

Abstract
Background
Lung cancer diagnosis in tissue material with commonly used histological techniques is sometimes inconvenient and in a number of cases leads to ambiguous conclusions. Frequently advanced immunostaining techniques have to be employed, yet they are both time consuming and limited. In this study a proteomic approach is presented which may help provide unambiguous pathologic diagnosis of tissue material.
Methods
Lung tissue material found to be pathologically changed was prepared to isolate proteome with fast and non selective procedure. Isolated peptides and proteins in ranging from 3.5 to 20 kDa were analysed directly using high resolution mass spectrometer (MALDI-TOF/TOF) with sinapic acid as a matrix. Recorded complex spectra of a single run were then analyzed with multivariate statistical analysis algorithms (principle component analysis, classification methods). In the applied protocol we focused on obtaining the spectra richest in protein signals constituting a pattern of change within the sample containing detailed information about its protein composition. Advanced statistical methods were to indicate differences between examined groups.
Results
Obtained results indicate changes in proteome profiles of changed tissues in comparison to physiologically unchanged material (control group) which were reflected in the result of principle component analysis (PCA). Points representing spectra of control group were located in different areas of multidimensional space and were less diffused in comparison to cancer tissues. Three different classification algorithms showed recognition capability of 100% regarding classification of examined material into an appropriate group.
Conclusion
The application of the presented protocol and method enabled finding pathological changes in tissue material regardless of localization and size of abnormalities in the sample volume. Proteomic profile as a complex, rich in signals spectrum of proteins can be expressed as a single point in multidimensional space and than analysed using advanced statistical methods. This approach seems to provide more precise information about a pathology and may be considered in futer evaluation of biomarkers for clinical applications in different pathology. Multiparameter statistical methods may be helpful in elucidation of newly expressed sensitive biomarkers defined as many factors "in one point".
Background
Cancer diagnosis based on standard histological methods is widely described and used in medicine. However, most of the procedures derive from a subjective assessment of observed changes and in some cases may be inconclusive. In practical terms, new, objective methods in cancer diagnosis are still needed especially in respect of their sensitive. Moreover, tumour growth in its early stages is restricted to only a part of the tissue and in some cases it may be overlooked. Recently, proteomics has proved to be a valuable approach in biomarker detection and finding sensitive parameters which indicate disease process [1-4]. The combination of spectroscopic methods of high resolution (mass spectroscopy, nuclear magnetic resonance) with advanced statistical methods leads to an increased likelihood of developing new applications for diagnostic purposes [5-9]. Molecular imaging techniques described in literature as well as profiling and detailed characterisation of biomarker methods tend to prove their usefulness [10-12]. However, many protocols while focusing on details and theoretical aspects, do not translate directly into practical applications since a large number of samples need to be tested to validate them. Molecular imaging with mass spectrometry may offer ample possibilities in practical usage yet in the future rather than at present, because of extent of time necessary for analysis, workload of instruments, and poor image resolution in comparison to other methods [13-15].
Our work suggests an option of proteomic profiling of biological materials which focuses on obtaining spectra rich in proteins that may reveal the proteomic composition of tissue. Using multivariate statistical methods each complex spectrum can be shown as a single point in multidimensional space (as a single parameter), then analysed in respect of localization in this space. The single point in multidimensional space become a specific biomarker if its location does not match the control region. Even a slight change in tissue structure and the nature of pathology of any origin should be reflected in proteomic profile, provided that profiles richest in functional proteome of mass range of 3.5 - 20 kDa (without high abundant proteins - large proteins) are obtained. These profiles may correspond to a type of the pathological change (cancer, inflammation) as well as to normal profiles. The obtained profiles form something like a "proteomic fingerprint" and allow for classification of tested materials into appropriate groups using statistical methods. Tissue models have to be built for a known lung tissue structure (normal, changed: cancer, inflammation) using supervised classification methods. These methods indicate also signals typical for both normal and pathological tissue (table 1). Once appropriate models are calculated, any unknown lung tissue material can be measured and classified into defined groups (previously built models). The appearance of signals typical for a pathology results in classification of examined tissue into an appropriate model. This appears to be a simple protocol without complex, time consuming and expensive procedures which seems to be relevant considering a large number of samples needed for future models validation.
Table 1.
Peak statistics - comparison of statistically important (p value of T-test/ANOVA < 0.05) peaks for data separation and recognition capability (biomarkers candidates).
| Mass (Da) | Mean (Control) | Mean (Pathological) | P value T-Test/ANOVA | P value Wilcoxon/Kruskal-Wallis | Difference Average |
|---|---|---|---|---|---|
| 8616.14 | 0.32 | 1.45 | < 0.000001 | 0.00000847 | < 0.000001 |
| 6228.05 | 1.55 | 4.07 | < 0.000001 | 0.00152 | < 0.000001 |
| 5759.35 | 1.54 | 2.65 | < 0.000001 | 0.00017 | 0.000773 |
| 3588.09 | 1.99 | 4.82 | < 0.000001 | 0.00017 | < 0.000001 |
| 7596.85 | 0.81 | 2.11 | < 0.000001 | 0.00154 | < 0.000001 |
| 5201.26 | 1.93 | 3.87 | < 0.000001 | 0.000366 | < 0.000001 |
| 4156.39 | 2.08 | 4.87 | < 0.000001 | 0.00017 | 0.0000021 |
| 9554.61 | 0.78 | 1.99 | 0.00000228 | 0.11 | < 0.000001 |
| 12350.75 | 0.25 | 1.22 | 0.00000228 | 0.0000289 | < 0.000001 |
| 3603.83 | 2.11 | 4.11 | 0.0000024 | 0.0162 | < 0.000001 |
| 6723.40 | 20.93 | 8.96 | 0.00000228 | 0.00000855 | 0.000101 |
| 11297.49 | 0.45 | 0.97 | 0.00000228 | 0.00807 | < 0.000001 |
| 10268.09 | 0.59 | 1.56 | 0.00000341 | 0.000314 | < 0.000001 |
| 10525.25 | 0.3 | 1.19 | 0.00000695 | 0.00017 | < 0.000001 |
| 5800.39 | 1.79 | 3.82 | 0.00000122 | 0.195 | < 0.000001 |
| 5910.05 | 1.78 | 3.59 | 0.00000596 | 0.00375 | < 0.000001 |
| 10403.69 | 0.47 | 1.43 | 0.000225 | 0.000331 | < 0.000001 |
| 8297.66 | 0.95 | 1.62 | 0.000225 | 0.176 | < 0.000001 |
| 4570.52 | 4.96 | 7.49 | 0.000296 | 0.106 | < 0.000001 |
| 9440.24 | 0.71 | 1.08 | 0.000296 | 0.11 | < 0.000001 |
| 6656.68 | 6.27 | 2.07 | 0.000608 | 0.00000986 | < 0.000001 |
| 11325.26 | 0.67 | 1.09 | 0.00168 | 0.488 | < 0.000001 |
| 9172.09 | 34.55 | 12.13 | 0.00171 | 0.00017 | < 0.000001 |
| 7042.18 | 23.45 | 5.42 | 0.00252 | 0.0000902 | < 0.000001 |
| 6020.90 | 5.33 | 2.28 | 0.00252 | 0.00017 | < 0.000001 |
| 11185.69 | 0.75 | 1.19 | 0.00252 | 0.891 | < 0.000001 |
| 4995.04 | 2.28 | 3.87 | 0.00409 | 0.162 | < 0.000001 |
| 8456.35 | 1.25 | 2.82 | 0.00517 | 0.31 | < 0.000001 |
| 9960.78 | 0.78 | 1.82 | 0.0059 | 0.211 | < 0.000001 |
| 7283.97 | 39.59 | 7.2 | 0.00715 | 0.000151 | < 0.000001 |
| 7249.02 | 2.69 | 1.77 | 0.00757 | 0.0042 | 0.00064 |
| 7173.62 | 4.21 | 7.46 | 0.00935 | 0.647 | < 0.000001 |
| 5711.21 | 4.23 | 6.21 | 0.00935 | 0.441 | < 0.000001 |
| 9378.33 | 1.82 | 1 | 0.00935 | 0.00238 | < 0.000001 |
| 4048.24 | 4.11 | 7.16 | 0.0141 | 0.176 | < 0.000001 |
| 5837.80 | 2.19 | 3.04 | 0.0197 | 0.423 | < 0.000001 |
| 3907.34 | 41.93 | 14.71 | 0.021 | 0.00771 | < 0.000001 |
| 10840.50 | 0.99 | 2.24 | 0.0231 | 0.149 | 0 |
| 6745.30 | 2.08 | 1.56 | 0.0392 | 0.0221 | 0.00817 |
Methods
Materials
Segments of specimens collected for an intraoperative consultation were examined. Only lung cancer suspected samples were taken into consideration. Diagnosis was made basing on segments prepared with a standard criostatic technique followed by revision on the basis of paraffin method from specimens stained with hematoxilin and eozine (HE) and complemented with immunohistochemical method. Pathological changes in tissues were classified according to The Word Health Organization classification. Types of pathology are listed in table 2.
Table 2.
Types f pathological change diagnosed in examined material.
| Diagnosis | Number of cases |
|---|---|
| Squamous cell carcinoma | 25 |
| Adenocarcinoma | 27 |
| Large cell carcinoma | 4 |
| Typical carcinoid | 1 |
| Large cell neuroendocrine carcinoma | 3 |
| Small cell carcinoma | 2 |
| Adenosquamous carcinoma | 4 |
| Sarconiatoid carcinoma | 4 |
| Bening tumors | 2 |
| Metastasis | 2 |
| Lymphoma | 2 |
| Ectopic tissue and tumor-like lesions | 9 |
All the material for proteomic examination was collected from tissue fragments which did not show necrosis in volume in a macroscopic examination.
The control group (15 different cases) consisted of pathologically unchanged fragments of the lung tissue obtained from lungs removed during surgery on trauma cases. The material did not show any histological changes in histological examination that followed.
Methods
Mass spectra were recorded in a linear mode of 3.1 - 20 kDa with a high resolution mass spectrometer of time of flight type (MALDI-TOF/TOF) Bruker version Autoflex III Smartbeam with 200 Hz laser technology. Sinapic acid (SA) of proteomic grade (Bruker) was used as a matrix. 400 mg of lung tissue was directly frozen at -80°C. Next, just before measurement, the material was defrozen, homogenized with 0.7 ml 0.9% NaCl solution and centrifuged at 13,000 rpm for 10 minutes. The obtained supernatant of 0.6 ml was treated with 50μl of 10% trifluoracetic acid (TFA) and centrifuged again and then it was transferred to Amicon Ultracel (Millipore) centrifugal filter of 3 kDa selectivity and centifuged at 13,000 rpm to decrease the volume to 50 μl. The concentrated solution containing proteins and peptides was desalted by twice repeated centrifugation in the same conditions with 450 μl of deionized water. Concentrated and desalted material of 10 μl was transferred to a conical tube and gently mixed with 20 μl of saturated SA (SA in water with 0.1% TFA and acetonitryl with 0.1% TFA at a ratio of 60:40). The obtained mixture was directly applied onto standard steel target and analyzed following solvent evaporation.
The instrument operated under control of FlexControl software (Bruker).
All the statistical calculations were done using ClinProtTols ver. 2.2 software (Bruker). Proteomic profiles were analyzed with principle component analysis algorithm (PCA) to observe differences in proteome profiles of particular groups. Supervised classification algorithms, Support Vector Machine (SVM), Genetic Algorithm (GA) and Supervised Neural Network (SNN) were used for classification of tissue signals into appropriate groups.
Results
MS analysis in 100 (15 control, 85 pathological) tissue isolates was done. Results as combined spectra in gel view mode are shown in figure 1.
Figure 1.
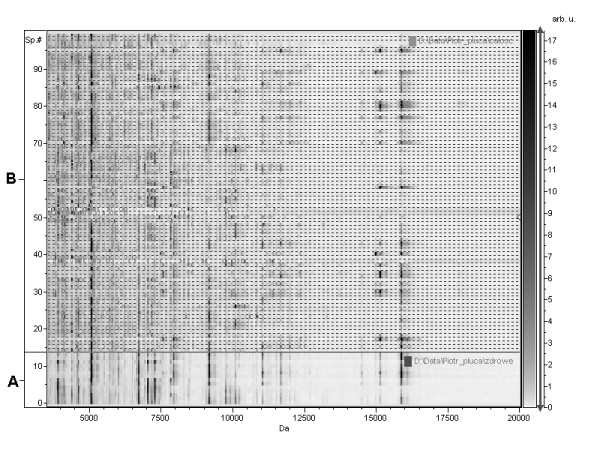
MS spectra in gel view mode: a) control group, b) pathologically changed tissues.
All the spectra were analyzed with PCA algorithm and resulting data distribution in three-dimensional space view is presented in figure 2. Rounded points are the control group profiles and are marked for reasons of clarity.
Figure 2.

The result of principle component analysis (PCA). Data distribution in three-dimensional space (PC1 - PC2- PC3). Points of control group were rounded.
Statistical analysis of all profiles for biomarker discovery revealed 39 masses of statistical importance (p < 0.005) which influenced division of investigated groups. Results are listed in table 1.
Proteome profiles were classified using three different supervised algorithms. Data for both (X1 - control, X2 - pathological) and recognition capability are listed in table 3.
Table 3.
Classification results.
| Algorithm | Validation | Recognition capability | ||
|---|---|---|---|---|
| XVal | X1 | X2 | ||
| Support Vector Machine (SVM) | 95.8% | 98.8% | 92.9% | 100% |
| Genetic Algorithm (GE) | 95.3% | 97.7% | 92.9% | 100% |
| Supervised Neural Network (SNN) | 88.7% | 98.8% | 78.6% | 100% |
Discussion
The proteomic investigation as a novel technique for biomarker discovery in disease process requires application of dedicated solutions including both advanced instruments and software. However, separation and concentration procedure are crucial for protein analysis [16]. Proteins of interest are a minority in comparison to a huge number of large structural proteins and other compounds which may negatively influence identification process especially in complex biological materials (blood, tissue) [17]. Moreover, mass spectroscopy with matrix associated laser desorption ionization, has its limitations: lack of highly abundant proteins in a sample, lack of salts, need for appropriate analytic concentrations, etc [18]. A number of protocols are described in literature for separation, concentration and desalting of proteins, however most of them are costly and time consuming [19-21]. We found that for peptides/proteins profiles purposes, considering the nature (composition - structure) of materials, it was enough to isolate only a limited range of proteins which could be presented as a single point in multidimensional space basic on statistical methods. A large number of mass signals recorded during measurement merge into one "signal" (a point located in multidimensional space) which can be treated as a specific biomarker. The obtained profiles (single points) become characteristic "fingerprints" for tissue composition. Any pathological changes in the examined material result in different localization in comparison to unchanged material what can be easy seen. Depending on the extent of pathological changes in tissue structure, proteins isolation procedure should include either more or fewer separation steps to obtain fractions which then can be successfully analyzed separately. In our approach we performed a simple isolation procedure based on precipitation of large proteins and then filtration using filters of cut-off level 3 kDa to concentrate and desalt peptides/proteins. This procedure allowed us to collect proteins in such a form that they can be measured directly in spectrometer without any expensive and time consuming procedures. As a detail examination of structure of particular peptides/proteins differentiating points distribution in multidimensional space was not our intent, we focused on reproducibility of this procedure. Our aim was to analyse profiles and compare them rather then to analyse a particular compound of which the profile was composed. Figure 1 presents spectra of all 100 samples, including control tissues (A) and pathologically changed (B) in gel view mode in order to combine all of them in one Figure 1. The data combination reveals that common signals may be noted in broad spectrum for both groups but pathological samples are even more complex in ranges between common bands. Signals in the same masses are most likely typical for the tissue origin (lung) and they reflect common to some extend structure for both types of the same tissues (pathological and normal). The multivariate statistical method enabled to distinguish normal samples from those diagnosed with cancer as well as those which indicate other pathological changes (table 2). As we were aware that signals typical for pathological tissue may overlap with signals coming from normal tissue which is present in material in every tumour growth in an unchanged form, we decided to examine a relatively large tissue segment of 400 mg, covering normal and pathological region. This facilitated testing specificity and sensitivity of the method with view to an early pathological change detection (in case the change cannot be seen macroscopically).
Recorded MS data typically comprise a large number of signals which makes it impossible to tell the difference between examined groups basing on direct raw MS spectra. However, the principle component analysis (PCA) algorithm enables to present complex spectrum in a single point located in multidimensional space. This location is strictly defined by sample composition, and the points representing spectra can be easily compared. The result of PCA analysis of all the samples shown in Figure 2 revealed different localization of normal and pathological samples. Moreover, pathological samples were more dispersed in space which could be explained by the fact that many different types of cancer were examined. We found 39 different masses (table 1) which are of statistical importance and discriminate all the data. The examination of compounds structure is a typical approach and has been widely described in references. We found that as much as 39 masses influence samples distribution in multidimensional space and all of them may be important for change identification. A typical approach based on a single, one-parametrical statistical investigation seems to be less efficient compared to multiparametrical methods especially in respect of sensitivity and selectivity. Also we attempted to point out that there was no need to precisely define (protein structure identification) each of 39 signals because all of them are strictly defined by the point located in multidimensional space (non - dimensional data x, y, z). Points obtained from PCA calculations can be then classified and compared using different classification algorithms (table 3). In our experiment the recognition capability of the tree most common models was 100% which may demonstrate a high specificity of the procedure.
Conclusions
The proteomic investigation of tissue samples using advanced statistical methods enabled differentiation of lung tissue with cancer and unchanged tissue material even in an early stage of growth (low pathologcial to unchanged tissue ratio in sample). Following ist validation, this method, supporting commonly used histological techniques, may be considered an objective, precise and fast procedure in an early cancer diagnosis. Our protocol may be useful in procedures that need a large number of samples due to the elimination of time consuming steps. Multiparameter statistical methods may be helpful in elaboration of newly defined biomarkers for clinical applications expressed as many factors "in one point".
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PW collected and registered all the samples, performed histological examinations, participated in the design of the study, MS measurements and data analysis, participated in data analysis and interpretation. TJ conceived of the study, developed the methodology, carried out MS measurements, performed the data analysis and interpretation, drafted the manuscript. JA and TG collected segments of specimens and participated in clinical diagnosis. KB participated in design of the study and its coordination. All authors read and approved the final manuscript.
Contributor Information
Piotr Waloszczyk, Email: piotrwaloszczyk@wp.pl.
Tomasz Janus, Email: tjanus@sci.pam.szczecin.pl.
Jacek Alchimowicz, Email: alchjacek@wp.pl.
Tomasz Grodzki, Email: grodzki_t@wp.pl.
Krzysztof Borowiak, Email: boroks@sci.pam.szczecin.pl.
References
- Palmblad M, Tiss A, Cramer R. Mass spectrometry in clinical proteomics - from the present to the future. Proteomics Clin Appl. 2009;3:6–17. doi: 10.1002/prca.200800090. [DOI] [PubMed] [Google Scholar]
- Munro NP, Cairns DA, Clarke P, Rogers M, Stanley AJ, Barrett JH, Harnden P, Thompson D, Eardley I, Banks RE, Knowles MA. Urinary biomarker profiling in transitional cell carcinoma. Int J Cancer. 2006;119:2642–2650. doi: 10.1002/ijc.22238. [DOI] [PubMed] [Google Scholar]
- Coombes KR, Morris JS, Hu J, Edmonson SR, Baggerly KA. Serum proteomics profiling - a young technology begins to mature. Nature Biotechnol. 2005;23:291–292. doi: 10.1038/nbt0305-291. [DOI] [PubMed] [Google Scholar]
- Check E. Proteomics and cancer: running before we can walk. Nature. 2004;429:496–497. doi: 10.1038/429496a. [DOI] [PubMed] [Google Scholar]
- Zhang X, Leung SM, Morris CR, Shigenaga MK. Evaluation of a novel, integrated approach using functionalized magnetic beads, bench-top MALDI-TOF-MS with prestructured sample supports, and pattern recognition software for profiling potential biomarkers in human plasma. J Biomol Technol. 2004;15:167–175. [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang Z, Rosenzweig J, Wang YY, Chan DW. Proteomics and bioinformatics approaches for identification of serum biomarkers to detect breast cancer. Clin Chem. 2002;48:1296–1304. [PubMed] [Google Scholar]
- Petricoin EF, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM, Mills GB, Simone C, Fishman DA, Kohn EC, Liotta LA. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359:572. doi: 10.1016/S0140-6736(02)07746-2. [DOI] [PubMed] [Google Scholar]
- Conrads TP, Zhou M, Petricoin EF, Liotta LA, Veenstra TD. Cancer diagnosis using proteomic patterns. Expert Rev Mol Diagn. 2003;3:411. doi: 10.1586/14737159.3.4.411. [DOI] [PubMed] [Google Scholar]
- Aebersold R, Goodlett DR. Mass spectrometry in proteomics. Chem Rev. 2001;101:269–295. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- Stoeckli M, Staab D, Schweitzer A. Compound and metabolite distribution measured by MALDI mass spectrometric imaging in whole-body tissue sections. Int J Mass Spectrom. 2007;260:195–202. doi: 10.1016/j.ijms.2006.10.007. [DOI] [Google Scholar]
- Andersson M, Groseclose MR, Deutch AY, Caprioli RM. Imaging mass spectrometry of proteins and peptides: 3 D volume reconstruction. Nat Methods. 2008;5:101–108. doi: 10.1038/nmeth1145. [DOI] [PubMed] [Google Scholar]
- Caldwell RL, Caprioli RM. Tissue profiling by mass spectrometry: A review of methodology and applications. Mol Cell Proteomics. 2005;4:394–401. doi: 10.1074/mcp.R500006-MCP200. [DOI] [PubMed] [Google Scholar]
- Chaurand P, Norris JL, Cornett DS, Mobley JA, Caprioli RM. New developments in profiling and imaging of proteins from tissue sections by MALDI mass spectrometry. J Proteome Res. 2006;5:2889–2900. doi: 10.1021/pr060346u. [DOI] [PubMed] [Google Scholar]
- Cornett DS, Reyzer ML, Chaurand P, Caprioli RM. MALDI imaging mass spectrometry: Molecular snapshots of biochemical systems. Na Methods. 2007;4:828–833. doi: 10.1038/nmeth1094. [DOI] [PubMed] [Google Scholar]
- Khatib-Shahidi S, Andersson M, Herman JL, Gillespie TA, Caprioli RM. Direct molecular analysis of whole-body animal tissue sections by imaging MALDI mass spectrometry. Anal Chem. 2006;78:6448–6456. doi: 10.1021/ac060788p. [DOI] [PubMed] [Google Scholar]
- Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat Biotechnol. 2006;24:971–983. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- Duncan R, McConkey EH. How many proteins are there in a typical mammalian cell? Clin Chem. 1982;28:749–755. [PubMed] [Google Scholar]
- Diamandis EP. Mass spectrometry as a diagnostic and a cancer biomarker discovery tool: opportunities and potential limitations. Mol Cell Proteomics. 2004;3:367–378. doi: 10.1074/mcp.R400007-MCP200. [DOI] [PubMed] [Google Scholar]
- Omenn GS. Strategies for plasma proteomic profiling of cancers. Proteomics. 2006;6:5662–5673. doi: 10.1002/pmic.200600331. [DOI] [PubMed] [Google Scholar]
- Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat Biotechnol. 2006;24:971–983. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- Görg A, Weiss W, Dunn MJ. Current two-dimensional electrophoresis technology for proteomics. Proteomics. 2004;4:3665–3685. doi: 10.1002/pmic.200401031. [DOI] [PubMed] [Google Scholar]