic acid (MOPS), pH 7.0, and 1 mM EDTA) for 30 min at 30°C with continuous shaking. Kinase reactions were resolved by SDS-PAGE, transferred to Immobilon-P membranes, and exposed to autoradiography. For preparation of phospho-Nrf2 substrate for in vitro ubiquitination assays, recombinant Nrf2 was submitted to the same conditions without inclusion of [γ-32P]ATP. The substrate (0.5 μg) was incubated with 5 ng of active recombinant GSK-3β (Upstate Biotechnology) per reaction in 25 μl of reaction buffer for 1 h at 30°C with continuous shaking. One microliter of these reaction mixtures was used for the in vitro ubiquitination assay.
Analysis of mRNA levels by real-time quantitative PCR.
Cells were plated on 60-mm dishes, and total cellular RNA was extracted using TRIzol reagent (Invitrogen). Equal amounts (1 μg) of RNA from each treatment were reverse transcribed for 75 min at 42°C using 5 U of avian myeloblastosis virus reverse transcriptase (Promega, Madison, WI) in the presence of 20 U of RNAsin (Promega). Quantitative PCR was performed with 20 ng of cDNA in a 25-μl reaction mixture that contained 0.3 μM primers and nucleotides, buffer, and Taq polymerase within the SYBR green I master mix (PE Applied Biosystems). Amplification was conducted in a 48-well Step One real-time PCR system (PE Applied Biosystems). PCR cycles proceeded as follows: initial denaturation for 10 min at 95°C and then 40 cycles of denaturation (15 s, 95°C), annealing (30 s, 60°C), and extension (30 s, 60°C). Primers were as follows: mNrf2 forward, 5′-ATCCAGACAGACACCAGTGGATC-3′, and reverse, 5′-GGCAGTGAAGACTGAACTTTCA-3′; hNrf2 forward, 5′ TCAGCATGCTACGTGATGAAG-3′, and reverse, 5′-TTTGCTGCAGGGAGTATT CA-3′; β-actin forward, 5′-TCCTTCCTGGGCATGGAG-3′, and reverse, 5′-AGGAGGAGCAATGATCTTGATCTT-3′. The gene expression primer and probe mixtures for β-TrCP1 and β-TrCP2 were Mm00477680_ml and Mm00460241_ml, respectively, purchased from ABI. The melting-curve analysis showed the specificity of the amplifications. The threshold cycle (CT), which inversely correlates with the target mRNA level, was measured as the cycle number at which the reporter fluorescent emission appeared above the background threshold (data not shown). To ensure that equal amounts of cDNA were added to the PCR mixture, the β-actin housekeeping gene was coamplified. Data analysis was based on the ΔCT method with normalization of the raw data to housekeeping genes as described in the manufacturer's manual (Applied Biosystems). All PCRs were performed in triplicate.
In vitro ubiquitination assay.
Purified recombinant proteins for β-TrCP-dependent ubiquitination were kindly provided by N. W. Pierce and R. J. Deshaies (Howard Hughes Medical Institute, Division of Biology, Pasadena, CA). Ubiquitination reactions were carried out as described previously (35) and contained ATP (2 mM), ubiquitin (30 μM), E1 (1 μM), Cdc34b (5 μM), SCFβ-TrCP (450 nM), and unphosphorylated or phosphorylated Nrf2 (20 ng) in ubiquitination buffer (30 mM Tris [pH 7.6], 5 mM MgCl2, 2 mM dithiothreitol [DTT], 100 mM NaCl). Prior to the ubiquitin reactions, E1, Cdc34b, and ubiquitin were incubated together for 2 min to allow E2 thioester formation. Reaction mixtures were incubated for 1 h at 25°C and quenched with SDS-PAGE buffer (62.5 mM Tris-HCl [pH 6.8], 12.5% [vol/vol] glycerol, 2% [wt/vol] SDS, 0.06% [wt/vol] bromophenol blue, 0.04% [vol/vol] 2-mercaptoethanol). Ubiquitination reactions were resolved by SDS-PAGE, followed by transfer to Immobilon-P membranes. Ubiquitinated proteins were detected with an antiubiquitin antibody (Chemicon, Millipore).
In vivo ubiquitination assay.
An in vivo ubiquitination assay was carried out using the method of Treier et al. (45). HEK293T cells were transfected with pHisUb along with the indicated plasmids. Approximately 24 h later, the transfected cells were washed with prewarmed phosphate-buffered saline and scraped into 0.4 ml of phosphate-buffered saline. A whole-cell lysate was prepared from 80 μl of the cell suspension and is referred to as the “input” fraction. His-tagged protein was purified from the remainder of the cell suspension as follows: the cell suspension was lysed by the addition of 1 ml of buffer A (6 M guanidine-HCl, 10 mM Tris in 0.1 M phosphate buffer, pH 8.0) supplemented with 5 mM imidazole. The resulting lysate was sonicated to reduce viscosity before 60 μl of Probond TM resin (Invitrogen, Carsband, CA) was added, and the mixture was rotated for 4 h at 25°C. Thereafter, the beads were washed sequentially with buffer A supplemented with 0.1% (vol/vol) Triton X-100, buffer B (8 M urea, 10 mM Tris in 0.1 M phosphate buffer, pH 8.0) supplemented with 0.1% Triton X-100, buffer C (8 M urea, 10 mM Tris in 0.1 M phosphate buffer, pH 6.5) supplemented with 0.2% Triton X-100, and finally buffer C supplemented with 0.1% Triton X-100. Bound material was eluted from the beads by suspension in 50 μl of modified Laemmli sample buffer (20 mM Tris-Cl, pH 6.8, 10% [vol/vol] glycerol, 0.8% [wt/vol] SDS, 0.1% [wt/vol] bromophenol blue, 0.72 M 2-mercaptoethanol, and 300 mM imidazole), followed by boiling for 4 min. The suspension was centrifuged (16,000 × g, 1 min, 20°C), and the resulting supernatant was collected and is referred to as the “pulldown” fraction.
siRNA assays.
The short interfering RNA (siRNA) used to knock down human GSK-3α or GSK-3β expression and the control scrambled siRNA sequence were purchased from Ambion Inc.: siRNA identifier (ID) s6236 for GSK-3α and siRNA ID s6241 for GSK-3β (Applied Biosystems). The siRNA used to knock down β-TrCP1 was from Thermo Scientific, Dharmacon (catalog no. L-044048-00-0005), and that used to knock down β-TrCP2 was from Applied Biosystems (catalog no. 175927). Briefly, HEK293T cells were seeded in 6-well plates (200,000 cells/well in 2 ml medium) before being transfected using calcium phosphate and the appropriate Nrf2 expression plasmids. To knock down GSK-3 isoforms, we performed siRNA transfection during two consecutive days. On the first day, we knocked down GSK-3 using 80 ng of Silencer Select validated siRNA with 30 μl of siPORT Amine reagent, and on the second day we used 40 ng of Silencer Select validated siRNA with 15 μl of siPORT Amine reagent. Twenty-four hours later, the cells were collected and Nrf2 and GSK-3 levels were analyzed.
Analyses of protein stability by pulse-labeling.
HEK293T cells were plated (2 × 106 cells/100-mm plate previously covered with poli-d-Lys) and transfected with the indicated plasmids. The following day, cells were washed (2×) with pulse-labeling medium (methionine-free, cysteine-free Dulbecco's modified Eagle's medium containing 10% dialyzed fetal bovine serum and 80 mg/ml gentamicin). Then, cells were incubated at 37°C for 30 min. The medium was removed and replaced with pulse-labeling medium containing 0.5 mCi/ml [35S]methionine (NEG 772 EasyTag Express protein labeling mix from Perkin Elmer). After proteins were labeled for 60 min, the medium was removed and the cells were washed twice with complete DMEM supplemented with 2 mM cysteine and 2 mM methionine (20-fold molar excess) for the appropriate times except for the “zero” samples. The zero points were rapidly washed with cold phosphate-buffered saline and lysed by the addition of radioimmune precipitation assay buffer (25 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin). The cell lysates were diluted 10-fold with IP buffer (Tris-HCl, pH 7.5, 20 mM; NaCl, 137 mM; NaF, 20 mM; sodium pyrophosphate, 1 mM; Na3VO4, 1 mM; Nonidet P-40, 1%; glycerol, 10%; phenylmethylsulfonyl fluoride, 1 mM; and leupeptin, 1 μg/ml) and sonicated (3× for 30 s each). Thereafter, the soluble portion of the lysates was prepared by centrifugation at 10,000 × g for 5 min at 4°C and retained for analysis. For immunoprecipitation, cell lysates were incubated with 3 μl of anti-V5 at 4°C with continuous rotation. After 2 h, a 20-μl slurry of protein G-Sepharose was added, and incubation was continued for an additional 1 h. Immunocomplexes were pelleted by centrifugation, washed three times in IP buffer, and resuspended in 40 μl of Laemmli sample buffer containing 1% β-mercaptoethanol. The samples were boiled, subjected to electrophoresis, and transferred to Immobilon-P membranes. After autoradiography, the blot was incubated with anti-V5 antibody to normalize the amount of Nrf2 protein per lane.
Image analyses and statistics.
Different band intensities (density arbitrary units), corresponding to immunoblot detection of protein samples, were quantified using the MCID software program (MCID, Cambridge, United Kingdom). Student's t test was used to assess differences between groups; a P value of <0.05 was considered significant. Unless indicated, all experiments were performed at least three times with similar results. The values in the graphs correspond to the means for at least three samples. Error bars indicate standard deviations.
RESULTS
GSK-3 modulates Nrf2 protein levels.
We first measured the levels of Nrf2 protein in HEK293T cells that had been treated with the two prototypic Nrf2 inducing agents, tBHQ (tert-butylhydroquinone) (15 μM, 6 h) and SFN (sulforaphane) (10 μM, 6 h), or the GSK-3 inhibitor SB216763 (20 μM, 6 h). As shown in Fig. 1A, under these conditions both tBHQ and SFN modestly increased the level of Nrf2 protein but had no effect on the level of β-catenin, which was used as a control for inhibition of GSK-3. Interestingly, SB216763, which increased β-catenin levels, also augmented the level of Nrf2 protein to an extent similar to that when HEK293T cells were treated with either tBHQ or SFN alone. Moreover, cotreatment with SB216763 and either of the two inducing agents resulted in a further elevation in the amount of the Nrf2 protein. mRNA levels for Nrf2 did not differ significantly among treatments (Fig. 1B), indicating that the increase in the Nrf2 protein produced by tBHQ, SFN, and SB216763 was not accompanied by upregulation of the Nrf2 gene (Fig. 1B).
FIG. 1.

GSK-3 modulates Nrf2 levels. (A) HEK293T cells were maintained in low-serum medium (0.5% FBS) for 16 h before finally being treated with 15 μM tBHQ, 10 μM SFN, or 20 μM SB216763 for 6 h. Upper blot, Nrf2 immunodetection in cell lysates; middle blot, β-catenin levels as a control for GSK-3 inhibition; lower blot, β-actin levels showing similar protein loads per lane. (B) Quantitative reverse transcriptase PCR (RT-PCR) determination of mRNA of Nrf2 normalized by β-actin from HEK293T cells treated as in panel A (expressed in arbitrary units). Variations are not statistically significant. (C) HEK293T cells were transfected with the 3×ARE-LUC and renilla control vectors, and after transfection the cells were treated with 3 μM tBHQ, 3 μM SFN, or 20 μM SB216763 for 16 h before luciferase activity was measured. Asterisks denote statistically significant differences between the untreated and SB216763-treated groups according to a Student t test.
The increase in the Nrf2 protein correlated with higher transactivation activity as determined with an ARE-driven luciferase reporter construct, 3×ARE-Luc, created from the mouse ho-1 promoter. HEK293T cells were transfected with 3×ARE-Luc, and after overnight recovery, they were stimulated for 16 h with tBHQ (3 μM), SFN (3 μM), or SB216763 (20 μM). As shown in Fig. 1C, tBHQ and SFN increased reporter gene activity between 2.5- and 3.0-fold. Inhibition of GSK-3 with SB216763 yielded a similar 2.5-fold increase. Moreover, combined treatment of either tBHQ or SFN with SB216763 led to a cooperative increase in luciferase activity of about 5.0-fold.
Nrf2 protein levels are regulated by GSK-3 in a Keap1-independent manner.
In order to determine if Keap1 might have a role in the GSK-3-dependent increase in the Nrf2 protein, we used MEFs derived from Keap1−/− mice and from Keap1+/+ littermates as a control. Treatment of Keap1+/+ MEFs with SB216763 (20 μM for 3 or 6 h) increased the basal level of the Nrf2 protein in a time-dependent manner (Fig. 2A); these results resemble those shown in Fig. 1. As expected, we found that the baseline Nrf2 protein level in Keap1−/− MEFs was higher than that in their wild-type counterparts. More importantly, SB216763 also increased the level of the Nrf2 protein in Keap1−/− MEFs, suggesting that an elevation of Nrf2 upon GSK-3 inhibition is not dependent on the Keap1 degradation pathway. The data reported for SB216763 (50% inhibitory concentration [IC50], 34 nM) were similar to those obtained using another potent and specific GSK-3 inhibitor, CT99021 (IC50, 10 nM) (data not shown).
FIG. 2.

GSK-3 inhibition promotes Nrf2 protein accumulation in a Keap1-independent manner. (A) MEFs from Keap1-deficient (Keap1−/−) or wild-type (Keap1+/+) littermates were maintained in low-serum medium for 16 h and then treated with 20 μM SB216763 for the times indicated. Upper blot, total Nrf2 protein levels; lower blot, β-actin levels showing that similar amounts of protein were loaded per lane. (B) MEFs were maintained in low-serum medium for 16 h and then treated with 20 μM SB216763 for 2 h prior to inhibition of protein synthesis with 40 μg/ml cycloheximide (CHX). Whole-cell lysates were prepared at the indicated times after addition of CHX. Upper blots, Nrf2 protein levels in Keap1+/+ and Keap1−/− fibroblasts. Lower blots, β-actin levels showing similar protein loaded per lane from Keap1+/+ and Keap1−/− fibroblasts. (C and D) Both graphs depict the natural logarithm of the relative levels of the Nrf2 protein as a function of CHX chase time in Keap1+/+ (C) or Keap1−/− (D) cells. The protein half-life has been determined in the linear range of the degradation curve.
In order to analyze if Nrf2 protein accumulation was due to an increase in its stability, we measured the half-life of Nrf2 in both MEF lines; densitometric analysis was validated using a sample dilution curve to ensure that the signal lay in the linear range (data not shown). MEFs were first treated with either SB216763 (20 μM) or vehicle for 2 h, and then protein synthesis was inhibited using CHX (40 μg/ml). Cells cotreated with SB216763 and CHX presented a delayed Nrf2 degradation curve compared to cells treated with CHX alone (Fig. 2B, C, and D). Thus, the half-life of Nrf2 increased from 26.5 min to 44.5 min in Keap1+/+ cells and from 24.4 min to 55.6 min in Keap1−/− fibroblasts in the presence of SB216763. Taken together, these results indicate that GSK-3 modulates Nrf2 levels through a Keap1-independent mechanism.
As an additional approach, we compared the levels of ectopically expressed V5-tagged wild-type Nrf2 with those of the V5-tagged mutant Nrf2ΔETGE, which cannot be turned over in a Keap1-dependent fashion. As shown in Fig. 3A and C, SB216763 produced a time-dependent accumulation of both Nrf2-V5 and Nrf2ΔETGE-V5 (at 3 h and 6 h), as well as β-catenin, which was used as an internal control for GSK-3 inhibition. To test whether GSK-3α and/or GSK-3β might be responsible for the increase in Nrf2, we knocked down each isoform using siRNA. HEK293T cells were first transfected with an expression vector for Nrf2-V5 or Nrf2ΔETGE-V5 before being transfected subsequently with siRNA against either GSK-3α, GSK-3β, or both isoforms. Knockdown of either isoform (by 70 to 80% according to densitometric analysis; not shown) resulted in a substantial increase in the levels of the Nrf2-V5 and Nrf2ΔETGE-V5 proteins (Fig. 3B and D). These results complement the observations made with the GSK-3 inhibitor and further indicate that Nrf2 stability is regulated by GSK-3.
FIG. 3.
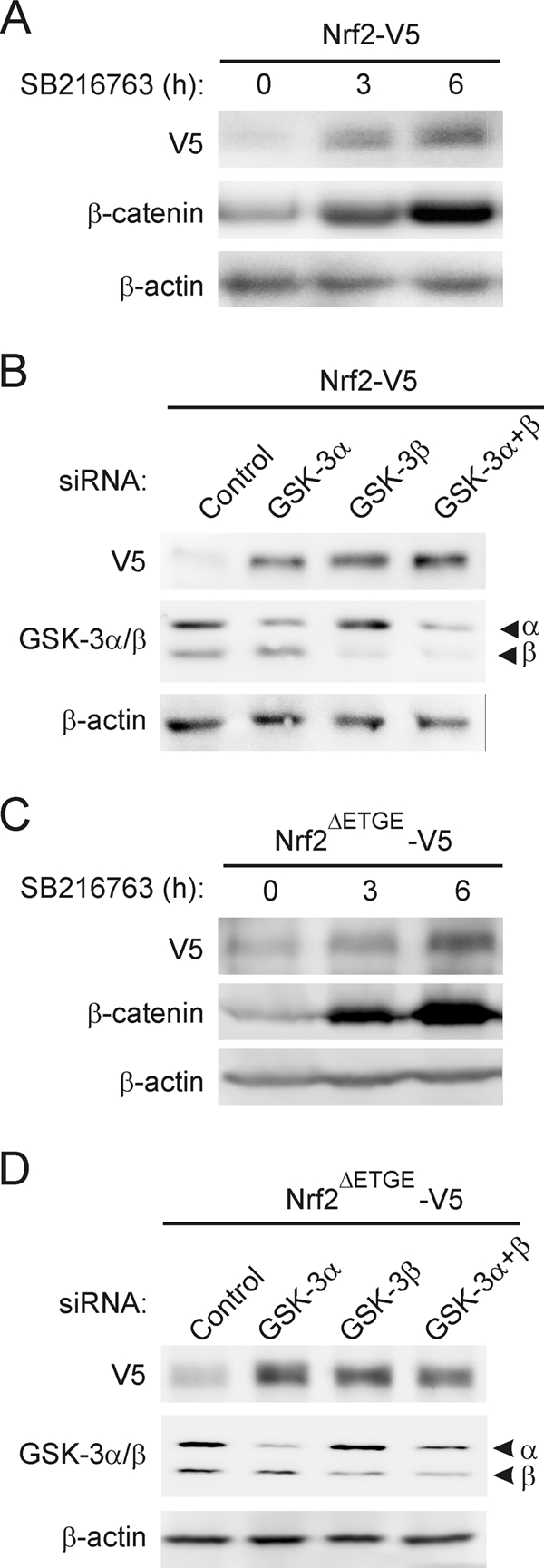
Modulation of Nrf2 protein levels by GSK-3 is independent of Keap1. (A and C) HEK293T cells were transfected with either V5-tagged Nrf2, the wild type, or the Keap1-insensitive Nrf2 version (Nrf2ΔETGE-V5), maintained in low-serum medium for 16 h, and then treated with 20 μM SB216763 for the indicated times. Upper blots, Nrf2-V5 (A) or Nrf2ΔETGE-V5 (C) protein levels; middle blots, β-catenin levels in the same cell lysates as a control for GSK-3 inhibition; lower blots, β-actin levels showing similar protein loads per lane. (B and D) HEK293T cells were transfected with either Nrf2-V5 (B) or Nrf2ΔETGE-V5 (D). After 24 h they were further transfected with siRNAs for GSK-3α, GSK-3β, or both or with a control scrambled siRNA as explained in Materials and Methods. Cells were lysed 24 h after siRNA transfection. Upper blots, Nrf2-V5 (B) or Nrf2ΔETGE-V5 (D) protein levels; middle blots, GSK-3α and -β protein levels; lower blots, β-actin levels showing that similar amounts of protein were loaded in each lane.
Degradation of Nrf2 by the E3 ubiquitin ligase SCF/β-TrCP complex.
Using bioinformatics, we found that Nrf2 contains an evolutionarily conserved sequence that resembles the consensus motif for substrate recognition by β-TrCP [K(X)nDSG(X)1-4S, where X is any residue]; in mouse Nrf2, this consensus sequence is located between amino acids 322 and 338 (Fig. 4A). It appears to be present in all vertebrate Nrf2 proteins except that of chicken, which contains the “DSG(X)1-4S” core sequence but lacks adjacent N-terminal lysine residues at the positions where it is found in mouse or human Nrf2. In this regard, chicken Nrf2 is similar to hSnail and hYAP. A comparison between this conserved DSGIS sequence in mammalian Nrf2 with those in proteins that are known to interact with β-TrCP indicates that it is most similar to those found in hEpoR and hYAP; these last two proteins possess just a Gly-Ile or Gly-Leu dipeptide between the two serine residues in the core consensus sequence.
FIG. 4.

Nrf2 is regulated by the E3 ligase β-TrCP complex through a destruction motif within its Neh6 domain. (A) Upper panel of sequences, primary structure of Nrf2 between residues 317 and 359 in the murine protein. The bold/underlined residues correspond to the putative site of phosphorylation by GSK-3, and the boxed ones correspond to the β-TrCP consensus motif. Lower panel of sequences, alignment of different well-known β-TrCP substrates showing the consensus sequence of the degradation motif. (B) Keap1−/− MEFs were transfected with siRNA against β-TrCP1 and/or β-TrCP2 as described in Materials and Methods. Upper blot, Nrf2 total protein levels; lower blot, glyceraldehyde-3-phosphate dehydrogenase (GADPH) levels showing similar protein loads per lane. (C) Quantitative RT-PCR determination of mRNA for β-TrCP1 and β-TrCP2 normalized by β-actin from MEFs transfected as in panel B. Asterisks denote statistically significant differences with P < 0.05. (D) HEK293T cells were cotransfected with the Nrf2-V5 or Nrf2ΔETGE-V5 expression vector and the indicated amounts of the β-TrCP dominant-negative (β-TrCPΔFbox-HA) mutant and then maintained in low-serum medium for 16 h. Whole-cell lysates were immunoblotted against anti-V5 antibody (upper blot) or anti-HA antibody (middle blot) or with anti-β-actin antibody showing similar amounts of protein per sample (lower blot). (E) In vitro ubiquinitation of Nrf2 by the β-TrCP complex. Bacterially expressed His-tagged Nrf2 was submitted to an in vitro kinase assay in the absence (Nrf2) or presence (phospho-Nrf2) of recombinant GSK-3β. Nrf2 and phospho-Nrf2 (20 ng) were incubated at 25°C for 1 h with purified ubiquitin, E1/cdc34b, β-TrCP/Skp1, and Cul1/Rbx1 as indicated. Polyubiquitinated Nrf2 was detected by immunoblotting with antiubiquitin antibody. Upper blot, antiubiquitin. E1-Ub, monoubiquitinated E1. Lower blot, anti-Nrf2 showing similar amounts of substrate loaded per lane. (F) p100 dishes of HEK293T cells were transfected with the indicated plasmids. After transfection (24 h), a whole-cell lysate (input) and an affinity-purified His-tagged fraction (His-pull-down) were blotted with anti-V5 antibody. Upper blot, anti-V5 input; middle blot, ectopically expressed HA-tagged GSK-3β; lower blot, anti-V5 detection in the His pulldown fraction. The bracket indicates the mobility of polyubiquitinated Nrf2-V5 (Nrf2-Ub) forms.
To determine the significance of the putative β-TrCP recognition motif in Nrf2, we measured the levels of the Nrf2 protein in Keap1−/− MEFs after knockdown of the β-TrCP1 and/or -2 isoform. As shown in Fig. 4B and C, fibroblasts transfected with siRNA against β-TrCP1 and/or -2 exhibited higher levels of the Nrf2 protein than cells transfected with control siRNA. These results indicate that both β-TrCP isoforms are involved in the regulation of Nrf2 in a Keap1-independent manner and support the hypothesis that a β-TrCP destruction motif exists in Nrf2.
In another set of experiments, we used a dominant-negative mutant of SCF/β-TrCP. We cotransfected HEK293T cells with expression constructs for Nrf2-V5 or Nrf2ΔETGE-V5, along with a dominant-negative β-TrCPΔFbox-HA mutant, which lacks the Fbox motif and cannot form functional SCF complexes. The presence of β-TrCPΔFbox-HA led to accumulation of both Nrf2-V5 and Nrf2ΔETGE-V5 (Fig. 4D), further indicating that β-TrCP participates in the degradation of Nrf2.
Initial evidence that Nrf2 is ubiquitinated in a β-TrCP-dependent manner was obtained from in vitro ubiquitination assays. Bacterially expressed His-tagged Nrf2 was subjected to an in vitro kinase assay (see below) in the absence or presence of recombinant GSK-3β and used as the substrate in an in vitro ubiquitination assay. As shown in Fig. 4E, Nrf2 was polyubiquitinated only in the presence of the complete β-TrCP-E3 ligase complex. Moreover, phospho-Nrf2 incorporated about 2-fold more ubiquitin than nonphosphorylated Nrf2. These results suggest that Nrf2 is a substrate for the β-TrCP ligase complex and pave the way for further analysis of this new mechanism of regulation.
Next, we studied the connection between β-TrCP-dependent regulation of Nrf2 stability and GSK-3. HEK293T cells were cotransfected with plasmids expressing Nrf2ΔETGE-V5 and either the control empty vector or an expression vector for His-tagged ubiquitin (His-Ub). These cotransfection experiments also included expression vectors for either the inactive kinase HA-GSK-3βY216F, containing a single Tyr216-to-Phe mutation in its activation loop, or the constitutively active HA-GSK-3βΔ9, which lacks the first 9 N-terminal residues, including Ser9, and is insensitive to downregulation by Akt phosphorylation. The amount of His-Ub bound to Nrf2 was measured in a pulldown assay that employed nickel columns (Fig. 4F). Protein recovered in the His-tagged fraction represented the polyubiquitinated transcription factor, since no such protein was recovered in this fraction unless Nrf2 and His-Ub were coexpressed. Interestingly, we could detect more highly polyubiquitinated Nrf2 forms in cells overexpressing HA-GSK-3βΔ9 than in HA-GSK-3βY216F-expressing cells. Overall, these results are consistent with the notion that β-TrCP plays a role in Nrf2 degradation.
GSK-3β participates in phosphorylation of serine residues within the β-TrCP destruction motif.
The β-TrCP recognition motif, along with adjacent C-terminal residues, contains a number of possible GSK-3 phosphorylation sites (Fig. 4A). To examine whether the β-TrCP motif is regulated by GSK-3β, we first analyzed whether bacterially expressed His-tagged mouse Nrf2ΔETGE-HisB and mouse Nrf2ΔETGE 6S/6A-HisB, in which all Ser residues at positions 335, 338, 342, 347, 351, and 355 had been changed to Ala, could be phosphorylated by GSK-3 in vitro. Kinase reactions were carried out using two constitutively active mutants of GSK-3β which lack pseudosubstrate inhibitory activity: the first mutant, HA-GSK-3βΔ9, contained a deletion of 9 N-terminal residues, including Ser9; the second mutant, HA-GSK-3βS9A, contained a single Ser9-to-Ala mutation. As shown in Fig. 5A, both Nrf2ΔETGE-HisB and Nrf2ΔETGE 6S/6A-HisB were phosphorylated in vitro by the two GSK-3β active mutants. However, Nrf2ΔETGE 6S/6A-HisB was phosphorylated to a much lesser extent than Nrf2ΔETGE-HisB, implying that GSK-3 phosphorylates at least some of the Ser residues in Nrf2 that are located immediately to the C-terminal side of the β-TrCP motif (see Discussion). Similar results were obtained with recombinant GSK-3β (data not shown); the inactive HA-GSK-3βY216F was unable to phosphorylate Nrf2 under the same purification conditions, thereby excluding the possibility that a contaminating kinase in the reaction mixture might be responsible for modifying Nrf2.
FIG. 5.

Mutation of 6S to A in the Neh6 domain of Nrf2 reduces its phosphorylation by GSK-3β. (A) Representative in vitro kinase assay on recombinant Nrf2 or Nrf26S/6A proteins with HA-tagged GSK-3β immunoprecipitated from HEK293T cells transfected with either the inactive HA-GSK-3βY216F mutant or the constitutively active HA-GSK-3βΔ9 or HA-GSK-3βS9A mutant. Upper blot, immunocomplex kinase assay; middle blot, immunodetection of Nrf2 to ensure similar quantity per reaction; lower blot, immunoblot with anti-HA antibody showing similar amount of GSK-3β per assay. (B) Basal levels of Nrf2ΔETGE-V5 or Nrf2ΔETGE 6S/6A-V5 in HEK293T-transfected cells. Upper blot, anti-V5; lower blot, β-actin. (C) Densitometric quantification of representative blots from panel B. (D) Nrf2 mRNA levels analyzed by quantitative RT-PCR from cells transfected as in panel B. (E) Graph depicts the ratio between Nrf2 protein and mRNA levels as shown in panels C and D.
To determine if phosphorylation of the serine cluster in Nrf2 between amino acids 335 and 355 affects its β-TrCP-directed proteasomal degradation, we compared the levels of Nrf2ΔETGE-V5 and Nrf2ΔETGE 6S/6A-V5 following their forced expression in HEK293 cells. Mutation of these Ser residues resulted in larger amounts of ectopic Nrf2 being observed without significant changes in their mRNA levels (Fig. 5B to E). Interestingly, the Nrf2ΔETGE 6S/6A-V5 mutant exhibited a faster mobility during SDS-PAGE than Nrf2ΔETGE-V5 (Fig. 5B); we attribute this variation to changes in the molecular mass of the polypeptide (104 Da for the six Ser-to-Ala substitutions) and a concomitant failure to phosphorylate the mutant protein.
By blocking translation of mRNA using CHX, we examined whether the 6-Ser cluster influenced the half-life of the Nrf2 protein. HEK293T cells were transfected with Nrf2ΔETGE-V5 or Nrf2ΔETGE 6S/6A-V5 and exposed to CHX (40 μg/ml) for various periods of time. As shown in Fig. 6A and B, mutation of the six Ser residues led to an approximately 2.0-fold increase in the half-life of Nrf2. Similar results were obtained with an Nrf2ΔETGEΔNeh6-V5 plasmid used as a control (29; also data not shown). Additional experiments were performed to investigate the stability of Nrf2 using the [35S]methionine pulse-chase assay. As shown in Fig. 6C and D, estimation of half-life for these mutants paralleled the results obtained with CHX experiments.
FIG. 6.

Mutation of the 6S cluster within the Neh6 domain increases Nrf2 protein stability. (A) HEK293T cells were transfected with Nrf2-ΔETGE-V5 or Nrf2ΔETGE 6S/6A-V5, serum starved for 16 h, and finally subjected to protein synthesis inhibition with 100 μg/ml CHX. Whole-cell lysates were prepared at the indicated times after addition of CHX. Upper blot, Nrf2-V5 protein levels. Lower blot, β-actin levels showing similar protein loads per lane. (B) The graph depicts the natural logarithm of the relative levels of the Nrf2-V5 protein as a function of CHX chase time. The protein half-life was determined using the linear part of the degradation curve. (C) HEK293T cells were transfected with Nrf2-ΔETGE-V5 or Nrf2-ΔETGE 6S/6A-V5, serum starved for 16 h, and then subjected to [35S]methionine/cysteine labeling for 1 h. Then, cells were incubated in high-methionine- and cysteine-containing medium and collected at the indicated times. For the 120-min time point, the cells were treated with MG132 as an internal control. Upper blot, 35S autoradiography; lower blot, anti-V5 antibody. (D) The graph shows the natural logarithm of the relative levels of Nrf2 as a function of 35S chase time. The half-life has been determined in the linear range of the degradation curve.
Nrf2 lacking the 6S cluster in its Neh6 domain is insensitive to GSK-3-induced degradation.
The relevance of the β-TrCP motif and its associated Ser residues to GSK-3-stimulated degradation of Nrf2 was analyzed by treating cells that had been transfected with either Nrf2ΔETGE-V5 or Nrf2ΔETGE 6S/6A-V5 with SB216763 for 3 h or 6 h. As expected, inhibition of GSK-3 resulted in the accumulation of Nrf2ΔETGE-V5 (Fig. 7A). In contrast, treatment with SB216763 did not increase the amount of the Nrf2ΔETGE 6S/6A-V5 protein. In addition, we used siRNAs against GSK-3α, GSK-3β, or both isoforms in HEK293T cells that had been transfected previously with expression vectors for either Nrf2ΔETGE-V5 or Nrf2ΔETGE 6S/6A-V5. Knockdown of either GSK-3α or GSK-3β (by 70 to 80% according to densitometric analysis; not shown) led to an increase in the amount of Nrf2ΔETGE-V5. In contrast, knockdown of GSK-3 did not alter the levels of the Nrf2ΔETGE 6S/6A-V5 protein (Fig. 7B). These data show that the six Ser residues within or adjacent to the β-TrCP destruction motif are necessary for the degradation of Nrf2 stimulated by GSK-3.
FIG. 7.

Mutation of the 6S cluster within the Neh6 domain renders the Nrf2 protein insensitive to GSK-3. (A) HEK293T cells were transfected with Nrf2-ΔETGE-V5 or Nrf2ΔETGE 6S/6A-V5, maintained in low-serum medium for 16 h, and then incubated with 20 μM SB216763 for the indicated times. Upper blot, anti-V5 immunoblot; middle blot, β-catenin levels as a control of GSK-3 inhibition; lower blot, β-actin levels showing the similar protein loads per lane. (B) HEK293T cells were transfected with either Nrf2ΔETGE-V5 or Nrf2ΔETGE 6S/6A-V5. After 24 h they were further transfected with siRNAs for GSK-3α, GSK-3β, or both or with a control scramble siRNA as described in Materials and Methods. Cells were lysed 24 h after siRNA transfection. Upper blot, anti-V5 antibody; middle blot, GSK-3α and -β protein levels; lower blot, β-actin levels showing similar protein loads per lane.
The SCF/β-TrCP complex does not promote ubiquitination of Nrf2ΔETGE 6S/6A-V5.
To gain further information about how β-TrCP negatively regulates Nrf2, we cotransfected HEK293T cells with expression vectors for Nrf2ΔETGE-V5 or Nrf2ΔETGE 6S/6A-V5 along with His-Ub and HA-GSK-3βY216F or HA-GSK-3βΔ9. The basal amount of ubiquitinated Nrf2ΔETGE 6S/6A-V5 was smaller than that of Nrf2ΔETGE-V5 (Fig. 8A). It is noteworthy that we still detected ubiquitination of Nrf2ΔETGE 6S/6A-V5, indicating the presence of other possible proteasome degradation motifs apart from those involving Keap1 and β-TrCP. As expected, HA-GSK-3βΔ9 promoted an increase in Nrf2ΔETGE-V5 ubiquitination, but, in contrast, it did not change the amount of ubiquitinated Nrf2ΔETGE 6S/6A-V5.
FIG. 8.

The 6S cluster within the Neh6 domain is a target of β-TrCP in a GSK-3-dependent manner. (A) p100 dishes of HEK293T cells were transfected with the indicated plasmids. After transfection (24 h), a whole-cell lysate (input) and an affinity-purified His-tagged fraction (His-Pull-down) were blotted with anti-V5 antibody. Upper panel, anti-V5 input detection; middle panel, ectopically expressed HA-tagged GSK-3β; lower panel, anti-V5 detection in His pulldown fractions. The bracket indicates the mobility of polyubiquitinated Nrf2-V5 (Nrf2-Ub) forms. (B) Keap1−/− or Nrf2−/− MEF cells were serum starved for 6 h before they were treated for a further 2 h with vehicle control, 10 μM LY294002, or 5 μM CT99021. Thereafter, the fibroblasts that had been subjected to different treatments were harvested separately, and each was lysed in 0.45 ml of buffer. A 50-μl portion of the lysate was retained as “input” (labeled on the left), and the remainder was immunoprecipitated with antibodies against either Nrf2 (labeled IP Nrf2) or β-TrCP (labeled IP β-TrCP) using Sepharose-protein G beads. The immunoprecipitated material was analyzed by Western blotting using antibodies against Nrf2 or β-TrCP as indicated to the left of the blots. (C) HEK293T cells were cotransfected with the indicated plasmids or with an empty vector (Mock). One-fifth of whole-protein lysate was used to control for protein expression as shown in the three upper panels. The rest of the protein lysates were immunoprecipitated with anti-Flag or anti-V5 antibodies and immunoblotted as indicated in the four lower panels. The arrow points the specific β-TrCP immunoreactive band. (D) After cotransfection of HEK293T cells with either Nrf2ΔETGE-V5 or Nrf2ΔETGE 6S/6A-V5 and β-TrCPΔFbox-HA, cells were maintained in low-serum medium for 16 h. Upper panel, anti-V5 antibody; middle panel, anti-HA antibody; lower panel, anti-β-actin antibody showing a similar protein load.
Since substrate phosphorylation increases the affinity of β-TrCP for its target proteins, we used a coimmunoprecipitation assay to examine whether the association of Nrf2 with β-TrCP is dependent on phosphorylation (Fig. 8B). GSK-3 activity was modulated in Keap1-null MEFs through indirect activation of this kinase by serum starvation for 6 h or by the use of LY294002 (10 μM, 2 h) and direct inhibition with the inhibitor CT99021 (5 μM, 2 h). LY294002 inhibits the phosphatidylinositol 3-kinase (PI3K)/Akt axis, which in these cells is a negative regulator of GSK-3, and therefore produces an increase in GSK-3 activity (data not shown). Nrf2-null MEFs were used as a control. As expected, basal Nrf2 levels were modified by these treatments, with slightly higher levels in CT99021-treated cells and lower levels in LY294002-treated and serum-starved cells. In pulldown experiments, equal immunoprecipitation of Nrf2 was achieved by using a limiting amount of Nrf2 antibody. Interestingly, when Nrf2 was immunoprecipitated under these conditions, we observed co-IP of β-TrCP in complexes from serum-starved and LY294002-treated cells, but less β-TrCP was observed in complexes from CT99021-treated cells. Conversely, when β-TrCP was immunoprecipitated, the amount of Nrf2 recovered in the complex was much larger under conditions of GSK-3 activation by either serum starvation or LY294002 treatment than under conditions where GSK-3 was inhibited with CT99021. These results indicate that association of Nrf2 and β-TrCP is enhanced by GSK-3-mediated phosphorylation.
Further evidence for the role of the Ser cluster of the Neh6 degron in interaction with β-TrCP was obtained in HEK293T cells cotransfected with expression vectors for Flag-tagged β-TrCP and either Nrf2ΔETGE-V5 or Nrf2ΔETGE 6S/6A-V5, together with either HA-GSK-3βY216F or HA-GSK-3βΔ9 (Fig. 8C). After 24 h of recovery from transfection, cellular lysates were immunoprecipitated with anti-V5 or anti-Flag antibodies. Immunoprecipitation with both antibodies revealed that an association was observed between β-TrCP and Nrf2ΔETGE-V5 but not between β-TrCP and Nrf2ΔETGE 6S/6A-V5. Furthermore, when HA-GSK-3βΔ9 was overexpressed, we could detect larger amounts of Nrf2ΔETGE-V5 bound to β-TrCP but not larger amounts of Nrf2ΔETGE 6S/6A-V5 bound to β-TrCP. These data correlate well with the results presented in Fig. 8A, and together they show that the lower ubiquitination of the 6S mutant is compatible with an inability to interact with β-TrCP.
As shown in Fig. 8D, the lack of association between β-TrCP and Nrf2 was accompanied by a failure of SCF/β-TrCP to regulate the transcription factor. HEK293T cells were cotransfected with expression vectors encoding either Nrf2ΔETGE-V5 or Nrf2ΔETGE 6S/6A-V5 and a dominant-negative β-TrCPΔFbox-HA mutant. Consistent with our previous findings (Fig. 4D), this dominant-negative version of the SCF/β-TrCP complex increased the level of the Nrf2ΔETGE-V5 protein. In contrast, the basal level of the Nrf2ΔETGE 6S/6A-V5 protein was higher than that of Nrf2ΔETGE-V5, but it was insensitive to further accumulation when coexpressed with β-TrCPΔFbox-HA. These findings suggest that the 6-Ser cluster within the Neh6 domain of Nrf2 is required in order to allow SCF/β-TrCP to negatively control the stability of Nrf2.
DISCUSSION
Since Nrf2 participates in multiple aspects of cell physiology, it is essential that its activity is stringently controlled. Herein, we have uncovered a novel pathway for Nrf2 degradation that is controlled by GSK-3 and the SCF/β-TrCP E3 ubiquitin ligase.
Some transcription factors, including Snail and β-catenin, are subject to dual regulation by GSK-3β in that the kinase controls both their subcellular location and their degradation. Thus, GSK-3β phosphorylates both Snail and β-catenin at 6-Ser clusters, thereby modulating their subcellular distribution and their β-TrCP-mediated ubiquitination (54). We speculated that Nrf2 might be similarly negatively regulated by GSK-3 and β-TrCP. In the present article, we have focused on the regulation of Nrf2 stability by GSK3/β-TrCP because Salazar et al. (39) have already described the nuclear exclusion of Nrf2 following its modification by GSK-3. Now we report that a potent and selective inhibitor of GSK-3, SB216367 (arylindolemaleimide; IC50, 34 nM) (also data not shown with CT99021, aminopyrimidine; IC50, 10 nM [30]), and siRNAs against GSK-3 isoforms produce an increase in the level of the Nrf2 protein. Stabilization of Nrf2 following GSK-3 inhibition occurred in Keap1-deficient MEFs and in an ectopically expressed Nrf2ΔETGE mutant that is insensitive to repression by Keap1. Therefore, regulation of Nrf2 protein stability by GSK-3 represents a previously unrecognized mechanism that is independent of the canonical Keap1 degradation pathway.
Within its Neh6 domain, Nrf2 contains a conserved region that conforms to the consensus motif recognized by β-TrCP. Thus, Neh6 contains a typical DSG motif followed by an isoleucine and a phosphorylatable Ser residue. This DSGIS sequence is identical to that found in the erythropoietin receptor (31) and the Yes-associated protein (52). Interestingly, using a completely different approach, McMahon et al. (29) reported a Keap1-independent degron in the same region of Nrf2 that β-TrCP recognizes and postulated that it limited the stability of the transcription factor during redox stress, when the E3 ubiquitin ligase substrate adaptor activity of Keap1 is inhibited. The region in mouse Nrf2 that was identified as a redox-independent degron by McMahon and colleagues was narrowed down to residues 329 to 339, but this was not reported to be a β-TrCP binding site, nor was it appreciated that it could be phosphorylated by GSK-3. We now propose that this redox-independent degron in the Neh6 domain also contains a cluster of Ser residues (in mouse Nrf2 these are Ser335, Ser338, Ser342, Ser347, Ser351, and Ser355) that are phosphorylated by GSK-3, probably in concert with other kinases.
GSK-3 provides a complex mode of regulation by protein phosphorylation, and presumably this applies to the phosphorylation of Nrf2. For the purpose of this discussion, we will exemplify GSK-3α and -β regulation of Nrf2 with the Ser/Thr protein kinase Akt. In cells exposed to growth and trophic factors, it is expected that GSK-3α and -β will be inactivated by Akt-mediated phosphorylation at Ser21 and Ser9, respectively. On the other hand, during prolonged oxidative stress when Akt is downregulated (for instance, by ceramide-activated phosphatase) (27, 40), it is likely that GSK-3 activity will increase. Moreover, the mechanism of action is also unusual in that GSK-3 shows a preference for substrates that have been phosphorylated previously by other kinases. Experiments under way will identify the residues in Nrf2 phosphorylated by GSK-3 and other cooperating kinases that cause activation of the Neh6 phosphodegron. Nevertheless, we can conclude from the data presented herein that phosphorylation of the Neh6 degron by GSK-3 generates a structural motif that is recognized by the β-TrCP E3 ligase.
Therefore, at least two mechanisms have evolved to control ubiquitin-proteasome-dependent degradation of Nrf2: the first, based on redox-dependent interactions between the Neh2 domain of Nrf2 and Keap1, leads to ubiquitination via the Keap1-Cul3/Rbx1 E3 ligase complex; the second, based on phosphorylation of the Neh6 domain of Nrf2, leads to ubiquitination via a β-TrCP/Cul1 E3 ligase complex. Both regulatory mechanisms must act in concert, and they may reflect differential regulation of Nrf2 under distinct physiological circumstances. In Fig. 9, we propose a “dual degradation” model that allows cross talk between both pathways under several physiological and pathological conditions, as follows.
FIG. 9.

Scheme showing the main participants in the “dual degradation” model. See Discussion for details.
(i) In a normal redox environment with typical growth and trophic support, a minimal amount of Nrf2 activity is required to maintain basal expression of some ARE-driven genes. Under these circumstances, the thiol groups of the redox-sensitive Cys residues in Keap1 are unmodified because electrophiles and prooxidants are present at low levels, and GSK-3α and -β should be inhibited by phosphorylation at Ser21 and Ser9, respectively, by active kinases such as Akt. Hence, under these conditions Nrf2 degradation occurs mainly via Keap1-Cul3/Rbx1 complexes.
(ii) Under the circumstances of strong oxidant or electrophilic injury with typical growth and trophic support, both GSK-3 and Nrf2 are subject to a temporal biphasic regulation. In the initial phase, oxidative stress leads to inhibition of several phosphatases, resulting in activation of Akt and further phosphorylation of GSK-3, causing inactivation of the kinase (27). At the same time, modification of thiols in Keap1 will lead to its inactivation and to stabilization of Nrf2, which will result in the induction of ARE-driven genes. In the late phase, Akt will be inhibited by ceramide-activated phosphatases or other mechanisms, and GSK-3 will become activated. Following its activation, GSK-3 will target Nrf2 for β-TrCP-mediated proteasomal degradation. Thus, we predict that under these conditions, GSK-3 and β-TrCP should provide the predominant mechanism by which Nrf2 is ubiquitinated and directed to the 26S proteasome for degradation. This biphasic model has a relevant corollary on cell fate: if GSK-3 is activated before Nrf2 restores redox homeostasis, cells should die as a result of oxidative stress.
(iii) Under conditions of modest redox perturbation and deprivation of growth or trophic support or signaling, leading to GSK-3 activation, the thiol groups in Keap1 will not be modified to a substantial degree, and it should target Nrf2 for degradation in a relatively efficient manner. At the same time, activation of GSK-3 will also target Nrf2 for degradation through the β-TrCP pathway. The suppression of Nrf2 activity to below normal levels will lead to a further diminution in the basal expression of antioxidant and detoxification genes, thereby sensitizing cells to oxidative stress. Under such circumstances, the ensuing accumulation of ROS will oxidize critical Cys residues in Keap1, leading to a loss of its substrate adaptor activity, which allows Nrf2 to escape Cul3/Rbx1-directed degradation and in turn enables redox homeostasis to be restored. In this scenario, Keap1 cycles between the reduced and oxidized state, and it compensates the redox fluctuations that result from Nrf2 being targeted for degradation by β-TrCP.
The above “dual degradation” model has implications in pathology and the design of new therapeutic strategies. For instance, low Keap1 activity has been reported in tumors of lung, gallbladder, breast, and head and neck origins (reviewed in reference 12), suggesting that increases in Nrf2 may, in certain circumstances, contribute to the development of neoplastic disease. We speculate that drugs that activate GSK-3, such as Akt inhibitors, might be of therapeutic value because they may decrease Nrf2 levels through stimulating β-TrCP-mediated degradation. In neurodegenerative pathologies, including Alzheimer's and Parkinson's diseases, or even during the normal aging process, where exacerbated GSK-3β activity has been reported, we speculate that β-TrCP-mediated degradation of Nrf2 may be responsible for oxidative stress that accompanies these conditions. In such instances, chemopreventive agents might block Keap1 activity and lead to increases in Nrf2 levels that could correct perturbations in redox homeostasis. Indeed, it has been reported that lipoic acid restores the decline in glutathione levels and Nrf2 transcriptional activity that occurs with aging (43). Similarly, SFN is neuroprotective in a model of Parkinsonism in mice exposed to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (A. Jazwa, A. I. Rojo, N. G. Innamorato, and A. Cuadrado, unpublished results). Moreover, under these pathological conditions GSK-3β inhibitors should also cooperate to increase Nrf2 levels.
In conclusion, the results presented here have revealed a Keap1-independent mechanism by which the stability of Nrf2 is controlled through GSK-3/β-TrCP. This newly identified mechanism must act in concert with the canonical Keap1 pathway. While the “dual degradation” model presented here is still speculative, it provides the basis to assess the contribution of each degron pathway in different physiological and pathological scenarios and to evaluate their potential use in therapy.
Acknowledgments
This work was supported by grants SAF2007-62646 and SAF2010-17822 from the Spanish Ministry of Science and Innovation (awarded to A.C.), by grants C4909/A5942 and C4909/A9990 from Cancer Research UK (awarded to J.D.H.), and by a grant from Tenovus Scotland (awarded to M.M. and J.D.H.). P.R. is a recipient of a Formación de Profesorado Universitario fellowship from the Spanish Ministry of Science and Innovation.
We thank Ronald T. Hay, Calum Sutherland, and Lindsay A. Allan for valuable discussions.
Footnotes
Published ahead of print on 18 January 2011.
REFERENCES
- 1.Aberle, H., A. Bauer, J. Stappert, A. Kispert, and R. Kemler. 1997. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 16:3797-3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Apopa, P. L., X. He, and Q. Ma. 2008. Phosphorylation of Nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. J. Biochem. Mol. Toxicol. 22:63-76. [DOI] [PubMed] [Google Scholar]
- 3.Calkins, M. J., et al. 2009. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid. Redox Signal. 11:497-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cuadrado, A., P. Moreno-Murciano, and J. Pedraza-Chaverri. 2009. The transcription factor Nrf2 as a new therapeutic target in Parkinson's disease. Expert Opin. Ther. Targets 13:319-329. [DOI] [PubMed] [Google Scholar]
- 5.Cullinan, S. B., J. D. Gordan, J. Jin, J. W. Harper, and J. A. Diehl. 2004. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 24:8477-8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cullinan, S. B., et al. 2003. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 23:7198-7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding, Q., et al. 2007. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol. Cell. Biol. 27:4006-4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dinkova-Kostova, A. T., et al. 2002. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. U. S. A. 99:11908-11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feldman, R. M., C. C. Correll, K. B. Kaplan, and R. J. Deshaies. 1997. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell 91:221-230. [DOI] [PubMed] [Google Scholar]
- 10.Furukawa, M., and Y. Xiong. 2005. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 25:162-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayakawa, M., H. Kitagawa, K. Miyazawa, M. Kitagawa, and K. Kikugawa. 2005. The FWD1/beta-TrCP-mediated degradation pathway establishes a ‘turning off switch’ of a Cdc42 guanine nucleotide exchange factor, FGD1. Genes Cells 10:241-251. [DOI] [PubMed] [Google Scholar]
- 12.Hayakawa, M., et al. 2008. Novel insights into FGD3, a putative GEF for Cdc42, that undergoes SCF(FWD1/beta-TrCP)-mediated proteasomal degradation analogous to that of its homologue FGD1 but regulates cell morphology and motility differently from FGD1. Genes Cells 13:329-342. [DOI] [PubMed] [Google Scholar]
- 13.Hayes, J. D., and M. McMahon. 2009. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 34:176-188. [DOI] [PubMed] [Google Scholar]
- 14.Homma, S., et al. 2009. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin. Cancer Res. 15:3423-3432. [DOI] [PubMed] [Google Scholar]
- 15.Huang, H. C., T. Nguyen, and C. B. Pickett. 2000. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc. Natl. Acad. Sci. U. S. A. 97:12475-12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Innamorato, N. G., I. Lastres-Becker, and A. Cuadrado. 2009. Role of microglial redox balance in modulation of neuroinflammation. Current Opin. Neurol. 22:308-314. [DOI] [PubMed] [Google Scholar]
- 17.Innamorato, N. G., et al. 2008. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. 181:680-689. [DOI] [PubMed] [Google Scholar]
- 18.Jain, A. K., and A. K. Jaiswal. 2006. Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J. Biol. Chem. 281:12132-12142. [DOI] [PubMed] [Google Scholar]
- 19.Kang, T., et al. 2008. GSK-3 beta targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3 beta inactivation correlates with Cdc25A overproduction in human cancers. Cancer Cell 13:36-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobayashi, A., et al. 2004. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24:7130-7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobayashi, M., and M. Yamamoto. 2006. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzyme Regul. 46:113-140. [DOI] [PubMed] [Google Scholar]
- 22.Latres, E., D. S. Chiaur, and M. Pagano. 1999. The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene 18:849-854. [DOI] [PubMed] [Google Scholar]
- 23.Li, X., J. Liu, and T. Gao. 2009. Beta-TrCP-mediated ubiquitination and degradation of PHLPP1 are negatively regulated by Akt. Mol. Cell. Biol. 29:6192-6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Limon-Mortes, M. C., et al. 2008. UV-induced degradation of securin is mediated by SKP1-CUL1-beta TrCP E3 ubiquitin ligase. J. Cell Sci. 121:1825-1831. [DOI] [PubMed] [Google Scholar]
- 25.Maher, J., and M. Yamamoto. 2010. The rise of antioxidant signaling—the evolution and hormetic actions of Nrf2. Toxicol. Appl. Pharmacol. 244:4-15. [DOI] [PubMed] [Google Scholar]
- 26.Martin, D., et al. 2004. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 279:8919-8929. [DOI] [PubMed] [Google Scholar]
- 27.Martin, D., M. Salinas, N. Fujita, T. Tsuruo, and A. Cuadrado. 2002. Ceramide and reactive oxygen species generated by H2O2 induce caspase-3-independent degradation of Akt/protein kinase B. J. Biol. Chem. 277:42943-42952. [DOI] [PubMed] [Google Scholar]
- 28.McMahon, M., N. Thomas, K. Itoh, M. Yamamoto, and J. D. Hayes. 2006. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 281:24756-24768. [DOI] [PubMed] [Google Scholar]
- 29.McMahon, M., N. Thomas, K. Itoh, M. Yamamoto, and J. D. Hayes. 2004. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 279:31556-31567. [DOI] [PubMed] [Google Scholar]
- 30.Meijer, L., M. Flajolet, and P. Greengard. 2004. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol. Sci. 25:471-480. [DOI] [PubMed] [Google Scholar]
- 31.Meyer, L., et al. 2007. Beta-Trcp mediates ubiquitination and degradation of the erythropoietin receptor and controls cell proliferation. Blood 109:5215-5222. [DOI] [PubMed] [Google Scholar]
- 32.Nioi, P., and T. Nguyen. 2007. A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem. Biophys. Res. Commun. 362:816-821. [DOI] [PubMed] [Google Scholar]
- 33.Pan, Y., C. B. Bai, A. L. Joyner, and B. Wang. 2006. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 26:3365-3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Papkoff, J., and M. Aikawa. 1998. WNT-1 and HGF regulate GSK3 beta activity and beta-catenin signaling in mammary epithelial cells. Biochem. Biophys. Res. Commun. 247:851-858. [DOI] [PubMed] [Google Scholar]
- 35.Petroski, M. D., and R. J. Deshaies. 2005. In vitro reconstitution of SCF substrate ubiquitination with purified proteins. Methods Enzymol. 398:143-158. [DOI] [PubMed] [Google Scholar]
- 36.Pi, J., et al. 2007. Molecular mechanism of human Nrf2 activation and degradation: role of sequential phosphorylation by protein kinase CK2. Free Radic. Biol. Med. 42:1797-1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rojo, A. I., et al. 2008. Functional interference between glycogen synthase kinase-3 beta and the transcription factor Nrf2 in protection against kainate-induced hippocampal cell death. Mol. Cell Neurosci. 39:125-132. [DOI] [PubMed] [Google Scholar]
- 38.Rojo, A. I., M. R. Sagarra, and A. Cuadrado. 2008. GSK-3beta down-regulates the transcription factor Nrf2 after oxidant damage: relevance to exposure of neuronal cells to oxidative stress. J. Neurochem. 105:192-202. [DOI] [PubMed] [Google Scholar]
- 39.Salazar, M., A. I. Rojo, D. Velasco, R. M. de Sagarra, and A. Cuadrado. 2006. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 281:14841-14851. [DOI] [PubMed] [Google Scholar]
- 40.Salinas, M., R. Lopez-Valdaliso, D. Martin, A. Alvarez, and A. Cuadrado. 2000. Inhibition of PKB/Akt1 by C2-ceramide involves activation of ceramide-activated protein phosphatase in PC12 cells. Mol. Cell Neurosci. 15:156-169. [DOI] [PubMed] [Google Scholar]
- 41.Shibata, T., et al. 2008. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl. Acad. Sci. U. S. A. 105:13568-13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skowyra, D., K. L. Craig, M. Tyers, S. J. Elledge, and J. W. Harper. 1997. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell 91:209-219. [DOI] [PubMed] [Google Scholar]
- 43.Suh, J. H., et al. 2004. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. U. S. A. 101:3381-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun, Z., Z. Huang, and D. D. Zhang. 2009. Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PloS One 4:e6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Treier, M., L. M. Staszewski, and D. Bohmann. 1994. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell 78:787-798. [DOI] [PubMed] [Google Scholar]
- 46.Varghese, B., et al. 2008. Polyubiquitination of prolactin receptor stimulates its internalization, postinternalization sorting, and degradation via the lysosomal pathway. Mol. Cell. Biol. 28:5275-5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wakabayashi, N., et al. 2004. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. U. S. A. 101:2040-2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang, B., and Y. Li. 2006. Evidence for the direct involvement of βTrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. U. S. A. 103:33-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu, X., and T. Kensler. 2005. Nrf2 as a target for cancer chemoprevention. Mutation Res. 591:93-102. [DOI] [PubMed] [Google Scholar]
- 50.Zhang, D. D., and M. Hannink. 2003. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 23:8137-8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang, D. D., S. C. Lo, J. V. Cross, D. J. Templeton, and M. Hannink. 2004. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 24:10941-10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao, B., L. Li, K. Tumaneng, C. Y. Wang, and K. L. Guan. 2010. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 24:72-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao, F., et al. 2009. Nrf2 promotes neuronal cell differentiation. Free Radic. Biol. Med. 47:867-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou, B. P., et al. 2004. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell biol. 6:931-940. [DOI] [PubMed] [Google Scholar]
- 55.Zhu, Z., and M. Kirschner. 2002. Regulated proteolysis of Xom mediates dorsoventral pattern formation during early Xenopus development. Dev. Cell 3:557-568. [DOI] [PubMed] [Google Scholar]